
Figure 5.
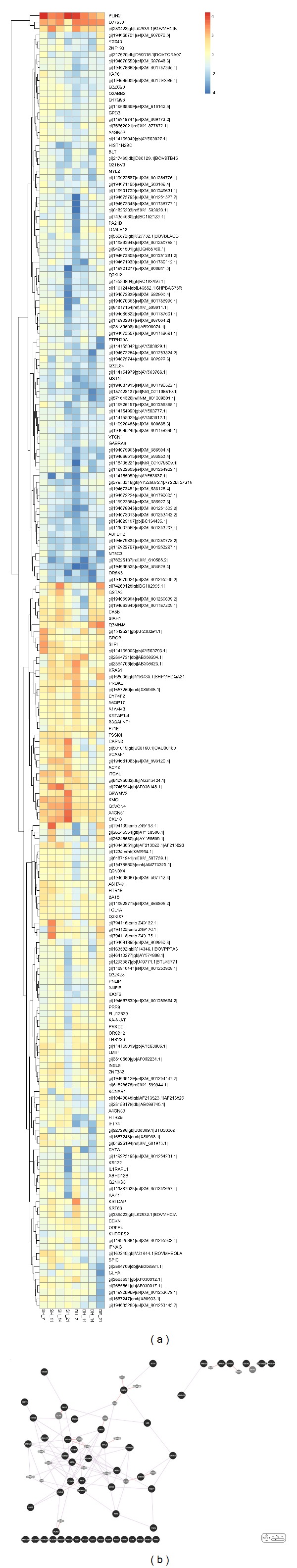
(a) Coexpression patterns from gene clustering in a sheep model data. 200 HMM-specific TDE genes are represented in heatmap. Each row contains a vector of time series expression profile in log2 scale; consequently the visualization in heatmap is originally made up of major three groups, high, moderate, and low expression levels with genens that are not detected by static methods but detected by HMM, of which we selected the most statistically significant 200 genes to present this heatmap. Interestingly, some genes at low expression levels were obviously differentially expressed at log2-scaled FC ~4 up to 5 and even some genes that significantly show temporal patterns at high expression levels were also detected, yet those genes were not detected by existing static methods suggesting that HMM method reassuringly has higher sensitivity and robustness than other existing static methods in identification of differential expression regardless of expression levels. (b) Gene functional pathway and network analysis with 528 HMM specific TDE genes in a sheep model data. To explore biological functions in this gene set further, whether or not those are genuinely differential expression or random noise by chance in terms of biological insights, gene ontology (GO) and KEGG pathway analysis were performed to identify meaningful functionalities and some meaningful functions related to developmental process (intermediate mesoderm formation, regulation of cell growth involved in regulation of muscle adaptation, intermediate mesoderm formation, etc.) and gender specific terms (granulosa cell development and maternal placenta development) are detected as we anticipated to confirm the sensitivity of dynamic HMM method. The purple and pink legends represent coexpression and physical interactions across genes, respectively, and black nodes are query genes in networks.