

Abstract
N-Acetylneuraminic acid is produced by alkaline epimerization of N-acetylglucosamine to N-acetylmannosamine and then subsequent condensation with pyruvate catalyzed by free N-acetylneuraminic acid aldolase. The high-alkaline conditions of this process result in the degradation of reactants and products, while the purification of free enzymes to be used for the synthesis reaction is a costly process. The use of N-acetylglucosamine 2-epimerase has been seen as an alternative to the alkaline epimerization process. In this study, these two enzymes involved in N-acetylneuraminic acid production were immobilized to biopolyester beads in vivo in a one-step, cost-efficient process of production and isolation. Beads with epimerase-only, aldolase-only, and combined epimerase/aldolase activity were recombinantly produced in Escherichia coli. The enzymatic activities were 32 U, 590 U, and 2.2 U/420 U per gram dry bead weight, respectively. Individual beads could convert 18% and 77% of initial GlcNAc and ManNAc, respectively, at high substrate concentrations and near-neutral pH, demonstrating the application of this biobead technology to fine-chemical synthesis. Beads establishing the entire N-acetylneuraminic acid synthesis pathway were able to convert up to 22% of the initial N-acetylglucosamine after a 50-h reaction time into N-acetylneuraminic acid.
INTRODUCTION
Derivatives of neuraminc acid frequently occur at the terminal position of cell-surface oligosaccharides (1). This high level of exposure correlates with their biological role in cellular interaction, bacterial and viral infection, and immune-system recognition (2–4). In addition, a derivative of N-acetylneuraminic acid (Neu5Ac), zanamivir, is used as a neurimidase inhibitor for the treatment of influenza virus infections (5). The biological roles and recent pharmaceutical interest in Neu5Ac make industrial-scale production of this chemical desirable. N-Acetylneuraminic acid aldolase (NanA; EC 4.1.3.3) catalyzes the conversion of Neu5Ac to N-acetyl-d-mannosamine (ManNAc) and pyruvate (6). Although the reverse reaction has an unfavorable equilibrium (Km = 28.71 mol−1) requiring a large excess of reactants, NanA is still the enzyme of choice, as pyruvate is readily available as a substrate. N-Acetylglucosamine 2-epimerase (Slr1975; E.C. 5.1.3.8) is able to catalyze the conversion of N-acetyl-d-glucosamine (GlcNAc) to ManNAc, with ATP required in catalytic amounts (7). Immobilization of enzymes onto a solid support facilitates removal from the product, often enhances stability, and allows reuse of the expensive reaction catalyst (8–10). Usually, enzymes are produced in a recombinant system, isolated, and then attached to a solid support system. This type of approach has been applied to both aldolase and epimerase enzymes (11, 12). Alternatively, the production and immobilization of the enzymes can occur in a single step, avoiding the potentially expensive attachment step and simplifying enzyme isolation from the recombinant system. For example, NanA has been immobilized in a spore-display system which allows one-step production (13).
Recently, the PhaC enzyme from Ralstonia eutropha was used to immobilize an N-terminally fused thermostable α-amylase (14) and an organophosphate hydrolase fused to the C terminus of PhaC (15) to self-assembled polyester beads in vivo. The polyhydroxyalkanoate (PHA) produced by PhaC aggregates formed spherical inclusions of 50 to 500 nm in diameter (16). Additionally, β-galactosidase has been successfully fused to the N terminus of class II PhaC from Pseudomonas aeruginosa, mediating recombinant production of β-galactosidase-displaying polyester beads (17). Expression of these proteins in a polyester (PHA) production host led to a one-step, self-assembled, and cost-effective enzyme display biobead system.
In this study, we utilized the previously identified properties of the PHA biobead system to enable efficient production, immobilization, and isolation of both NanA from Escherichia coli and Slr1975 from Synechocystis sp. strain PCC 6803 on both individual- and dual-enzyme-displaying PHA beads. The aim of this study was to demonstrate enzymatic synthesis of Neu5Ac by the PHA enzyme-immobilization biobead system and the wider applicability of this platform technology to fine-chemical synthesis.
MATERIALS AND METHODS
Chemicals.
GlcNAc (≥99% purity), ManNAc (≥98% purity), sodium pyruvate, and ATP were purchased from Sigma-Aldrich (St. Louis, MO). Neu5Ac was purchased from GlycoFineChem (New Zealand).
Bacteria, plasmids, and growth conditions.
Escherichia coli XL1-Blue was grown at 37°C and 200 rpm and E. coli BL21(DE3) at 25°C and 200 rpm. When required, ampicillin (75 μg/ml), chloramphenicol (50 μg/ml), and tetracycline (12.5 μg/ml) were added to the Luria-Bertani medium. Bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material.
Plasmid construction.
General cloning procedures and DNA isolation were performed as described elsewhere (18). Primers were purchased from Integrated DNA Technologies (Coraville, IA), while Taq and Platinum Pfx polymerases were purchased from Invitrogen (Carlsbad, CA). The slr1975 and nanA genes were synthesized by Genscript Corporation (Piscataway, NJ) and inserted into pUC57 vectors with flanking restriction endonuclease sites for subsequent cloning.
The construction of the pET14b:NanA-PhaC-l-Slr1975 plasmid was based on the pET14b:ZZ-PhaC-l-green fluorescent protein (GFP) plasmid from previous studies (19). The plasmid pET14b:ZZ-PhaC-l-GFP was hydrolyzed with the XhoI and BamHI restriction enzymes, cleaving the gfp region from the vector. The resulting backbone was ligated to the purified slr1975 fragment. This gave rise to the plasmid pET14b:ZZ-PhaC-l-Slr1975. The plasmid pET14b:ZZ-PhaC-l-Slr1975 was hydrolyzed with the NdeI restriction enzyme, cleaving the zz region from the vector. The resulting backbone was purified and ligated to the purified nanA fragment (see Fig. S1 in the supplemental material).
Additionally, the plasmid pET14b:ZZ-PhaC-l-Slr1975 was hydrolyzed with the NdeI restriction enzyme, cleaving the zz region from the vector, and religated to produce the plasmid pET14b:PhaC-l-Slr1975.
The pET14b:NanA-PhaC construct was assembled by hydrolyzing the plasmid pET14b:AmyS-PhaC (14) with the restriction enzymes XbaI and NotI, and the resulting backbone was ligated with the nanA fragment to produce the plasmid pET14b:NanA-PhaC (see Fig. S2 in the supplemental material). DNA sequencing was performed to confirm the expected sequence.
The pET14b:PhaC-NanA construct was assembled by amplification of the nanA gene from pUC57:NanA, adding XhoI and BamHI restriction sites. The plasmid pET14b:PhaC-l-Slr1975 was hydrolyzed with the restriction enzymes XhoI and BamHI, and the resulting backbone was ligated with the nanA fragment to produce the plasmid pET14b:PhaC-NanA.
Additional plasmid constructs pET14b:Slr1975-PhaC and pET14b:Slr1975-PhaC-NanA were created in a similar manner, although they were not tested extensively past the initial immobilized-enzyme screening process.
DNA sequencing was performed to confirm the expected plasmid sequence and was carried out by the Massey Genome Service (Palmerston North, New Zealand) on a capillary ABI3730 Genetic Analyzer (Applied Biosystems, Foster City, CA).
Overexpression of phaC genetic constructs.
Cells of E. coli BL21/pMCS69 were transformed with pET14b plasmids containing the nanA-phaC, phaC-nanA, phaC-l-slr1975, or nanA-phaC-l-slr1975 construct. The plasmid pMCS69 contained the phaA and phaB genes that mediate the synthesis of the precursor (R)-3-hydroxybutyryl coenzyme A (3HB-CoA) required for polyester synthesis. Cultures were grown at 37°C until an optical density (OD) of 0.3 was reached, gene expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG), and the cultures were transferred to 25°C for an additional 48 h of growth. Cells were harvested by centrifugation (4,000 × g, 20 min).
Determination of PhaC functionality.
Initial screening of PhaC functionality was performed by staining cell pellets with Nile Red and observing cells with fluorescence microscopy as previously described (20). Ultimately, PHA production was quantified using gas chromatography-mass spectrometry (GC/MS) after conversion of the PHA into 3-hydroxymethylester by acid-catalyzed methanolysis (21). GC/MS was performed by The New Zealand Institute for Plant & Food Research (Palmerston North, New Zealand).
PHA bead isolation.
E. coli cell pellets containing polyester were resuspended in 50 mM potassium phosphate buffer (pH 7.5) and mechanically disrupted using a constant cell disruption system at 20 kpsi with a one-shot head adapter (Constant Systems Ltd., Northants, United Kingdom), and the lysate was subjected to centrifugation (4,000 × g, 20 min, 4°C) to sediment the PHA beads. Final separation of the beads from the bacterial lysate was achieved by ultracentrifugation (150,000 × g, 2 h, 10°C) on a glycerol gradient (22). Isolated PHA beads were stored at 4°C in 50 mM potassium phosphate buffer (pH 7.5) supplemented with 20% (vol/vol) ethanol until required.
Protein analysis.
Bead-associated proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as described elsewhere (23). Protein bands of interest were excised from an SDS-PAGE gel and subjected to tryptic digest and mass spectrometry performed by the Centre for Protein Research (Otago, New Zealand).
Activity assays.
Slr1975 epimerase activity in 1 ml of 50 mM potassium phosphate buffer (pH 7.5) was determined at 37°C. Isolated beads were reacted with 20 mM ManNAc (with 5 mM ATP and 10 mM MgCl2) for 30 min, the resulting mixture was centrifuged (6,000 × g, 4 min), and an aliquot was boiled at 100°C for 5 min and subjected to high-performance liquid chromatography (HPLC) (12). One unit was defined as the amount of Slr1975 needed to catalyze the production of 1 μmol GlcNAc per min. NanA aldolase activity was determined at 37°C in 1 ml of 50 mM potassium phosphate buffer (pH 7.5). Isolated beads were reacted with 4 mM Neu5Ac for 10 min, the resulting mixture was centrifuged (6,000 × g, 4 min), and an aliquot was boiled at 100°C for 5 min and subjected to HPLC (13). One unit of enzyme activity was defined as the amount of NanA needed to catalyze the production of 1 μmol pyruvate per min.
Synthesis of Neu5Ac.
The reaction conditions for GlcNAc epimerization to ManNAc were 100 mM GlcNAc, 10 mM ATP, and 10 mM MgCl2 at 50°C for 20 h. PHA beads were present at 5 mg (wet bead mass) in 50 mM potassium phosphate buffer (pH 7.5). For the synthesis of Neu5Ac from ManNAc, 1 M sodium pyruvate and 400 mM ManNAc were reacted with 5 mg PHA beads in 50 mM potassium phosphate buffer (pH 7.5) at 50°C for 44 h (13, 24). Recycling reactions for PhaC-l-Slr1975 beads were identical to the reaction conditions described above. The conditions for NanA-PhaC bead-recycling reactions were 250 mM pyruvate and 100 mM ManNAc for 20 h. The full biosynthesis of Neu5Ac from GlcNAc and pyruvate was initially carried out with 100 mM sodium pyruvate, 250 mM GlcNAc, 10 mM ATP, and 10 mM MgCl2 at 50°C for 44 h. PHA beads were present at 10 mg (wet bead weight) in 50 mM potassium phosphate buffer (pH 7.5). The final conditions were as described above, except the reactions were carried out at 30°C for 50 h using a GlcNAc starting concentration of 100 mM (12).
Analytical procedures.
ManNAc, pyruvate, and Neu5Ac concentrations were determined by a Dionex Summit HPLC system using two Phenomenex (Torrance, CA) RHM columns (300 by 7.8 mm) in series. The flow rate was 0.5 ml/min of 6 mM H2SO4, and the columns were maintained at 60°C. The injection volume was 25 μl, and the UV detector was set at 205 nm. Chromatographic data were analyzed with Chromeleon software (v 6.3).
Neu5Ac levels were additionally determined by a resorcinol-based colorimetric assay. Briefly, 100 μl of 40 mM periodic acid was vortex mixed into 500 μl of sample and placed in an ice bath for 20 min. Then 1.25 ml of resorcinol solution (0.6 g resorcinol, 250 μl 100 mM CuSO4, 45 ml 37% HCl, adjusted to 100 ml with H2O) was added, and the samples were vortex mixed and returned to the ice bath for 5 min. Samples were boiled in a 100°C oil bath for 15 min, and 1.25 ml of tert-butanol was added and vortex mixed vigorously to achieve a single phase. Finally, samples were incubated for 3 min at 37°C and cooled to room temperature, and absorbance at 630 nm was measured.
RESULTS
Bioengineering for the recombinant production of PHA beads displaying selected enzymes relevant to Neu5Ac synthesis.
PhaC, a 64-kDa protein which forms a dimer, was used as an anchoring protein for the display of the 46-kDa dimeric Slr1975 from Synechocystis sp. PCC 6803 (25) and the 33-kDa tetrameric NanA from E. coli on the surface of PHA beads. NanA was fused to the N terminus of PhaC, while Slr1975 was fused to the C terminus via a glycine linker. Both single- and double-enzyme fusions corresponding to hybrid genes were constructed. The T7 promoter was used to induce high-level expression of the respective fusion proteins.
The fusion proteins NanA-PhaC, PhaC-l-Slr1975, and NanA-PhaC-l-Slr1975 all mediated the formation of PHA beads in the production host BL21(DE3)/pMCS69 as indicated by the Nile Red staining procedure (see Fig. S3 in the supplemental material). GC/MS analysis of the dried cells producing the respective PHA beads confirmed a high level of PHA production, contributing to 45% to 47% of cellular dry weight (see Table S2 in the supplemental material).
Proteins associated with isolated PHA beads were analyzed by SDS-PAGE. NanA-PhaC, PhaC-l-Slr1975, and NanA-PhaC-l-Slr1975 had theoretical molecular masses of 97, 112, and 145 kDa, respectively, and appear as the predominant proteins with the respective apparent molecular masses (Fig. 1). The identity of each fusion protein was confirmed by tryptic peptide fingerprinting using matrix-assisted laser desorption ionization–time of flight tandem mass spectrometry (MALDI-TOF MS/MS) (see Table S3 in the supplemental material).
Fig 1.
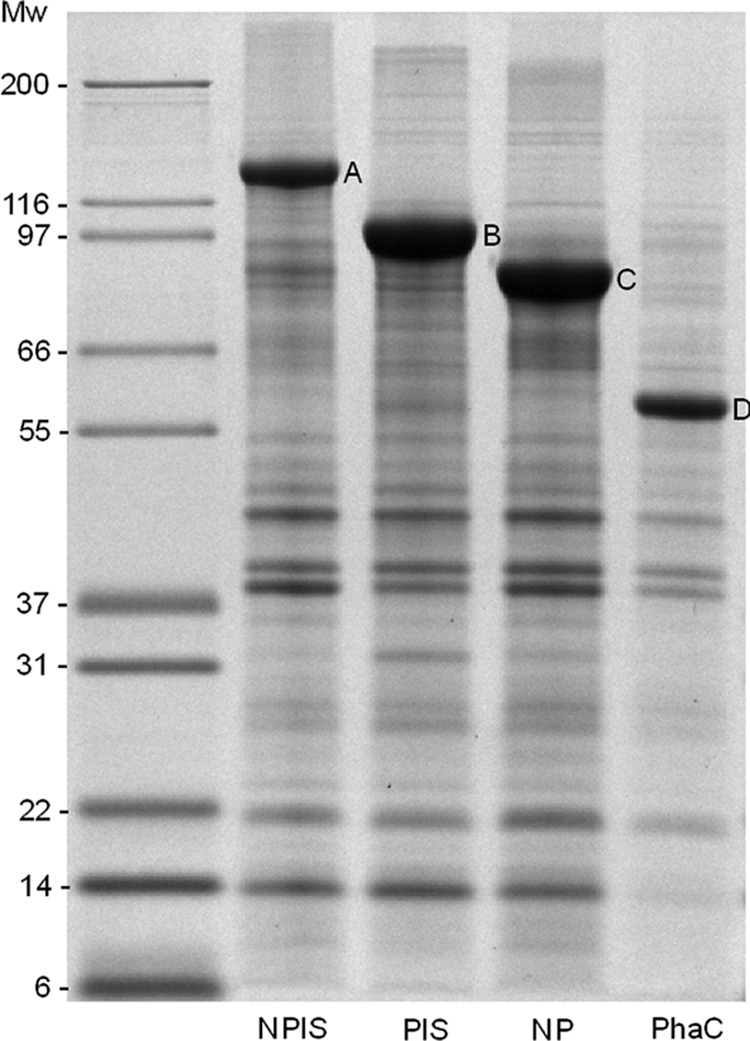
Protein profile of PHA beads as demonstrated by SDS-PAGE. Mw, molecular mass standard Mark 13 ladder (Invitrogen, CA); NPlS, NanA-PhaC-l-Slr1975 (A); PlS, PhaC-l-Slr1975 (B); NP, NanA-PhaC (C); PhaC, PhaC (D). The letters A, B, C, and D correspond to the fusion proteins of interest overproduced on their respective PHA beads.
Screening for successful enzyme immobilization of PhaC-fusion constructs.
Reaction conditions for immobilized NanA were confirmed using PHA beads, the formation of which was mediated by the use of pET14b:PhaC-NanA. Neu5Ac production was measured using the resorcinol assay. PHA bead concentrations could range between 1 and 15 mg/ml wet bead mass (see Fig. S4 in the supplemental material), and the optimal temperature for NanA was found to be 50°C (see Fig. S5 in the supplemental material), agreeing with other reports (13). Enzyme activity was sensitive to trypsin treatment, indicating successful enzyme immobilization on the bead surface (not shown). For PHA beads with immobilized PhaC-l-Slr1975, a reaction temperature between 37 and 50°C had negligible impact on Neu5Ac production; therefore, all further production reactions were carried out at 50°C (not shown). In addition, the immobilized fusion proteins NanA-PhaC, PhaC-NanA, Slr1975-PhaC, PhaC-Slr1975, NanA-PhaC-l-Slr1975, and Slr1975-PhaC-NanA were screened for activity with the resorcinol assay. Slr1975-PhaC and Slr1975-PhaC-NanA were found not to possess epimerase activity and were not studied further (data not shown). Further, NanA-PhaC had yield superior to that of PhaC-NanA under the previously determined aldolase conditions; therefore, PhaC-NanA was not studied further (not shown).
Activity assays of enzymes displayed on PHA beads.
N-Acetylglucosamine 2-epimerase activity tests were used for beads displaying Slr1975 fusions. PhaC-only beads served as a negative control and showed some background activity which corresponded to 0.2 U/g dry bead mass. Beads displaying PhaC-Slr1975 had 32 U/g dry bead mass, and beads displaying NanA-PhaC-l-Slr1975 had 2.2 U/g dry bead mass (Table 1). Beads displaying immobilized NanA fusions were subjected to N-acetylneuraminic acid aldolase activity tests using Neu5Ac as a substrate. PhaC-only beads had no detectable aldolase activity, whereas beads displaying NanA-PhaC had 590 U/g dry bead mass. Beads displaying NanA-PhaC-l-Slr1975 had 420 U/g dry bead mass (Table 2). Specific enzyme activity was determined by quantifying bead protein concentrations using densitometry from SDS-PAGE and known standards of bovine serum albumin (BSA) (see Fig. S6 in the supplemental material) and calculating the fusion enzyme concentration present in each activity reaction.
Table 1.
Specific epimerase activity of PHA beads with immobilized Slr1975
| Fusion protein (n = 3) | Specific activity (mean ± SD) |
|
|---|---|---|
| No. of enzyme units per dry bead wt (U/g) | No. of enzyme units per protein wt (U/mg) | |
| PhaC-Slr1975 | 32.3 ± 1.1 | 1.76 ± 0.38 |
| NanA-PhaC-l-Slr1975 | 2.17 ± 0.18 | 0.58 ± 0.07 |
| PhaC | 0.20 ± 0.19 | 0.04 ± 0.04 |
Table 2.
Specific aldolase activity of PHA beads with immobilized NanA
| Fusion protein (n = 3) | Specific activity (mean ± SD) |
|
|---|---|---|
| No. of enzyme units per dry bead wt (U/g) | No. of enzyme units per protein wt (U/mg) | |
| NanA-PhaC | 585 ± 49 | 42.6 ± 6.9 |
| NanA-PhaC-l-Slr1975 | 418 ± 53 | 81.9 ± 19 |
| PhaC | NDa | ND |
ND, not determined.
Production of ManNAc by Slr1975-displaying beads.
The enzymatic epimerization of GlcNAc to ManNAc was carried out at 50°C, and the reaction conditions were 100 mM GlcNAc, 10 mM MgCl2, and 10 mM ATP in 50 mM phosphate buffer (pH 7.5). Reactions were stopped after 20 h, and a sample was taken to be measured by HPLC. For the PhaC-l-Slr1975 fusion, the concentration of ManNAc reached 21 mM (18% conversion), the likely equilibrium point of the reaction. The NanA-PhaC-l-Slr1975 fusion reached only 4 mM (4% conversion) after 20 h, while the PhaC beads had no detectable production of ManNAc (Table 3). The ability of the PhaC-l-Slr1975 beads to be recycled was tested over five cycles under conditions identical to those described above. After an initial yield of 22% ManNAc, the conversion rate slowly declined from 19% to 17% over the next four cycles (Fig. 2).
Table 3.
Production of N-acetylmannosamine and N-acetylneuraminic acid using single-bead systems
| Fusion protein (n = 3) | Production (mean ± SD) (% conversion)a |
|
|---|---|---|
| ManNAc (mM) | Neu5Ac (mM) | |
| NanA-PhaC | NA | 282 ± 8.0 (77) |
| PhaC-l-Slr1975 | 21.4 ± 1.0 (18) | NA |
| NanA-PhaC-l-Slr1975 | 4.47 ± 0.10 (4) | 257 ± 11 (70) |
| PhaC | ND | ND |
NA, not available; ND, not determined.
Fig 2.

Recycling of PhaC-l-Slr1975-displaying PHA beads. Reaction conditions were 100 mM GlcNAc with 10 mM (both) ATP and MgCl2 as cofactors. The reaction was performed at 50°C for 20 h. The yield of ManNAc from GlcNAC was measured by HPLC. Error bars represent ± 1 standard deviation; n = 3.
Production of Neu5Ac under optimal conditions using enzyme-immobilized beads.
The enzymatic synthesis of ManNAc (400 mM) and pyruvate (1 M) to Neu5Ac was carried out at 50°C. The amount of PHA beads was 5 mg wet bead mass per ml (0.63 U/ml). After a 44-h reaction, the concentrations of ManNAc and Neu5Ac were measured using HPLC. NanA-PhaC and NanA-PhaC-l-Slr1975 beads were able to convert approximately 77% and 70% of the initial ManNAc to Neu5Ac, respectively. PhaC-only beads showed no detectable production of Neu5Ac (Table 3). Recycling of the NanA-PhaC beads was tested under conditions of lower substrate concentrations (100 mM ManNAc and 250 mM pyruvate) and over a shorter time frame (20 h). The initial reaction produced a yield of 73% Neu5Ac. However, in the second reaction, yield increased to 97%, with subsequent reactions showing decreasing yields (Fig. 3). After four reaction cycles, the overall yield had decreased back to 70%.
Fig 3.

Recycling of NanA-PhaC-displaying PHA beads. Reaction conditions were 100 mM ManNAc and 250 mM pyruvate at 50°C for 20 h. The yield of Neu5Ac from ManNAc was measured by HPLC. Error bars represent ± 1 standard deviation; n = 3.
Enzymatic synthesis of GlcNAc and pyruvate to Neu5Ac was initially performed at 50°C using substrate concentrations of 250 mM and 100 mM, respectively. The cofactors of 10 mM MgCl2 and 10 mM ATP were also added. Overall, each reaction mixture contained 10 mg (wet bead mass) of PHA beads. The weights of wet beads added to the reaction were as follows: PhaC only, 10 mg; NanA-PhaC-l-Slr1975, 10 mg; and NanA-PhaC plus PhaC-l-Slr1975, 5 mg each. Samples were taken at 44 h and subjected to analysis by HPLC. PhaC-only beads showed no detectable levels of ManNAc or Neu5Ac. NanA-PhaC plus PhaC-l-Slr1975 showed some production of 41 mM ManNAc and 14 mM Neu5Ac and 14% and 4.7% conversion of the starting GlcNAc concentration, respectively. NanA-PhaC-l-Slr1975 had slight production of 6.0 mM ManNAc and 5.1 mM Neu5Ac and 2% and 1.7% conversion of the starting GlcNAc concentration, respectively (Fig. 4). Finally, the combination of NanA-PhaC plus PhaC-l-Slr1975 was tested further under conditions favoring the conversion of GlcNAc to ManNAc. The initial concentration of substrate was 100 mM (both) pyruvate and GlcNAc with 10 mM (both) MgCl2 and ATP added. The weights of wet beads added were as follows: NanA-PhaC, 2.5 mg; and PhaC-l-Slr1975, 7.5 mg. The reaction was carried out at 30°C for 50 h. While the PhaC-only control had no detectable production of ManNAc or Neu5Ac, the final yield of the two-bead system with respect to the starting GlcNAc concentration was 19% ManNAc and 22% Neu5Ac (Fig. 4).
Fig 4.

(A) Production of Neu5Ac from 250 mM GlcNAc and 100 mM pyruvate over 44 h at 50°C using 10 mg (wet weight) of NanA- and Slr1975-displaying beads. NanA-PhaC plus PhaC-l-Slr1975 beads (5 mg [wet weight] each) yielded 14% ManNAC and 4.7% Neu5Ac conversion of the starting GlcNAc concentration. NanA-PhaC-l-Slr1975 beads (10 mg wet weight) yielded 2% ManNAc and 1.7% Neu5Ac conversion of the starting GlcNAc concentration. (B) Production of Neu5Ac from 100 mM GlcNAc and 100 mM pyruvate over 50 h at 30°C using 10 mg (wet weight) of NanA- and Slr1975-displaying beads. NanA-PhaC plus PhaC-l-Slr1975 beads (2.5 mg and 7.5 [wet weight], respectively) yielded 19% ManNAC and 22% Neu5Ac conversion of the starting GlcNAc concentration. Error bars represent ± 1 standard deviation; n = 3.
DISCUSSION
Previous research has demonstrated that PHA beads are a useful platform for protein immobilization (16). Enzymes immobilized to PHA beads have shown high density display, high activity, enhanced stability, and improved kinetic properties (14), suggesting a role for this technology in fine-chemical synthesis. The one-step production and immobilization of the target enzymes NanA and Slr1975 offer significant advantages over previous multistep immobilization techniques (e.g., 7).
The PhaC fusion proteins were overproduced using the pET vector expression system in E. coli, leading to a high level of protein production on the PHA bead surface as exhibited by the NanA-PhaC and PhaC-l-Slr1975 enzyme immobilizations of 4.5 μg/mg and 7.8 μg/mg wet bead weight, respectively (Fig. 1; see also Fig. S3 in the supplemental material). In contrast, the NanA-PhaC-l-Slr1975 double fusion displayed 1.6 μg/mg wet bead weight (i.e., less fusion protein on the bead surface). It has been previously suggested that the N terminus of PhaC has an important interaction with its C terminus (19). Therefore, the lower level of protein immobilization seen on the NanA-PhaC-l-Slr1975 PHA beads may be explained by the steric hindrance of this interaction due to attaching fusion partners on both termini of the PhaC enzyme.
The epimerase activity of the PhaC-l-Slr1975 fusion protein was 1.8 U/mg protein compared to 0.58 U/mg for the NanA-PhaC-l-Slr1975 fusion protein (Table 1). Reported activities of immobilized GlcNAc-2 epimerases range from 3.4 to 29 U/mg protein (11, 12). The C terminus of PhaC has been recognized as important for PhaC attachment to the bead surface (19), and disruption of the PhaC C terminus may explain both the lower level of fusion protein attachment seen in the double fusion protein and its lower epimerase activity.
In contrast, the aldolase activity of NanA-PhaC was 43 U/mg protein compared with 82 U/mg for the NanA-PhaC-l-Slr1975 fusion protein (Table 2). The assembly of the tetrameric structure of NanA may be assisted by the lower levels of protein expression found on the NanA-PhaC-l-Slr1975 bead surface.
The equilibrium point for the epimerization of GlcNAc to ManNAc has been reported as 1:4 (11), matching the observed 18% conversion of 100 mM GlcNAc after 20 h by the beads displaying only the epimerase. In contrast, the double-enzyme bead was able to convert only 4% of the initial GlcNAc over the same reaction time (Table 3). The low reaction yield of the epimerase portion of the double fusion can be explained by its low activity measurement. Additionally, the PhaC-l-Slr1975 beads were able to produce this conversion rate over at least five reaction cycles, showing that these beads can retain their catalytic activity for sustained periods (100 h) at relatively high temperature (Fig. 2).
The production of Neu5Ac using high concentrations of ManNAc and pyruvate was particularly successful, with conversion rates of 77% and 70% from the aldolase-only and double-enzyme beads, respectively (Table 3). Although the fusion protein for the double-enzyme bead exhibits higher aldolase activity than beads displaying only the aldolase, the abundance of fusion protein on the surface of the aldolase-only beads means that the two beads have similar aldolase activities as a function of bead weight. As the two beads have high aldolase activities, the activity of PHA-immobilized NanA compares favorably with the previously reported activities of 2.5 to 36 U/mg protein (11, 12), and they performed similarly over the 44-h reaction time. Recycling reactions showed that NanA-PhaC beads retain at least 70% conversion of ManNAc over four reaction cycles (Fig. 3). The increase in yield seen in the second reaction may have been due to carryover of reactants associated with the NanA enzyme to the next reaction.
Ultimately, the levels of applicability of the two systems to the production of Neu5Ac from GlcNAc and pyruvate were compared by equalizing the amounts of enzyme beads added to the reaction on a per-mass basis. As pyruvate is an inhibitor of Slr1975, its concentration must be kept below 100 mM (26) even though it is driving the reaction toward Neu5Ac. Given these constraints, the production system composed of 5 mg aldolase-only beads with 5 mg of epimerase-only beads produced 14 mM Neu5Ac (4.7% conversion of GlcNAc) whereas 10 mg of the double-enzyme beads produced only 5.1 mM Neu5Ac (1.7% conversion of GlcNAc). Despite the higher aldolase activity of the dual-enzyme beads, a Neu5Ac production system composed of single-enzyme immobilization is the more effective option (Fig. 4). Both the activity measurements of Slr1975-displaying beads and the low yield of ManNAc in the full biosynthesis pathway suggested that the Slr1975-displaying bead was not performing optimally in the double-bead system. Therefore, a further set of reaction conditions hypothesized to favor conversion of GlcNAc to ManNAc by Slr1975 was tested. The production system consisting of single-enzyme immobilizations (7.5 mg of PhaC-l-Slr1975 beads and 2.5 mg of NanA-PhaC beads) was able to convert 22% of the starting GlcNAc to Neu5Ac (Fig. 4); this compares to the traditional approach utilizing alkaline epimerization and free NanA, which has been reported to yield 33% N-acetylneuraminic acid (27).
Conclusion.
This study demonstrated that NanA and Slr1975 can be successfully immobilized on the PHA biobead system by fusion to PhaC. Both NanA and Slr1975 retained enzymatic activity in both single and double fusions to PhaC, and the resulting functionalized beads could be produced in a one-step, cost-efficient bacterial fermentation that is amenable to industrial-scale production (28). Recycling experiments showed that the single-fusion beads could be recovered and reused several times before losing their initial yield potential, and the overall biosynthesis pathway from GlcNAc to Neu5Ac was shown to perform at a relatively high level (22% yield), which may make the PHA biobead platform suitable for use as a biocatalytic production system in fine-chemical synthesis.
ACKNOWLEDGMENTS
This study was supported by the New Zealand Foundation for Research Science and Technology, the Institute of Molecular Biosciences at Massey University, and PolyBatics Ltd. (Palmerston North, New Zealand). D.O.H. received a Ph.D. scholarship from Massey University. The purchase of the Phenomenex RHM HPLC columns was funded by New Zealand Pharmaceuticals, Ltd. (Palmerston North, New Zealand).
Footnotes
Published ahead of print 1 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03947-12.
REFERENCES
- 1. Traving C, Schauer R. 1998. Structure, function and metabolism of sialic acids. Cell. Mol. Life Sci. 54:1330–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brusés JL, Rutishauser U. 2001. Roles, regulation, and mechanism of polysialic acid function during neural development. Biochimie 83:635–643 [DOI] [PubMed] [Google Scholar]
- 3. Mahdavi J, Sondén B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, ltraja S, Wadström T, Kersulyte D, Berg DE, Dubois A, Petersson C, Magnusson KE, Norberg T, Lindh F, Lundskog BB, Arnqvist A, Hammarström L, Borén T. 2002. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 297:573–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schauer R. 2004. Sialic acids: fascinating sugars in higher animals and man. Zoology 107:49–64 [DOI] [PubMed] [Google Scholar]
- 5. von Itzstein M. 2007. The war against influenza: discovery and development of sialidase inhibitors. Nat. Rev. Drug Discov. 6:967–974 [DOI] [PubMed] [Google Scholar]
- 6. Rodríguez-Aparicio LB, Ferrero MA, Reglero A. 1995. N-Acetyl-D-neuraminic acid synthesis in Escherichia coli K1 occurs through condensation of N-acetyl-D-mannosamine and pyruvate. Biochem. J. 308(Pt 2):501–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Luchansky SJ, Yarema KJ, Takahashi S, Bertozzi CR. 2003. GlcNAc 2-epimerase can serve a catabolic role in sialic acid metabolism. J. Biol. Chem. 278:8035–8042 [DOI] [PubMed] [Google Scholar]
- 8. Katchalski-Katzir E. 1993. Immobilized enzymes—learning from past successes and failures. Trends Biotechnol. 11:471–478 [DOI] [PubMed] [Google Scholar]
- 9. Novick SJ, Rozzell JD. 2005. Immobilization of enzymes by covalent attachment, p 247–271. In Barredo JL. (ed), Microbial enzymes and biotransformations. Springer, Secaucus, NJ [Google Scholar]
- 10. Sheldon RA. 2007. Enzyme immobilization: the quest for optimum performance. Adv. Synth. Catal. 349:1289–1307 [Google Scholar]
- 11. Hu S, Chen J, Yang Z, Shao L, Bai H, Luo J, Jiang W, Yang Y. 2010. Coupled bioconversion for preparation of N-acetyl-D-neuraminic acid using immobilized N-acetyl-D-glucosamine-2-epimerase and N-acetyl-D-neuraminic acid lyase. Appl. Microbiol. Biotechnol. 85:1383–1391 [DOI] [PubMed] [Google Scholar]
- 12. Wang T-H, Chen Y-Y, Pan H-H, Wang F-P, Cheng C-H, Lee W-C. 2009. Production of N-acetyl-D-neuraminic acid using two sequential enzymes overexpressed as double-tagged fusion proteins. BMC Biotechnol. 9:63 doi:10.1186/1472-6750-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu X, Gao C, Zhang X, Che B, Ma C, Qiu J, Tao F, Xu P. 2011. Production of N-acetyl-D-neuraminic acid by use of an efficient spore surface display system. Appl. Environ. Microbiol. 77:3197–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rasiah IA, Rehm BHA. 2009. One-step production of immobilized alpha-amylase in recombinant Escherichia coli. Appl. Environ. Microbiol. 75:2012–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blatchford PA, Scott C, French N, Rehm BHA. 2012. Immobilization of organophosphohydrolase OpdA from Agrobacterium radiobacter by overproduction at the surface of polyester inclusions inside engineered Escherichia coli. Biotechnol. Bioeng. 109:1101–1108 [DOI] [PubMed] [Google Scholar]
- 16. Grage K, Jahns AC, Parlane N, Palanisamy R, Rasiah IA, Atwood JA, Rehm BHA. 2009. Bacterial polyhydroxyalkanoate granules: biogenesis, structure, and potential use as nano-/micro-beads in biotechnological and biomedical applications. Biomacromolecules 10:660–669 [DOI] [PubMed] [Google Scholar]
- 17. Peters V, Rehm BHA. 2006. In vivo enzyme immobilization by use of engineered polyhydroxyalkanoate synthase. Appl. Environ. Microbiol. 72:1777–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sambrook J, Fritsch E, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 19. Jahns AC, Rehm BHA. 2009. Tolerance of the Ralstonia eutropha class I polyhydroxyalkanoate synthase for translational fusions to its C terminus reveals a new mode of functional display. Appl. Environ. Microbiol. 75:5461–5466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peters V, Rehm BHA. 2005. In vivo monitoring of PHA granule formation using GFP-labeled PHA synthases. FEMS Microbiol. Lett. 248:93–100 [DOI] [PubMed] [Google Scholar]
- 21. Brandl H, Gross RA, Lenz RW, Fuller RC. 1988. Pseudomonas oleovorans as a source of poly(beta-hydroxyalkanoates) for potential applications as biodegradable polyesters. Appl. Environ. Microbiol. 54:1977–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jahns AC, Haverkamp RG, Rehm BHA. 2008. Multifunctional inorganic-binding beads self-assembled inside engineered bacteria. Bioconjug. Chem. 19:2072–2080 [DOI] [PubMed] [Google Scholar]
- 23. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 24. Dawson M, Noble D, Mahmoudian M. December 2000. Process for the preparation of N-acetylneuraminic acid. US patent 6,156,544. [Google Scholar]
- 25. Wang T-H, Lee W-C. 2006. Expression and characterization of the N-acetyl-D-glucosamine 2-epimerase as a tagged protein for the conversion of N-acetyl-D-glucosamine to N-acetyl-D-mannosamine. J. Chin. Inst. Chem. Eng. 37:131–137 [Google Scholar]
- 26. Lee J-O, Yi J-K, Lee S-G, Takahashi S, Kim B-G. 2004. Production of N-acetylneuraminic acid from N-acetylglucosamine and pyruvate using recombinant human renin binding protein and sialic acid aldolase in one pot. Enzyme Microb. Technol. 35:121–125 [Google Scholar]
- 27. Blayer S, Woodley JM, Dawson MJ, Lilly MD. 1999. Alkaline biocatalysis for the direct synthesis of N-acetyl-D-neuraminic acid (Neu5Ac) from N-acetyl-D-glucosamine (GlcNAc). Biotechnol. Bioeng. 66:131–136 [DOI] [PubMed] [Google Scholar]
- 28. Rehm BHA. 2010. Bacterial polymers: biosynthesis, modifications and applications. Nat. Rev. Microbiol. 8:578–592 [DOI] [PubMed] [Google Scholar]