

Abstract
Objective
We aimed to test the fetal overnutrition hypothesis by comparing the associations of maternal and paternal adiposity (sum of skinfolds) with adiposity and cardiovascular risk factors in children.
Design
Children from a prospective birth cohort had anthropometry, fat percentage (bio-impedance), plasma glucose, insulin and lipid concentrations and blood pressure measured at 9·5 years of age. Detailed anthropometric measurements were recorded for mothers (at 30 ± 2 weeks’ gestation) and fathers (5 years following the index pregnancy).
Setting
Holdsworth Memorial Hospital, Mysore, India.
Subjects
Children (n 504), born to mothers with normal glucose tolerance during pregnancy.
Results
Twenty-eight per cent of mothers and 38 % of fathers were overweight/obese (BMI ≥ 25·0 kg/m2), but only 4 % of the children were overweight/obese (WHO age- and sex-specific BMI ≥ 18·2 kg/m2). The children's adiposity (BMI, sum of skinfolds, fat percentage and waist circumference), fasting insulin concentration and insulin resistance increased with increasing maternal and paternal sum of skinfolds adjusted for the child's sex, age and socio-economic status. Maternal and paternal effects were similar. The associations with fasting insulin and insulin resistance were attenuated after adjusting for the child's current adiposity.
Conclusions
In this population, both maternal and paternal adiposity equally predict adiposity and insulin resistance in the children. This suggests that shared family environment and lifestyle, or genetic/epigenetic factors, influence child adiposity. Our findings do not support the hypothesis that there is an intra-uterine overnutrition effect of maternal adiposity in non-diabetic pregnancies, although we cannot rule out such an effect in cases of extreme maternal obesity, which is rare in our population.
Keywords: Adiposity, Cardiovascular risk factors, Children, India, Insulin resistance, Intergeneration, Maternal and paternal effects
Excessive adiposity leading to overweight or obesity is an increasing problem in both developed and developing countries( 1 ). Childhood overweight/obesity is associated with long-term adverse health consequences because of tracking of obesity to adult life( 2 ) and is thought to contribute to the rising burden of chronic diseases like CVD and diabetes( 3 ).
It is well established that adiposity in children is related to parental adiposity. This association could reflect genetic effects and/or shared family lifestyle factors such as diet and physical activity patterns( 4 ). There has been recent interest in the fetal overnutrition hypothesis, which proposes that maternal obesity may contribute to increased offspring adiposity through intra-uterine exposure to excess fuels (for example glucose and lipids)( 5 – 9 ). Maternal gestational diabetes mellitus (GDM) is associated with an increased risk of obesity in children( 10 ) and this is thought to occur because of materno-fetal transfer of excess glucose and other fuels during pregnancy. If similar effects occur with maternal obesity in the absence of frank GDM, the prevention of maternal obesity before pregnancy could be an important public health target.
The fetal overnutrition hypothesis can be tested indirectly by comparing the relative effects of mothers’ and fathers’ adiposity on offspring adiposity. Assuming that genetic effects and shared lifestyle effects act equally through both parents, these would predict similar effects of maternal and paternal adiposity. The fetal overnutrition hypothesis would predict a stronger maternal than paternal effect( 11 , 12 ). Stronger maternal than paternal effects could also arise because of the fact that mothers tend to have a greater role in the feeding and general care of children. Studies examining childhood adiposity in relation to the parents’ adiposity have come mainly from high-income countries, have been limited to BMI or weight as the measure of adiposity, and have reported conflicting findings( 6 , 7 , 11 – 20 ). Some found a greater influence of the mother's than the father's BMI on offspring BMI( 7 , 11 – 13 ) or fat mass( 14 ), while others have reported that both predict offspring BMI( 6 , 15 – 20 ) or fat mass equally( 19 ). Only a few studies have reported associations between maternal adiposity and childhood CVD risk factors such as blood pressure( 8 , 21 , 22 ), lipid profile( 23 ) and insulin resistance( 24 ). Studies examining associations between paternal adiposity and CVD risk factors in children are scarce.
Although Indians are not an obese population in BMI terms, they are known to have a high body fat percentage relative to their BMI( 25 ). Therefore examining the associations between parental adiposity and offspring adiposity in this population may be helpful in understanding the factors influencing the increasing prevalence of CVD and its risk factors in this population. The Mysore Parthenon study in south India( 26 ), a prospective birth cohort study, enabled us to investigate offspring adiposity and cardiovascular risk factors in relation to both maternal and paternal adiposity. That study has collected direct measures of body fat (skinfolds) in both parents and the children, as well as body fat percentage (fat %) measured by bio-impedance, blood pressure and plasma glucose, insulin and lipids in the children. We compared the strength of associations of maternal and paternal adiposity with these measurements in the children.
Methods
The Mysore Parthenon study (Fig. 1) has been described earlier( 26 ). In brief, during 1997–1998, 830 women booking into the antenatal clinic at the Holdsworth Memorial Hospital (HMH), Mysore, India and satisfying the eligibility criteria (no known history of diabetes, singleton pregnancy of <32 weeks’ gestation and planning to deliver at HMH) participated in the study. A total of 663 women delivered live, normal babies at HMH. All available children (n 630), excluding twenty-five who died and eight with medical conditions, were measured annually until the age of 5 years and every 6 months thereafter. At 9·5 years, excluding ninety-one (fifty-six refused, twenty-six had moved away, nine were untraceable), 539 children participated in the study. The HMH research ethics committee approved the study and informed written consent was obtained from the parents and assent from the children.
Fig. 1.

Flow diagram of the study cohort included for analysis (HMH, Holdsworth Memorial Hospital; OGTT, oral glucose tolerance test; GDM, gestational diabetes mellitus)
Detailed maternal anthropometry including height, weight and skinfold thicknesses (triceps, biceps, subscapular and suprailiac) were measured at 30 ± 2 weeks’ gestation by one of two trained observers using standardized methods. Height was measured to the nearest 0·1 cm using a Harpenden stadiometer (CMS Instruments, London, UK); weight was measured to the nearest 500 g using a Seca weighing scale (Seca, Hamburg, Germany); skinfold thicknesses were measured to the nearest 0·1 mm using Harpenden callipers (CMS Instruments; average of three readings). In inter-observer variation studies (carried out 6-monthly during the study), the ratio of the standard deviation of between-observer differences to the standard deviation of between-subject differences was 0·05, 0·04 and 0·05 for maternal height, weight and sum of skinfolds, respectively. For fathers and also for mothers, similar anthropometric data were collected at the child's 5th year follow-up. Maternal measurements in pregnancy, a preferable measure for testing the fetal overnutrition hypothesis, were used for the main analysis.
At 9·5 years children were measured by one of five trained observers. Weight was measured to the nearest 100 g using digital scales (Salter, Tonbridge, UK). Height was measured to the nearest 0·1 cm using a wall-mounted stadiometer (Microtoise; CMS Instruments). Waist circumference (WC; midpoint between the lower border of the last rib and the iliac crest in the mid-axillary line) was measured to the nearest 0·1 cm using an anthropometric tape. Triceps and subscapular skinfold thicknesses were measured to the nearest 0·1 mm using Harpenden callipers (CMS Instruments). In inter-observer variation studies (carried out twice during the study), the ratio of the standard deviation of between-observer differences to the standard deviation of between-subject differences was 0·02, 0·02, 0·33, 0·24 and 0·05 for weight, height, WC, triceps and subscapular skinfold thickness measurements, respectively. Fat % was measured by bio-impedance (Bodystat QuadScan 4000; Isle of Mann, UK). Blood pressure, systolic (SBP) and diastolic (DBP), was measured in the left arm using a cuff size appropriate for the mid-upper-arm circumference and after the child had been seated at rest for at least 5 min (Dinamap, Criticon, FL, USA). After an overnight fast, blood samples were collected fasting and 120 min post glucose load (1·75 g/kg body weight). Plasma glucose concentrations and fasting total cholesterol (TC), TAG and HDL-cholesterol (HDL-C) concentrations were measured by standard enzymatic methods (Alcyon 3000 AutoAnalyzer; Abbott Laboratories, Abbott Park, IL, USA). Inter-assay CV were <5 % for all. LDL-cholesterol (LDL-C) was calculated using the formula:
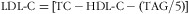 (
27
). Insulin was measured using a time-resolved fluoroimmunoassay (Delfia; Wallac QY, Turku, Finland). Intra-assay and inter-assay CV were 12·5 % at <45 pmol/l and <10 % at >45 pmol/l. Insulin resistance was estimated using the homeostasis model assessment equation (HOMA-IR)(
28
). According to WHO age- and sex-specific growth standards, 9·5-year-old children are considered overweight if BMI is >18·2 and <20·9 kg/m2 for boys or >18·7 and <22·0 kg/m2 for girls, and obese if BMI is ≥20·9 kg/m2 for boys and ≥22·0 kg/m2 for girls(
29
). Socio-economic status (SES) was assessed using the Standard of Living Index designed by the National Family Health Survey-2(
30
). It is a standard questionnaire based on type and size of the house, household sanitary facilities, source of water and power supply, cooking fuel used, ownership of house/property, land and livestock and household assets. A score was assigned to each question and finally the scores from each question were summed to obtain the final score. None of the mothers had ever smoked.
(
27
). Insulin was measured using a time-resolved fluoroimmunoassay (Delfia; Wallac QY, Turku, Finland). Intra-assay and inter-assay CV were 12·5 % at <45 pmol/l and <10 % at >45 pmol/l. Insulin resistance was estimated using the homeostasis model assessment equation (HOMA-IR)(
28
). According to WHO age- and sex-specific growth standards, 9·5-year-old children are considered overweight if BMI is >18·2 and <20·9 kg/m2 for boys or >18·7 and <22·0 kg/m2 for girls, and obese if BMI is ≥20·9 kg/m2 for boys and ≥22·0 kg/m2 for girls(
29
). Socio-economic status (SES) was assessed using the Standard of Living Index designed by the National Family Health Survey-2(
30
). It is a standard questionnaire based on type and size of the house, household sanitary facilities, source of water and power supply, cooking fuel used, ownership of house/property, land and livestock and household assets. A score was assigned to each question and finally the scores from each question were summed to obtain the final score. None of the mothers had ever smoked.
Statistical methods
Excluding thirty-five children born to GDM mothers, data were analysed for 504 children (504 mother–offspring pairs; 441 father–offspring pairs; Fig. 1). Variables with skewed distributions were log-transformed (maternal BMI, parents’ and children's skinfold thicknesses, children's fasting insulin and TAG concentrations and HOMA-IR). Exposures were maternal and paternal adiposity measures (sum of skinfolds (SS)) and overweight/obesity (BMI ≥ 25·0 kg/m2). BMI was also used as a continuous variable. Outcomes were children's adiposity measures (BMI, SS, fat % and WC) and CVD risk factors (fasting and 120 min post-load glucose, fasting insulin, TAG, HDL-C and LDL-C concentrations, HOMA-IR and SBP). Associations of the parents’ adiposity with offspring adiposity and CVD risk factors were examined by multiple linear regression analysis in a series of models. Model 1 was unadjusted; model 2 was adjusted for covariates (the child's sex and age, SES, maternal age, parity and breast-feeding duration); model 3 included model 2 parameters and both maternal and paternal adiposity measures; model 4 included model 2 parameters, plus maternal glucose concentrations during pregnancy (only for maternal effects); and model 5 included model 2 parameters plus the child's current SS as a potential mediating factor (for CVD risk markers only). To facilitate interpretation, internally standardized Z-scores of parents’ SS and BMI were used in regression models. We compared regression coefficients for maternal and paternal effects by comparing the difference to its standard error using Z tests. Differences in parental associations with adiposity between boys and girls were tested using interaction tests. A P value of <0·05 was considered significant. Data were analysed using the STATA statistical software package version 10.
Power calculation
To assess the difference in the effects of maternal and paternal adiposity (exposures, SS and BMI) on child adiposity and risk factors (outcomes) we used regression analysis, with standardized versions of the exposures and outcomes, and adjusted for the child's age, sex and the mother's age, parity and SES. In our study (n 441) the standard error of the difference between the regression coefficients for mother's and father's adiposity was 0·07 sd per sd. Therefore, using a test at the 5 % level of statistical significance, our study had 80 % power to detect a difference between these regression coefficients of 0·20 sd or more in the child's adiposity or risk factors per sd of parental adiposity.
Results
Characteristics of the parents and the children are shown in Table 1. During pregnancy, 120 (23·8 %) mothers were overweight (BMI ≥ 25·0 and <30·0 kg/m2), twenty-four (4·8 %) were obese (BMI ≥ 30·0 kg/m2) and none (0 %) were extremely obese (BMI ≥ 40·0 kg/m2). Corresponding figures for mothers and fathers 5 years following the index pregnancy were 123 (25·7 %), thirty-six (7·5 %) and zero (0 %) and 149 (33·8 %), twenty (4·5 %) and zero (0 %), respectively. Among the children (244 boys and 260 girls), ten (3·9 %) girls were overweight and one (0·4 %) was obese. Corresponding figures for boys were seven (2·9 %) and three (1·2 %), respectively. There was a positive correlation (r = 0·2–0·4) between the mother's and the father's height, weight, BMI and all of the skinfold thicknesses. Maternal age at pregnancy was positively correlated with maternal BMI, SS, child's SS, fat %, WC, fasting insulin and HOMA-IR, and negatively correlated with TAG concentrations (r = 0·1–0·2; P < 0·05 for all). Maternal parity was positively associated with maternal BMI (r = 0·2, P = 0·0001).
Table 1.
Descriptive statistics (demographic characteristics, anthropometric measures and cardiovascular risk markers) of the study participants, the Mysore Parthenon study
| n | Mean, n or Median | sd, % or IQR | |
|---|---|---|---|
| Mother's characteristics during pregnancy | |||
| Age (years) | 504 | 23·5 | 4·0 |
| Parity (n, %) | 504 | ||
| 0 | 263 | 52·2 | |
| 1 | 159 | 31·6 | |
| 2+ | 82 | 16·2 | |
| Weight (kg)† | 504 | 55·0 | 49·5, 61·3 |
| Height (cm) | 504 | 154·3 | 5·4 |
| BMI (kg/m2)† | 504 | 23·0 | 20·7, 25·6 |
| Triceps skinfold thickness (mm)† | 504 | 16·7 | 12·1, 23·2 |
| Biceps skinfold thickness (mm)† | 504 | 8·6 | 6·3, 12·4 |
| Subscapular skinfold thickness (mm)† | 504 | 24·3 | 17·6, 31·6 |
| Suprailiac skinfold thickness (mm)† | 504 | 31·2 | 22·6, 41·4 |
| SS (mm)† | 504 | 82·1 | 60·4, 107·1 |
| Glucose area under the curve (mmol/l) | 479 | 1108·7 | 154·4 |
| Mother's characteristics at 5-year follow-up | |||
| Weight (kg) | 478 | 55·6 | 10·8 |
| Height (cm) | 478 | 154·4 | 5·3 |
| BMI (kg/m2) | 478 | 23·3 | 4·4 |
| Triceps skinfold thickness (mm) | 478 | 22·5 | 9·6 |
| Biceps skinfold thickness (mm)† | 478 | 12·4 | 7·3, 19·5 |
| Subscapular skinfold thickness (mm) | 469 | 30·4 | 12·5 |
| Suprailiac skinfold thickness (mm) | 475 | 27·7 | 14·4 |
| SS (mm) | 469 | 93·9 | 39·8 |
| Father's characteristics at 5-year follow-up | |||
| Weight (kg) | 441 | 66·9 | 11·3 |
| Height (cm) | 441 | 167·6 | 6·1 |
| BMI (kg/m2) | 441 | 23·8 | 3·7 |
| Triceps skinfold thickness (mm)† | 441 | 12·5 | 8·9, 15·9 |
| Biceps skinfold thickness (mm)† | 441 | 6·8 | 5·0, 9·5 |
| Subscapular skinfold thickness (mm)† | 436 | 24·2 | 17·3, 32·8 |
| Suprailiac skinfold thickness (mm) | 437 | 24·0 | 10·5 |
| SS (mm) | 435 | 70·2 | 27·0 |
| Children's characteristics at 9·5 years | |||
| Age (years) | 504 | 9·4 | 0·1 |
| Breast-feeding duration (months) | 485 | 12·2 | 5·2 |
| Height (cm) | 504 | 130·7 | 5·7 |
| BMI (kg/m2) | 504 | 14·5 | 1·9 |
| Triceps skinfold thickness (mm)† | 504 | 9·3 | 7·6, 11·4 |
| Subscapular skinfold thickness (mm)† | 504 | 7·1 | 5·7, 9·1 |
| SS (mm)† | 504 | 16·3 | 13·4, 20·5 |
| Fat % | 504 | 27·3 | 6·5 |
| WC (cm) | 503 | 54·2 | 5·5 |
| Glucose0 (mmol/l) | 498 | 4·7 | 0·4 |
| Glucose120 (mmol/l) | 476 | 5·1 | 0·9 |
| Insulin0 (pmol/l)† | 498 | 22·5 | 14·4, 33·0 |
| HOMA-IR† | 498 | 0·8 | 0·5, 1·2 |
| TAG (mmol/l)† | 498 | 0·8 | 0·7, 1·1 |
| HDL-C (mmol/l) | 498 | 1·1 | 0·2 |
| LDL-C (mmol/l) | 498 | 2·3 | 0·5 |
| SBP (mmHg) | 504 | 101·0 | 8·5 |
| SES (score) | 504 | 34 | 29, 38 |
IQR, interquartile range; SS, sum of skinfolds; fat %, body fat percentage; WC, waist circumference; glucose0, fasting glucose concentration; glucose120, 120 min post-load glucose concentration; insulin0, fasting insulin concentration; HOMA-IR, homeostasis model assessment–insulin resistance; HDL-C, HDL-cholesterol; LDL-C, LDL-cholesterol; SBP, systolic blood pressure; SES, socio-economic status.
†Transformed variable.
Maternal age, SS and SES were higher among those children who were not studied (n 91) compared with those who took part in the study (n 504; maternal age (mean 24·6 v. 23·5 years; P < 0·03), maternal SS (95·1 v. 86·4 mm; P = 0·02), SES (37·02 v. 33·70; P < 0·0001)). Maternal BMI and the children's gender ratio and birth weight were similar among those who were not studied compared with those who took part (maternal BMI (23·9 v. 23·3 kg/m2; P = 0·2), gender (50 % boys v. 48 % boys; P = 0·9) and birth weight (2·899 v. 2·842 kg; P = 0·3)).
The children's current adiposity measures (BMI, SS, fat % and WC) were positively correlated with their CVD risk factors (negatively correlated with HDL-C; r = 0·1–0·4). The strongest associations were seen with SS. There were no associations between maternal glucose concentrations (0, 30, 60, 120 and 180 min post load) and the children's adiposity or CVD risk factors (data not shown).
Relationships of maternal and paternal adiposity with offspring adiposity
Higher maternal and paternal SS were associated with higher BMI, SS, fat % and WC in the children (Table 2), and this appeared to hold true across the full range of parental SS (Fig. 2). A 1 sd increase in parental adiposity was associated with approximately a 25 % sd increase in child adiposity. The results were similar after adjusting for maternal age and parity, and the child's sex, age, breast-feeding duration and SES (model 2); when maternal and paternal SS were mutually adjusted (model 3); and additionally adjusted for maternal glucose concentrations in pregnancy (only for maternal effects, model 4; Table 2). Maternal effects were stronger than paternal effects; however, Z tests indicated no significant differences between maternal and paternal effects, except on the children's fat % where maternal effects were stronger than paternal effects (P for difference is 0·02).
Table 2.
Associations of maternal and paternal adiposity (sum of skinfolds) with offspring adiposity and cardiovascular risk factors, the Mysore Parthenon study
| Model 1 | Model 2 | Model 3 | Model 4 | Model 5 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Children's adiposity and cardiovascular risk factors | β† | 95 % CI | β† | 95 % CI | β† | 95 % CI | β† | 95 % CI | β† | 95 % CI | P‡ |
| Maternal SS | |||||||||||
| BMI (kg/m2) | 0·52*** | 0·36, 0·68 | 0·49*** | 0·32, 0·65 | 0·36*** | 0·20, 0·53 | 0·50*** | 0·33, 0·68 | 0·3 | ||
| SS (mm)§ | 0·10*** | 0·07, 0·13 | 0·09*** | 0·06, 0·11 | 0·07*** | 0·04, 0·10 | 0·09*** | 0·06, 0·12 | 0·6 | ||
| Fat % | 1·65*** | 1·09, 2·22 | 1·35*** | 0·83, 1·87 | 1·28*** | 0·73, 1·82 | 1·37*** | 0·82, 1·91 | 0·02 | ||
| WC (cm) | 1·35*** | 0·88, 1·83 | 1·20*** | 0·71, 1·69 | 0·81** | 0·31, 1·31 | 1·23*** | 0·72, 1·75 | 0·9 | ||
| Glucose0 (mmol/l) | −0·02 | −0·05, 0·02 | −0·02 | −0·05, 0·02 | −0·03 | −0·06, 0·01 | −0·01 | −0·05, 0·03 | −0·03 | −0·07, 0·01 | 0·045 |
| Glucose120 (mmol/l) | −0·03 | −0·11, 0·05 | −0·03 | −0·12, 0·05 | −0·05 | −0·15, 0·04 | −0·05 | −0·14, 0·03 | −0·07 | −0·16, 0·02 | 0·054 |
| Insulin0 (pmol/l)§ | 0·08** | 0·03, 0·13 | 0·06* | 0·01, 0·11 | 0·04 | −0·02, 0·10 | 0·06* | 0·01, 0·12 | 0·02 | −0·04, 0·07 | 0·4 |
| HOMA-IR§ | 0·08** | 0·02, 0·13 | 0·06* | 0·002, 0·11 | 0·04 | −0·02, 0·09 | 0·06* | 0·005, 0·12 | 0·01 | −0·05, 0·06 | 0·3 |
| TAG (mmol/l)§ | −0·04* | −0·07, −0·004 | −0·04* | −0·07, −0·01 | −0·03 | −0·07, 0·002 | −0·05* | −0·08, −0·01 | −0·05** | −0·09, −0·02 | 0·3 |
| HDL-C (mmol/l) | 0·003 | −0·02, 0·02 | 0·004 | −0·02, 0·02 | 0·004 | −0·02, 0·03 | 0·003 | −0·02, 0·02 | 0·007 | −0·01, 0·03 | 0·7 |
| LDL-C (mmol/l) | −0·005 | −0·05, 0·04 | −0·007 | −0·06, 0·04 | −0·001 | −0·05, 0·05 | −0·004 | −0·06, 0·05 | −0·02 | −0·07, 0·03 | 0·2 |
| SBP (mmHg) | 0·53 | −0·23, 1·28 | 0·67 | −0·11, 1·46 | 0·46 | −0·39, 1·31 | 0·65 | −0·17, 1·47 | −0·27 | −1·02, 0·49 | 0·9 |
| Paternal SS | |||||||||||
| BMI (kg/m2) | 0·38*** | 0·22, 0·54 | 0·30** | 0·13, 0·47 | 0·23** | 0·06, 0·40 | |||||
| SS (mm)§ | 0·08*** | 0·05, 0·11 | 0·07*** | 0·04, 0·10 | 0·06*** | 0·03, 0·08 | |||||
| Fat % | 0·74* | 0·15, 1·34 | 0·52 | −0·02, 1·06 | 0·28 | −0·26, 0·83 | |||||
| WC (cm) | 1·26*** | 0·78, 1·73 | 1·00*** | 0·46, 1·45 | 0·81** | 0·30, 1·31 | |||||
| Glucose0 (mmol/l) | 0·03 | −0·01, 0·06 | 0·03 | −0·01, 0·07 | 0·04 | −0·003, 0·08 | 0·02 | −0·02, 0·06 | |||
| Glucose120 (mmol/l) | 0·08 | −0·01, 0·16 | 0·07 | −0·02, 0·16 | 0·08 | −0·01, 0·18 | 0·06 | −0·03, 0·15 | |||
| Insulin0 (pmol/l)§ | 0·09** | 0·04, 0·15 | 0·08** | 0·03, 0·14 | 0·08** | 0·02, 0·13 | 0·05 | −0·002, 0·11 | |||
| HOMA-IR§ | 0·10** | 0·04, 0·15 | 0·09** | 0·03, 0·15 | 0· 08** | 0·02, 0·14 | 0·06 | −0·0005, 0·12 | |||
| TAG (mmol/l)§ | −0·02 | −0·05, 0·02 | −0·01 | −0·05, 0·02 | −0·01 | −0·04, 0·03 | −0·02 | −0·05, 0·02 | |||
| HDL-C (mmol/l) | −0·001 | −0·02, 0·02 | −0·002 | −0·02, 0·02 | −0·003 | −0·02, 0·02 | −0·0002 | −0·02, 0·02 | |||
| LDL-C (mmol/l) | −0·05 | −0·10, 0·001 | −0·06* | −0·11, −0·003 | −0·06* | −0·11, −0·001 | −0·06* | −0·11, −0·01 | |||
| SBP (mmHg) | 0·46 | −0·33, 1·25 | 0·49 | −0·34, 1·33 | 0·41 | −0·44, 1·26 | −0·24 | −1·04, 0·56 | |||
SS, sum of skinfolds; fat %, body fat percentage; WC, waist circumference; glucose0, fasting glucose concentration; glucose120, 120 min post-load glucose concentration; insulin0, fasting insulin concentration; HOMA-IR, homeostasis model assessment–insulin resistance; HDL-C, HDL-cholesterol; LDL-C, LDL-cholesterol; SBP, systolic blood pressure; SES, socio-economic status.
*P < 0·05; **P < 0·01; ***P < 0·001.
†β is the effect size derived using maternal or paternal SS as a continuous variable (standardized) in multiple linear regression analysis. For logged variables, the effect size indicates the percentage change in the outcome per unit change in the predictor. Model 1: unadjusted; model 2: adjusted for the child's sex, age, breast-feeding duration, and maternal age, parity and SES; model 3: model 2+maternal and paternal SS mutually adjusted; model 4: model 2+maternal glucose area under the curve at pregnancy; model 5: model 2+the child's SS at 9·5 years.
‡P value for difference between maternal and paternal effects derived using the Z test (model 3).
§Logged variables.
Fig. 2.

Association of maternal (—●—) and paternal (– –●– –) sum of skinfolds and BMI with child's sum of skinfolds (SS), the Mysore Parthenon study. Values are means with 95 % confidence intervals represented by vertical bars
The results were similar when we used maternal and paternal overweight or obesity (BMI ≥ 25·0 kg/m2) as the exposure measure (Table 3) and also when BMI was used as a continuous variable (Supplementary Materials; Fig. 2). There was no significant difference between maternal and paternal effects on the children's fat %. The findings were also similar if we used maternal adiposity measures 5 years following the index pregnancy, rather than the pregnancy values (data not shown). Interaction tests indicated no differences between boys and girls in any of the associations described.
Table 3.
Associations of maternal and paternal adiposity (overweight/obesity, BMI ≥ 25·0 kg/m2) with offspring adiposity and cardiovascular risk factors, the Mysore Parthenon study
| Model 1 | Model 2 | Model 3 | Model 4 | Model 5 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Children's adiposity and cardiovascular risk factors | β† | 95 % CI | β† | 95 % CI | β† | 95 % CI | β† | 95 % CI | β† | 95 % CI | P‡ |
| Maternal overweight/obesity | |||||||||||
| BMI (kg/m2) | 0·94*** | 0·58, 1·29 | 0·96*** | 0·60, 1·33 | 0·74*** | 0·37, 1·11 | 0·95*** | 0·57, 1·33 | 0·6 | ||
| SS (mm)§ | 0·13*** | 0·07, 0·20 | 0·11** | 0·05, 0·17 | 0·09** | 0·03, 0·16 | 0·11** | 0·05, 0·18 | 1·0 | ||
| Fat % | 1·73** | 0·47, 2·98 | 1·12 | −0·03, 2·27 | 1·24* | 0·03, 2·45 | 1·14 | −0·05, 2·34 | 0·4 | ||
| WC (cm) | 2·16*** | 1·12, 3·20 | 2·18*** | 1·10, 3·25 | 1·60** | 0·48, 2·71 | 2·06*** | 0·95, 3·18 | 0·9 | ||
| Glucose0 (mmol/l) | −0·04 | −0·12, 0·03 | −0·05 | −0·13, 0·03 | −0·04 | −0·12, 0·05 | −0·02 | −0·10, 0·06 | −0·06 | −0·14, 0·02 | 0·08 |
| Glucose120 (mmol/l) | −0·02 | −0·20, 0·16 | −0·03 | −0·22, 0·15 | 0·01 | −0·19, 0·21 | −0·07 | −0·27, 0·12 | −0·07 | −0·26, 0·12 | 0·5 |
| Insulin0 (pmol/l)§ | 0·16** | 0·04, 0·28 | 0·12* | 0·01, 0·24 | 0·09 | −0·03, 0·21 | 0·12* | 0·005, 0·24 | 0·07 | −0·05, 0·18 | 0·5 |
| HOMA-IR§ | 0·15* | 0·03, 0·27 | 0·11 | −0·01, 0·23 | 0·08 | −0·05, 0·21 | 0·12 | −0·01, 0·24 | 0·05 | −0·07, 0·17 | 0·4 |
| TAG (mmol/l)§ | −0·04 | −0·11, 0·03 | −0·04 | −0·12, 0·03 | −0·03 | −0·11, 0·05 | −0·05 | −0·12, 0·03 | −0·05 | −0·13, 0·02 | 1·0 |
| HDL-C (mmol/l) | 0·005 | −0·04, 0·05 | 0·003 | −0·04, 0·05 | −0·001 | −0·05, 0·05 | 0·002 | −0·04, 0·05 | 0·01 | −0·04, 0·05 | 0·9 |
| LDL-C (mmol/l) | 0·06 | −0·05, 0·16 | 0·05 | −0·06, 0·16 | 0·06 | −0·05, 0·18 | 0·04 | −0·07, 0·15 | 0·04 | −0·07, 0·15 | 0·056 |
| SBP (mmHg) | 1·41 | −0·22, 3·05 | 1·28 | −0·43, 3·00 | 1·21 | −0·63, 3·05 | 1·41 | −0·36, 3·18 | 0·10 | −1·50, 1·70 | 1·0 |
| Paternal overweight/obesity | |||||||||||
| BMI (kg/m2) | 0·74*** | 0·41, 1·08 | 0·70*** | 0·35, 1·04 | 0·61*** | 0·27, 0·95 | |||||
| SS (mm)§ | 0·11** | 0·05, 0·17 | 0·10** | 0·04, 0·16 | 0·09** | 0·03, 0·15 | |||||
| Fat % | 0·95 | −0·29, 2·19 | 0·69 | −0·41, 1·79 | 0·54 | −0·56, 1·65 | |||||
| WC (cm) | 2·07*** | 1·06, 3·08 | 1·93*** | 0·91, 2·95 | 1·73** | 0·71, 2·75 | |||||
| Glucose0 (mmol/l) | 0·06 | −0·01, 0·14 | 0·07 | −0·01, 0·14 | 0·07 | −0·01, 0·15 | 0·05 | −0·03, 0·13 | |||
| Glucose120 (mmol/l) | 0·11 | −0·07, 0·29 | 0·12 | −0·06, 0·30 | 0·12 | −0·07, 0·30 | 0·09 | −0·10, 0·27 | |||
| Insulin0 (pmol/l)§ | 0·17** | 0·05, 0·28 | 0·16** | 0·05, 0·27 | 0·15** | 0·04, 0·26 | 0·11* | 0·005, 0·22 | |||
| HOMA-IR§ | 0·18** | 0·06, 0·30 | 0·18** | 0·06, 0·29 | 0·17** | 0·05, 0·29 | 0·13 * | 0·01, 0·24 | |||
| TAG (mmol/l)§ | −0·03 | −0·10, 0·04 | −0·04 | −0·11, 0·04 | −0·03 | −0·10, 0·04 | −0·04 | −0·11, 0·03 | |||
| HDL-C (mmol/l) | −0·005 | −0·05, 0·04 | −0·003 | −0·05, 0·04 | −0·003 | −0·05, 0·04 | −0·0003 | −0·04, 0·04 | |||
| LDL-C (mmol/l) | −0·08 | −0·19, 0·02 | −0·09 | −0·20, 0·01 | −0·10 | −0·21, 0·01 | −0·10 | −0·21, 0·01 | |||
| SBP (mmHg) | 1·36 | −0·26, 2·99 | 1·37 | −0·30, 3·04 | 1·22 | −0·46, 2·91 | 0·31 | −1·27, 1·90 | |||
SS, sum of skinfolds; fat %, body fat percentage; WC, waist circumference; glucose0, fasting glucose concentration; glucose120, 120 min post-load glucose concentration; insulin0, fasting insulin concentration; HOMA-IR, homeostasis model assessment–insulin resistance; HDL-C, HDL-cholesterol; LDL-C, LDL-cholesterol; SBP, systolic blood pressure; SES, socio-economic status.
*P < 0·05; **P < 0·01; ***P < 0·001.
†β is the effect size derived using maternal or paternal overweight/obesity as a continuous variable (standardized) in multiple linear regression analysis. For logged variables, the effect size indicates the percentage change in the outcome per unit change in the predictor. Model 1: unadjusted; model 2: adjusted for the child's sex, age, breast-feeding duration, and maternal age, parity and SES; model 3: model 2+maternal and paternal SS mutually adjusted; model 4: model 2+maternal glucose area under the curve at pregnancy; model 5: model 2+the child's SS at 9·5 years.
‡P value for difference between maternal and paternal effects derived using the Z test (model 3).
§Logged variables.
Relationships of maternal and paternal adiposity with offspring cardiovascular risk factors
Higher maternal and paternal SS were associated with higher fasting insulin concentrations and HOMA-IR in the children (Table 2). The associations remained similar after adjusting for potential confounders in a series of models. However, the effects of maternal SS were not significant when maternal and paternal SS were mutually adjusted (model 3; Table 2). Again, Z tests showed no significant difference between maternal and paternal effects. The associations were attenuated after adjusting for the children's current adiposity (model 5; Table 2).
Higher maternal SS and BMI were associated with lower TAG concentrations in the children (Table 2 and Supplementary Materials). The results were similar after adjusting for potential confounders. Higher paternal SS was associated with lower LDL-C concentrations in the children which was significant after adjusting for potential confounders (Table 2). Neither maternal nor paternal adiposity predicted glucose concentrations, HDL-C concentrations or blood pressure in the children. Interaction tests showed no differences in any of the associations described between boys and girls.
The results were similar when maternal and paternal overweight or obesity were used as the exposure in the analysis (Table 3), when BMI was used as a continuous variable (Supplementary Materials) and also when we used maternal adiposity measures 5 years following the index pregnancy, rather than the pregnancy values (data not shown).
Discussion
Summary of the main findings
We examined associations between both parents’ adiposity and measures of adiposity and CVD risk factors in a cohort of Indian children whose mothers were non-diabetic during pregnancy. Both maternal and paternal adiposity were positively associated with adiposity (BMI, SS, fat % and WC) and fasting insulin concentration and HOMA-IR in the children. Although maternal effects were generally stronger than paternal effects, the differences were small and non-significant except for parental SS in relation to the child's fat %. We conclude that maternal and paternal effects were essentially similar in magnitude in our study.
Strengths and weakness
Strengths of the study were its prospective design, with good characterization of maternal glucose tolerance during pregnancy and continuous follow-up of a large number of children from birth. We collected detailed measurements of the parents’ as well as the children's adiposity (not just BMI) and CVD risk factors. A limitation was that we did not collect the fathers’ anthropometric data at the same time as the mothers’ (during pregnancy), and used the fathers’ data collected at the child's 5-year follow-up to compare maternal and paternal effects. This might have led to differences in mother–offspring and father–offspring associations. However, our findings were similar if we used the 5-year follow-up data for both parents.
Parent–child adiposity; comparison with other studies
Our results are consistent with a number of other studies in Western populations that have found similar effects of maternal and paternal BMI on childhood BMI( 6 , 14 – 19 ) or fat mass (SS, dual-energy X-ray absorptiometry)( 18 , 20 ). However, others have reported stronger maternal than paternal effects( 11 – 13 ). Another recent study from the USA found that maternal pre-gravid BMI and gestational weight gain predicted fat mass (dual-energy X-ray absorptiometry) in the children at 8 years while there was no association with paternal BMI( 7 ). In the UK Avon Longitudinal Study of Pregnancy and Childhood, maternal and paternal BMI was associated with offspring fat mass at 9–11 years and maternal effects were stronger than paternal. However, by using FTO genotype and a Mendelian randomization approach, the authors concluded that the difference between maternal and paternal effects was very small and that fetal overnutrition related to higher maternal BMI is unlikely to cause childhood obesity( 12 ).
Some of the best evidence for the fetal overnutrition hypothesis comes from studies of children born before or after maternal bariatric surgery. Kral et al. found a higher prevalence of obesity among children born to extremely obese mothers (mean BMI 48·0 kg/m2) before they underwent biliopancreatic bypass surgery, compared with children born after maternal surgery( 31 ). These findings were confirmed in a follow-up study limited to siblings born before and after maternal bariatric surgery( 32 ). Although these were small studies, they suggest an intra-uterine effect of extreme maternal obesity on adiposity in the children. The lack of a difference between maternal and paternal effects in Mysore could be because we do not have any extremely obese mothers, and also because our children are thin. Taken together, the evidence from our own and the above studies suggests that, in the absence of maternal GDM, fetal exposure to maternal adiposity during pregnancy does not increase adiposity in children over and above the effects of genes and/or shared family lifestyle, except at very high levels of maternal obesity.
Parents’ adiposity–child cardiovascular risk factors
In our study, both maternal and paternal SS was positively associated with fasting insulin concentrations and HOMA-IR in the children at 9·5 years. These associations were attenuated after adjusting for the child's current adiposity, suggesting that this is a mediating factor. Few other studies have reported insulin concentrations or insulin resistance in children in relation to the parents’ adiposity. Consistent with our findings, however, fasting insulin and HOMA-IR were positively related to maternal BMI in 11-year-old Afro-Jamaican children. The associations were non-significant after adjusting for the children's WC( 24 ). In contrast, a recent study among Estonian and Swedish children showed no associations between the parents’ current BMI and offspring HOMA-IR( 19 ).
We found no associations between parental adiposity and blood pressure in the children. Other studies have shown inconsistent results. Among Australian( 33 ) and UK( 8 ) children, there were positive associations of maternal BMI and/or weight with offspring SBP. In the Australian study, there was a similar association with paternal BMI; in the UK, fathers were not studied. However, among Jamaican( 21 ) and Philippine( 22 ) children, there were inverse associations between maternal triceps skinfold thickness during pregnancy and SBP (again, fathers were not studied). Since all these findings were in children of similar age, the differences between studies are likely to result from variation in the level of parent and child adiposity; in the latter two studies, parents were much thinner than in Mysore and the European studies.
In our study, maternal but not paternal adiposity was inversely related to the children's TAG concentrations and paternal but not maternal adiposity was inversely related to the children's LDL-C concentrations. The Estonia/Sweden study showed no associations between either parent's BMI and any of the children's lipid concentrations( 19 ), but a recent study from the UK reported that greater maternal weight was associated with lower HDL-C in children at 9 years( 8 ). Among adolescent Filipinos maternal arm fat area was positively associated with HDL-C in boys and LDL-C in girls, and inversely associated with LDL-C in boys( 23 ). A mechanism for the inverse associations in our study is not clear.
Conclusions
In this urban Indian population both maternal and paternal adiposity were related equally to adiposity and insulin resistance in the children at age 9·5 years. Our findings suggest that genetic or shared family environmental factors are most likely to underlie these associations, although we cannot draw any conclusions regarding the relative contributions of these. We cannot rule out another possibility that equal maternal and paternal effects on the child's adiposity could occur because of epigenetic changes in gametic DNA, which could theoretically result from nutritional exposures in either or both parents( 34 ). Our study, in a population with low levels of childhood obesity, did not support the hypothesis that maternal adiposity, in the absence of GDM, increases adiposity and cardiovascular risk in the children through a fetal overnutrition effect. This lack of evidence is echoed by the absence of a relationship between maternal glucose concentrations (a fetal fuel) and adiposity in the children. Therefore, while there is good evidence that fetal overnutrition caused by GDM or extreme maternal obesity is an important cause of childhood obesity, it does not seem likely that lesser levels of maternal adiposity are fuelling the worldwide epidemic of obesity through this intergenerational mechanism.
Acknowledgements
This work was funded by Parthenon Trust, Switzerland (no grant number), The Wellcome Trust, UK (grant number 079877/Z/06/Z) and The Medical Research Council, UK (grant number: G0400519). The authors declare that there is no duality of interest associated with this manuscript. The authors’ contributions are as follows: S.R.V., conception and design, analysis and interpretation of data, drafting the article and approval of final version; G.V.K., conception and design, revising the manuscript critically for intellectual content and approval of final version; S.C.K., conception and design, revising the manuscript critically for intellectual content and approval of final version; C.O., analysis and interpretation of data and approval of final version; C.H.D.F., conception and design, interpretation of data, revising the manuscript critically for intellectual content and approval of final version. The authors are grateful to the families for their participation, and thank all staff of HMH obstetric department, the research team members for their contributions and Sneha-India for its support.
Supplementary material
For supplementary material accompanying this paper visit https://doi.org/10.1017/S1368980012003795.
click here to view supplementary material
References
- 1. World Health Organization (2000) Obesity: Preventing and Managing A Global Epidemic. WHO Technical Report Series no. 899, pp. 1–4. Geneva: WHO. [PubMed] [Google Scholar]
- 2. Daniels SR (2009) Complications of obesity in children and adolescents. Int J Obes (Lond) 33, Suppl. 1, S60–S65. [DOI] [PubMed] [Google Scholar]
- 3. Bridger T (2009) Childhood obesity and cardiovascular disease. Paediatr Child Health 14, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harrap SB, Stebbing M, Hopper JL et al. (2000) Familial patterns of covariation for cardiovascular risk factors in adults: The Victorian Family Heart Study. Am J Epidemiol 152, 704–715. [DOI] [PubMed] [Google Scholar]
- 5. Boney CM, Verma A, Tucker R et al. (2005) Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics 115, e290–e296. [DOI] [PubMed] [Google Scholar]
- 6. Kivimaki M, Lawlor DA, Smith GD et al. (2007) Substantial intergenerational increases in body mass index are not explained by the fetal overnutrition hypothesis: the Cardiovascular Risk in Young Finns Study. Am J Clin Nutr 86, 1509–1514. [DOI] [PubMed] [Google Scholar]
- 7. Catalano PM, Farrell K, Thomas A et al. (2009) Perinatal risk factors for childhood obesity and metabolic dysregulation. Am J Clin Nutr 90, 1303–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fraser A, Tilling K, Macdonald-Wallis C et al. (2010) Association of maternal weight gain in pregnancy with offspring obesity and metabolic and vascular traits in childhood. Circulation 121, 2557–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gale CR, Javaid MK, Robinson SM et al. (2007) Maternal size in pregnancy and body composition in children. J Clin Endocrinol Metab 92, 3904–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reece EA (2010) The fetal and maternal consequences of gestational diabetes mellitus. J Matern Fetal Neonatal Med 23, 199–203. [DOI] [PubMed] [Google Scholar]
- 11. Lawlor DA, Smith GD, O'Callaghan M et al. (2007) Epidemiologic evidence for the fetal overnutrition hypothesis: findings from the mater-university study of pregnancy and its outcomes. Am J Epidemiol 165, 418–424. [DOI] [PubMed] [Google Scholar]
- 12. Lawlor DA, Timpson NJ, Harbord RM et al. (2008) Exploring the developmental overnutrition hypothesis using parental offspring associations and FTO as an instrumental variable. PLoS Med 5, e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Whitaker KL, Jarvis MJ, Beeken RJ et al. (2010) Comparing maternal and paternal intergenerational transmission of obesity risk in a large population-based sample. Am J Clin Nutr 91, 1560–1567. [DOI] [PubMed] [Google Scholar]
- 14. Salbe AD, Weyer C, Lindsay RS et al. (2002) Assessing risk factors for obesity between childhood and adolescence: I. Birth weight, childhood adiposity, parental obesity, insulin and leptin. Pediatrics 110, 299–306. [DOI] [PubMed] [Google Scholar]
- 15. Li L, Law C, Lo Conte R et al. (2009) Intergenerational influences on childhood body mass index: the effect of parental body mass index trajectories. Am J Clin Nutr 89, 551–557. [DOI] [PubMed] [Google Scholar]
- 16. Davey Smith G, Steer C, Leary S et al. (2007) Is there an intrauterine influence on obesity? Evidence from parent child associations in the Avon Longitudinal Study of Parents and Children (ALSPAC). Arch Dis Child 92, 876–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Safer DL, Agras WS, Bryson S et al. (2001) Early body mass index and other anthropometric relationships between parents and children. Int J Obes Relat Metab Disord 25, 1532–1536. [DOI] [PubMed] [Google Scholar]
- 18. Burke V, Beilin LJ, Simmer K et al. (2005) Predictors of body mass index and associations with cardiovascular risk factors in Australian children: a prospective cohort study. Int J Obes (Lond) 29, 15–23. [DOI] [PubMed] [Google Scholar]
- 19. Labayen I, Ruiz JR, Ortega FB et al. (2010) Intergenerational cardiovascular disease risk factors involve both maternal and paternal BMI. Diabetes Care 33, 894–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Patel R, Martin RM, Kramer MS et al. (2011) Familial associations of adiposity: findings from a cross-sectional study of 12,181 parental–offspring trios from Belarus. PLoS One 6, e14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Godfrey KM, Forrester T, Barker DJ et al. (1994) Maternal nutritional status in pregnancy and blood pressure in childhood. Br J Obstet Gynaecol 101, 398–403. [DOI] [PubMed] [Google Scholar]
- 22. Adair LS, Kuzawa CW & Borja J (2001) Maternal energy stores and diet composition during pregnancy program adolescent blood pressure. Circulation 104, 1034–1039. [DOI] [PubMed] [Google Scholar]
- 23. Kuzawa CW & Adair LS (2003) Lipid profiles in adolescent Filipinos: relation to birth weight and maternal energy status during pregnancy. Am J Clin Nutr 77, 960–966. [DOI] [PubMed] [Google Scholar]
- 24. Boyne MS, Osmond C, Fraser RA et al. (2010) Developmental origins of cardiovascular risk in Jamaican children: The Vulnerable Windows Cohort Study. Br J Nutr 104, 1026–1033. [DOI] [PubMed] [Google Scholar]
- 25. Banerji MA, Faridi N, Atluri R et al. (1999) Body composition, visceral fat, leptin and insulin resistance in Asian Indian men. J Clin Endocrinol Metab 84, 137–144. [DOI] [PubMed] [Google Scholar]
- 26. Krishnaveni GV, Hill JC, Leary SD et al. (2005) Anthropometry, glucose tolerance and insulin concentrations in Indian children: relationships to maternal glucose and insulin concentrations during pregnancy. Diabetes Care 28, 2919–2925. [DOI] [PubMed] [Google Scholar]
- 27. Friedewald WT, Levy RI & Fredrickson DS (1972) Estimation of the concentration of low-density lipoprotein cholesterol in plasma without use of the preparative ultracentrifuge. Clin Chem 18, 499–502. [PubMed] [Google Scholar]
- 28. Matthews DR, Hosker JP, Rudenski AS et al. (1985) Homeostasis model assessment: insulin resistance and β-cell function from fasting glucose and insulin concentrations in man. Diabetologia 28, 412–419. [DOI] [PubMed] [Google Scholar]
- 29. World Health Organization (2007) WHO Child Growth Standards. http://www.who.int/growthref/en/ (accessed January 2012).
- 30. International Institute for Population Sciences & ORC Macro (2001) National Family Health Survey (NFHS-2), India 1998–1999. Mumbai/Calverton, MD: IIPS/ORC Macro. [Google Scholar]
- 31. Kral JG, Biron S, Simard S et al. (2006) Large maternal weight loss from obesity surgery prevents transmission of obesity to children who were followed for 2 to 18 years. Pediatrics 118, e1644–e1649. [DOI] [PubMed] [Google Scholar]
- 32. Smith J, Cianfione K, Biron S et al. (2009) Effects of maternal surgical weight loss in mothers on intergenerational transmission of obesity. J Clin Endocrinol Metab 94, 4275–4283. [DOI] [PubMed] [Google Scholar]
- 33. Lawlor DA, Najman JM, Sterne J et al. (2004) Associations of parental, birth, and early life characteristics with systolic blood pressure at 5 years of age: findings from the Mater-University study of pregnancy and its outcomes. Circulation 110, 2417–2423. [DOI] [PubMed] [Google Scholar]
- 34. Ng SF, Lin RC, Laybutt DR et al. (2010) Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature 467, 963–966. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
For supplementary material accompanying this paper visit https://doi.org/10.1017/S1368980012003795.
click here to view supplementary material