

Abstract
Human immunodeficiency virus type 1 (HIV-1) infection of most human cells is dependent on cyclophilin A (CypA); however, the opposite phenomenon, known as CypA-dependent inhibition, is also observed in the combination of some capsid (CA) mutations and cell lines. Here, we identified a CA N121K mutant whose infection of 293T, Jurkat, and HeLa cells was impaired by CypA. The N121K mutant could be a useful tool for analyzing the mechanisms underlying CypA-dependent restriction.
TEXT
The host cellular protein cyclophilin A (CypA) binds to the proline-rich loop in the N-terminal region of the human immunodeficiency virus type 1 (HIV-1) capsid (CA) protein (1, 2). Inhibition of the CA-CypA interaction by using competitive chemicals such as cyclosporine (CsA), introduction of mutations into the CypA-binding loop within CA, or RNA interference (RNAi)-mediated knockdown of endogenous CypA in target cells leads to reduced HIV-1 infectivity (3, 4). An initial model assumed that the incorporation of CypA into the virions facilitates uncoating of the HIV-1 core. However, recent studies show that the CA-CypA interaction in target cells, rather than that of virus-incorporated CypA, is important for HIV-1 infection (5, 6). CypA catalyzes the cis-trans isomerization of proline peptide bonds, leading to conformational changes in CA and, in turn, the core structure. Although the CA-CypA interaction has been well characterized in vitro, the precise role played by CypA during HIV-1 replication is still unclear (3, 4). Recent reports indicate that the CA-CypA interaction is involved not only in uncoating but also in nuclear import and integration (7, 8).
HIV-1 infection of most human cells is dependent on CypA; however, the opposite phenomenon, known as CypA-dependent inhibition, is observed for some CA mutant viruses. A previous study showed that the passage of wild-type (WT) HIV-1 in HeLa-CD4 cells (when CypA was inhibited by CsA) resulted in the generation of two CA escape mutants, A92E and G94D (9). These mutations tolerate the inhibition of CA-CypA interaction, but their replication in HeLa and H9 cells (although not in other cell lines such as 293T and Jurkat cells) becomes dependent on CsA (6, 10). Several CA mutants, such as R132K and T54A, showed similar CsA-dependent phenotypes in a target cell-contingent manner (11–13). These results imply the bidirectional function of CypA during HIV-1 replication. To better understand the function of CypA during HIV-1 replication, the viral regions that modulate CypA dependence must be identified. To this end, we tried to isolate HIV-1 mutants that can replicate in Jurkat cells in the absence of CypA. The newly isolated CA mutant N121K was generated by serial passage of a CypA-nonbinding HIV-1 mutant (NL4-3.P90A.A92E) in Jurkat cells (14). In this paper, we report that N121K infection is inhibited in a CypA-dependent manner not only in HeLa and H9 cells but also in 293T and Jurkat cells.
We attempted to isolate HIV-1 mutants that replicate in Jurkat cells in the absence of CypA; HIV-1 infection of Jurkat cells is CypA dependent. Jurkat cells were infected with either WT virus or a CypA-nonbinding mutant, NL4-3.P90A.A92E, in the presence or absence of 1 μM cyclosporine (CsA) as follows: culture I, NL4-3 without CsA; culture II, NL4-3 plus 1 μM CsA; culture III, NL4-3.P90A.A92E without CsA; and culture IV, NL4-3.P90A.A92E plus 1 μM CsA. After repeated passage (47 times), we succeeded in obtaining replication-competent viruses under all culture conditions. Sequence analysis of the entire gag region identified a virus from culture II that harbored a mutation in MA (V34I). The CA N121K and CA R132K mutants arose from cultures III and IV, respectively. The mutation in MA overlapped with a mutation reported to increase viral fitness in T-cell cultures (15). Therefore, we studied two CA mutant viruses in detail. Since the R132K substitution identified in the virus from culture IV had also been identified previously (11, 16), we focused on the N121K mutant (which harbored a mutation in the loop between helix 6 and helix 7), which arose from culture III (Jurkat cells infected with NL4-3.P90A.A92E in the absence of CsA).
We constructed an N121K substitution virus on a WT (NL4-3 or NL4-3-luc) and the CypA-nonbinding mutant (NL4-3.P90A.A92E or NL4-3.P90A.A92E-luc) backbone. Viral replication was then examined in Jurkat and H9 cells, and infection efficiency was examined in Jurkat, 293T, and HeLa cells. Figure 1A to D shows the replication kinetics of the WT and CA mutant viruses in Jurkat and H9 cells in the presence or absence of 1 μM CsA. The triple mutant virus (N121K.P90A.A92E) replicated as efficiently as did the WT virus in the presence or absence of CsA in both cell lines. However, the replication of the N121K single mutant virus in both Jurkat and H9 cells increased in the presence of CsA (Fig. 1B and D). Similar results were observed in the single-round infection experiments (Fig. 1E to G). Infection by the N121K.P90A.A92E mutant was not impaired in any of the cell lines, in either the presence or the absence of CsA. However, the infection of Jurkat, 293T, and HeLa cells by N121K increased in the presence of 1 μM CsA.
Fig 1.

Inhibition of the CA-CypA interaction rescues the infectivity of N121K HIV-1. (A to D) Jurkat (A and B) or H9 (C and D) cells were infected with 1 ng of p24 of wild-type (WT) or CA mutant (P90A.A92E, A92E, N121K, and N121K.P90A.A92E) HIV-1 in the presence or absence of 1 μM CsA. Virus replication was determined by enzyme-linked immunosorbent assay of p24 in the culture supernatant. (E to G) Jurkat (E), 293T (F), or HeLa (G) cells were infected (in the presence or absence of CsA) with vesicular stomatitis virus G protein-pseudotyped WT or CA mutant viruses carrying the luciferase gene. After 48 h, cellular luciferase activity was determined using the Steady-Glo luciferase assay system (Promega). (H) HeLa cells were transduced with vectors expressing a nontarget (nonT) or CypA-targeted short hairpin RNA (CypAsh) in the absence or presence of 10 μg/ml of aphidicolin (APC; Sigma-Aldrich) and then infected with the pseudotyped WT or CA mutant viruses. Error bars represent the standard deviations (from parallel cultures performed in triplicate). The results are representative of at least three independent experiments.
Previous studies show that CypA-dependent inhibition of infection by some CA mutants is greater in nondividing cells (16, 17). Therefore, to investigate whether the mechanism underlying the CypA-dependent inhibition of infection by N121K mutants is similar to that observed for other CA mutants such as A92E, we tested whether cell growth arrest of CypA- or nontarget short hairpin RNA (shRNA)-transduced HeLa cells further inhibited infection by N121K. CypA knockdown by shRNA restored the infection efficiency of the N121K mutant (Fig. 1H), suggesting that N121K is inhibited by CypA in the cytoplasm of the target cells as previously observed for other CsA-dependent mutant viruses (5, 18). Aphidicolin-induced growth arrest markedly increased the level of N121K inhibition in HeLa cells (as was observed in A92E mutants) (16, 17).
The A105T mutation is suppressive in CsA-dependent CA mutants, such as A92E (12). Therefore, we next asked whether the A105T mutation suppressed the inhibition of N121K infectivity in 293T and HeLa cells. The infectivity of a dual mutant (N121K.A105T) virus was approximately 10-fold higher than that of a single mutant N121K virus in both 293T and HeLa cells. The A105T mutation fully restored the infection efficiency of the N121K mutant virus in 293T cells (Fig. 2A). In contrast, the A105T-mediated rescue of infection efficiency in HeLa cells was only moderate, and it was increased by the addition of CsA (Fig. 2B). Interestingly, the infection efficiency of the single A105T mutant decreased in both HeLa cells and 293T cells in the presence of CsA. Yang and Aiken suggested two possible explanations for why the A105T mutation may compensate for the structural changes caused by CypA or block the potent inhibitory factor activated by CypA-dependent manner (12).
Fig 2.
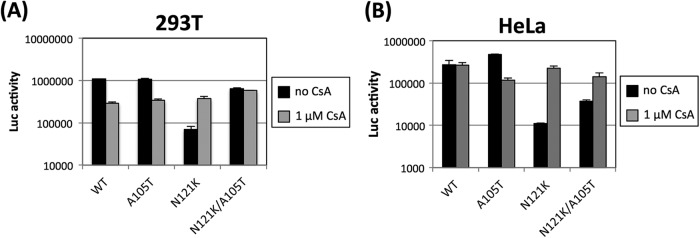
Rescue of N121K HIV-1 infection by the CA A105T mutation. Single-cycle infectivity was determined by infection with the vesicular stomatitis virus G protein-pseudotyped CA mutant virus carrying the luciferase gene. A total of 5 × 104 293T (A) or HeLa (B) cells were inoculated with 1 ng of p24 from each virus. After 48 h, the luciferase activity was measured. Error bars represent the standard deviations (for parallel cultures performed in triplicate). The results are representative of at least three independent experiments.
We next determined which N121K replication step(s) is regulated by cellular CypA by measuring the synthesis of new reverse transcription (RT) products, or the production of two-long-terminal-repeat (2-LTR) circular DNA products, by quantitative real-time PCR (19) in the presence or absence of 1 μM CsA. Viral infectivity was also measured in parallel (Fig. 3A). There was no clear difference between the early and late N121K RT products in the presence or absence of CsA (Fig. 3B and C). However, the addition of CsA increased the amount of N121K 2-LTR circular DNA products (Fig. 3D). These results indicate that the replication of N121K is impaired at the nuclear transport stage. Although it is thought that the inhibition of these viruses occurs at either the RT, nuclear import, or integration stages, the replication step impaired by CypA (with regard to the A92E and G94D mutant viruses) is still controversial (12, 16, 17, 20). Our quantitative PCR data show that the inhibition of N121K replication occurs at the nuclear import stage, supporting previous data showing that the CA-CypA interaction is involved in a series of viral replication steps at the nuclear import stage (8, 17).
Fig 3.

The replication defect observed in N121K virus occurs at the nuclear import stage. HeLa cells (2 × 105) were infected with 4 ng of p24 from vesicular stomatitis virus G protein-pseudotyped WT, A92E, or N121K virus (harboring the luciferase gene). The infection was performed under the following conditions: no drug, 1 μM CsA, 1 μM azidothymidine (AZT; Sigma-Aldrich), and 1 μM raltegravir (RAL). (A) Virus infectivity was determined by measuring the luciferase activity 2 days after infection. Total DNA was extracted 24 h (B and C) or 36 h (D) after infection, and 200 ng of the DNA was subjected to quantitative PCR (19). The error bars represent the standard deviations from duplicate assays performed in a single experiment. The results are representative of two independent experiments. UD, undetectable.
Next, we conducted infection experiments by titrating the dose of CsA and examining the effects on the infectivity of WT, A92E, and N121K viruses. 293T or HeLa cells were incubated with 0.5 ng of virus p24 in the presence of increasing concentrations of CsA. Along with the increase of the CsA concentration, the infectivity of the WT virus gradually decreased to approximately one-fifth of that of the virus in the control infection without CsA in 293T cells (Fig. 4A). The infectivity of the A92E virus was not affected by the CsA concentration in 293T cells. In contrast, the infectivity of the N121K virus gradually increased with increasing CsA concentration (up to 0.15 μM CsA). The infection efficiencies of N121K and the WT virus were comparable at CsA concentrations above 1.25 μM. In HeLa cells, the titration curve for the N121K virus was similar to that of the A92E virus; however, the infectivity of N121K was less than that of A92E at most CsA concentrations tested (Fig. 4B). The dual mutant virus (N121K.A92E) also showed increased infectivity in line with the increasing CsA concentration, although the infectivity was about 10-fold less than that of N121K in 293T cells and that of A92E and N121K in HeLa cells. This suggests that CypA-dependent restriction of the N121K.A92E dual mutant is increased in both cell types. Although the underlying mechanisms are still unclear, these data suggest that the CypA-dependent restriction factor(s) is expressed at different levels in 293T and HeLa cells. If the sensitivity of N121K to this hypothetical factor is higher than that of A92E, this may account for the different infectivities of A92E in 293T and HeLa cells. N121K might be very sensitive to this factor, and the A92E.N121K mutant is even more sensitive because A92E is potently restricted by the as-yet-unknown factor. This hypothesis can explain the different levels of N121K toleration shown by the introduction of an additional A105T mutation shown in Fig. 2.
Fig 4.
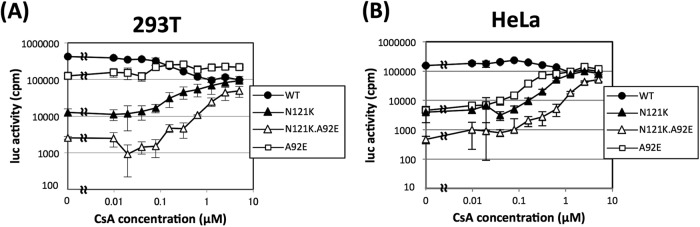
Effects of titrating CsA on the infectivity of WT, A92E, N121K, and N121K.A92E HIV-1. In the presence of different concentrations of CsA, 4 × 104 293T cells (A) or HeLa cells (B) were infected with 0.5 ng of p24 from vesicular stomatitis virus G protein-pseudotyped HIV-1 carrying the luciferase gene as a marker. Cellular luciferase activity was measured after 48 h. The error bars represent the standard deviations from parallel cultures performed in triplicate. The results are representative of two independent experiments.
In this report, we infected 293T, Jurkat, and HeLa cells with a CA mutant virus, N121K, which has a CsA-dependent phenotype. Although a similar CsA-dependent mutant virus, such as A92E, was previously identified, the CsA dependence was observed only in high-CypA-expressing cells, such as HeLa and H9 (9, 11, 12, 21). Two hypotheses have been proposed to explain CsA-dependent infection by CA mutants. The first is that high levels of cellular CypA alter the stability and uncoating of the mutant viral core; the second is that a potent, as-yet-unknown HIV-1-inhibitory factor(s) interrupts the proper timing of uncoating or disrupts nuclear import or integration (5, 12, 18, 20). In the present study, N121K replication was disrupted after the RT step and the magnitude of this inhibition varied in a cell-type-dependent manner. Inhibition of N121K virus infection appears to be stronger in HeLa cells than in 293T cells, and this can be explained by the potent as-yet-unknown restrictive factor differentially expressed in 293T and HeLa cells. Further work is needed to validate this model. The N121K mutation is located in the loop between helices 6 and 7. Two different groups reported that human TRIM5α inhibits N-tropic murine leukemia virus (N-MLV) infection in human cells by recognizing a comparable region within the viral CA (in the loop between helix 6 and helix 7) (22, 23). Considering the conformational homology between N-MLV and HIV-1, this region might be accessible to human TRIM5α. We knocked down endogenous TRIM5α in HeLa cells but found no significant difference in the infectivities of the A92E and N121K mutants (data not shown). This suggests that TRIM5α does not play a role in the CypA-dependent restriction of A92E and N121K. Still, the possibility that potent, as-yet-unknown HIV-1-inhibitory factors, which associate with CypA, may be able to access this region cannot be ruled out (5).
Although CypA supports HIV-1 infection, some CA mutants showed CypA-dependent impairment of infection. Our data indicate that CypA impairs N121K infection, even in 293T and Jurkat cells that do not express CypA at very high levels. One of the proposed mechanisms for CypA-dependent restriction was that the high CypA expression level and mutation in CA modulate the stability of viral core (21). In our data, addition of CsA rescues the infection of N121K virus in 293T cells, but the infection efficiency of N121K does not exceed that of the WT virus infection even at the highest CsA concentration. It could be said that the infection of N121K might require CypA for its infection, and CypA facilitates the HIV-1 infection at different stage of replication. Indeed, CsA treatment reduced the infectivity of the WT virus in 293T cells, and restriction was observed at the early RT production step (data not shown). The stability of viral core alone cannot explain why both CypA-dependent infection and CypA-mediated inhibition are observed for the same virus. Therefore, we could assume that a potent HIV-1-inhibitory factor(s) is also involved in the CypA-dependent restriction and that each CA mutant has a different sensitivity for it. Thus, the N121K CA mutant could be a useful tool for analyzing the mechanism(s) underlying CypA-dependent restriction of HIV-1 infection. Understanding this mechanism may also help to determine the exact role played by CypA during HIV-1 replication.
ACKNOWLEDGMENTS
This work was supported by grants-in aid from the Ministry of Health, Labor, and Welfare, Japan, awarded to T.M. and T.T.
We thank Harumi Saida and Yuya Mitsuki for technical support. The following reagent was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: raltegravir (catalog number 11680) from Merck & Company, Inc.
Footnotes
Published ahead of print 16 January 2013
REFERENCES
- 1. Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, Hill CP. 1996. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87:1285–1294 [DOI] [PubMed] [Google Scholar]
- 2. Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP. 1993. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73:1067–1078 [DOI] [PubMed] [Google Scholar]
- 3. Goff SP. 2004. Genetic control of retrovirus susceptibility in mammalian cells. Annu. Rev. Genet. 38:61–85 [DOI] [PubMed] [Google Scholar]
- 4. Mascarenhas AP, Musier-Forsyth K. 2009. The capsid protein of human immunodeficiency virus: interactions of HIV-1 capsid with host protein factors. FEBS J. 276:6118–6127 [DOI] [PubMed] [Google Scholar]
- 5. Hatziioannou T, Perez-Caballero D, Cowan S, Bieniasz PD. 2005. Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J. Virol. 79:176–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Song C, Aiken C. 2007. Analysis of human cell heterokaryons demonstrates that target cell restriction of cyclosporine-resistant human immunodeficiency virus type 1 mutants is genetically dominant. J. Virol. 81:11946–11956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman DR, Coffin JM, Hughes SH, Unutmaz D, Engelman A, KewalRamani VN. 2010. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7:221–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hué S, Fletcher AJ, Lee K, KewalRamani VN, Noursadeghi M, Jenner RG, James LC, Bushman FD, Towers GJ. 2011. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 7:e1002439 doi:10.1371/journal.ppat.1002439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aberham C, Weber S, Phares W. 1996. Spontaneous mutations in the human immunodeficiency virus type 1 gag gene that affect viral replication in by the presence of cyclosporins. J. Virol. 70:3536–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Towers GJ, Hatziioannou T, Cowan S, Goff SP, Luban J, Bieniasz PD. 2003. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat. Med. 9:1138–1143 [DOI] [PubMed] [Google Scholar]
- 11. Schneidewind A, Brockman MA, Yang R, Adam RI, Li B, Le Gall S, Rinaldo CR, Craggs SL, Allgaier RL, Power KA, Kuntzen T, Tung CS, LaBute MX, Mueller SM, Harrer T, McMichael AJ, Goulder PJ, Aiken C, Brander C, Kelleher AD, Allen TM. 2007. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J. Virol. 81:12382–12393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang R, Aiken C. 2007. A mutation in alpha helix 3 of CA renders human immunodeficiency virus type 1 cyclosporine A resistant and dependent: rescue by a second-site substitution in a distal region of CA. J. Virol. 81:3749–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. 2003. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 77:5439–5450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Braaten D, Franke EK, Luban J. 1996. Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J. Virol. 70:3551–3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murakami T, Freed EO. 2000. Genetic evidence for an interaction between human immunodeficiency virus type 1 matrix and alpha-helix 2 of the gp41 cytoplasmic tail. J. Virol. 74:3548–3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qi M, Yang R, Aiken C. 2008. Cyclophilin A-dependent restriction of human immunodeficiency virus type 1 capsid mutants for infection of nondividing cells. J. Virol. 82:12001–12008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamashita M, Emerman M. 2009. Cellular restriction targeting viral capsids perturbs human immunodeficiency virus type 1 infection of nondividing cells. J. Virol. 83:9835–9843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sokolskaja E, Sayah DM, Luban J. 2004. Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J. Virol. 78:12800–12808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Urano E, Kuramochi N, Ichikawa R, Murayama SY, Miyauchi K, Tomoda H, Takebe Y, Nermut M, Komano J, Morikawa Y. 2011. Novel postentry inhibitor of human immunodeficiency virus type 1 replication screened by yeast membrane-associated two-hybrid system. Antimicrob. Agents Chemother. 55:4251–4260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Y, Kar AK, Sodroski J. 2009. Target cell type-dependent modulation of human immunodeficiency virus type 1 capsid disassembly by cyclophilin A. J. Virol. 83:10951–10962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ylinen LM, Schaller T, Price A, Fletcher AJ, Noursadeghi M, James LC, Towers GJ. 2009. Cyclophilin A levels dictate infection efficiency of human immunodeficiency virus type 1 capsid escape mutants A92E and G94D. J. Virol. 83:2044–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maillard PV, Zoete V, Michielin O, Trono D. 2011. Homology-based identification of capsid determinants that protect HIV1 from human TRIM5α restriction. J. Biol. Chem. 286:8128–8140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ohkura S, Goldstone DC, Yap MW, Holden-Dye K, Taylor IA, Stoye JP. 2011. Novel escape mutants suggest an extensive TRIM5α binding site spanning the entire outer surface of the murine leukemia virus capsid protein. PLoS Pathog. 7:e1002011 doi:10.1371/journal.ppat.1002011 [DOI] [PMC free article] [PubMed] [Google Scholar]