

Abstract
Gene therapy is the modification of gene expression to treat a disease. However, efficient intracellular delivery and monitoring of gene therapeutic agents is an ongoing challenge. Use of theranostic agents with suitable targeted, controlled delivery and imaging modalities has the potential to greatly advance gene therapy. Inorganic nanoparticles including magnetic nanoparticles, gold nanoparticles, and quantum dots have been shown to be effective theranostic agents for the delivery and spatiotemporal tracking of oligonucleotides in vitro and even a few cases in vivo. Major concerns remain to be addressed including cytotoxicity, particularly of quantum dots; effective dosage of nanoparticles for optimal theranostic effect; development of real-time in vivo imaging; and further improvement of gene therapy efficacy.
Keywords: gene delivery, quantum dots, gold nanoparticles, magnetic nanoparticles, imaging
1. Introduction
Gene therapy, or the modification of gene expression via nucleic acids to treat inherited or acquired disease, has become a tremendous area of research over the past 20 years. It has been demonstrated as a potential treatment for an extensive range of diseases including cystic fibrosis, age-related macular degeneration, cardiovascular disease, and various cancers, among many others. [1-3] Gene therapy necessitates delivery of nucleic acids, such as DNA, antisense oligonucleotides (ASON), small interfering RNA (siRNA), micro RNA (mRNA), or small hairpin RNA (shRNA), not only to the target tissue but also into the cytosol of the cells.[4] This has proven to be no easy feat; nucleic acids are highly susceptible to degradation by nucleases and harsh conditions found within the body, and once entrapped within the endosome of a cell the cargo must escape to the cytosol to initiate gene therapy.[5-7] Therefore, copious research has focused on the development of gene transfection vectors, which are designed to transport the nucleic acids to specific tissue and facilitate intracellular delivery into the cytosol.
Gene transfection vectors are categorized as viral or nonviral, also known as synthetic, vectors. While viral vectors have very high transfection efficiency, they also present undesirable or even fatal side effects such as endogenous virus recombination, oncogenic effects, and unanticipated immune response.[8] Synthetic vectors, such as lipids, peptides, and polymers, avoid these deleterious responses and also have advantages such as ease of use and scalable production.[8] Further, a synthetic vector which not only delivers the genetic material but also possesses a targeting or imaging modality has monumental value.
Such theranostic agents are advantageous to polymer or liposome vectors alone because they offer improved disease treatment by their ability to target delivery as well as monitor therapeutic localization in a noninvasive manner.[9] Theranostic agents are generally comprised of a nanoscale inorganic component, such as gold or iron oxide particles, that can be modified with a lipid or polymeric coating, functional group, and/or targeting moiety, and then loaded with a therapeutic agent. This review highlights recent advances in the use of theranostic, inorganic-based particles for the delivery and spatiotemporal tracking of oligonucleotide transfection and gene therapy, which are summarized in Table 1.
Table 1.
Summary of inorganic theranostic particles used in gene therapy.
| Particle Type | Imaging | Delivery/release method | Coating | In vitro or in vivo | Gene Therapy | Ref. |
|---|---|---|---|---|---|---|
| MNP | Fluoresence | Magnetofection | Acidic/Neutral PEI | In vitro | DNA | [98] |
| MNP | Fluoresence | Magnetofection | Chitosan | In vitro | DNA | [99] |
| MNP | Fluoresence | Magnetofection | PEI | In vitro | DNA/siRNA | [37] |
| MNP | Fluoresence | Magnetofection | PPI dendrimer | In vitro/In vivo | siRNA | [100] |
| MNP | Fluoresence | Magnetofection | PAMAM dendrimer | In vitro | asODN | [101] |
| MNP | Fluoresence | Magnetofection | Chitosan/PEI/PEG | In vitro | DNA | [23] |
| MNP | Fluoresence | Magnetofection | ODN | In vitro | DNA | [102] |
| MNP | Fluoresence | Magnetofection | PEI/DNA | In vitro | DNA | [29] |
| MNP | MRI/Fluoresence | Magnetofection | Dextran | In vitro | shRNA | [103] |
| MNP | Fluoresence | Magnetofection | Cationic Lipid | In Vivo | siRNA/DNA | [104] |
| MNP | Fluoresence | Magnetofection | Cationic Lipid/PEI | In Vitro | pDNA | [105] |
| MNP | Fluoresence | Magnetofection | Cationic Lipid | In Vitro | DNA | [33] |
| MNP | Fluoresence | Magnetofection | Clustered PEI MNPs | In Vitro | siRNA | [35] |
| MNP | Fluoresence | Magnetofection | deacylated PEI | In vitro | pDNA | [106] |
| MNP | Fluoresence | Magnetofection | DNA | In vitro | DNA | [107] |
| MNP | Fluoresence | Magnetofection | DNA | In vitro | DNA | [108] |
| MNP | Fluoresence | Magnetofection | Cationic Lipospheres | In vitro | siRNA | [109] |
| AuNR | SERS, Fluorescence microscopy, Maestro | N/A | PEG | In vivo (mouse) | N/A | [55] |
| AuNP | SERS, Dark-field microscopy, TEM | Electrostatic | PEI/chitosan | In vitro | shRNA | [52] |
| AuNS | Fluorescence microscopy, Flow cytometry | Laser-induced | In vitro | ssDNA | [56] | |
| AuNS | Fluorescence microscopy, Dark-field microscopy | Laser-induced | L-lysine | In vitro | siRNA, ssDNA | [57] |
| AuNS | Fluorescence microscopy, TEM | Laser-induced | PEG, lipid bilayer, Tat peptide | In vitro | siRNA | [53] |
| AuNP | Fluorescence microscopy | Dicer cleavage | In vitro | siRNA | [58] | |
| AuNP | Light microscopy | Dicer cleavage | In vitro | miRNA | [59] | |
| AuNP | TEM, Dark-field microscopy | Electrostatic | PEI, Hyaluronic acid | In vitro | siRNA | [21] |
| AuNR | Dark-field microscopy | Electrostatic | Polyelectrolytes | In vitro | siRNA | [60] |
| AuNP | TEM, Fluorescent microscopy | N/A | N/A | In vitro | N/A | [50] |
| AuNP | TEM, Fluorescent microscopy | N/A | N/A | In vitro | N/A | [49] |
| AuNS | TEM, Contrast microscopy | N/A | DMSA | In vitro | N/A | [110] |
| QD | Flow cytometry | Lipofectamine 2000 | In vitro | siRNA | [71] | |
| QD | Flow cytometry | Disulfide reduction | F3 peptide | In vitro | siRNA | [72] |
| QD | Fluorescent imaging, TEM | Disulfide reduction | Lipofectamine, RGD and Tat peptides | In vitro | siRNA | [73] |
| QD | Time-lapse fluorescent imaging | In vitro | ASON | [74] | ||
| QD | Confocal microscopy | Electrostatic | PEI-g-PEG | In vitro | [76] | |
| QD | Flow cytometry, Fluorescent imaging | Electrostatic | PEG-amine | In vitro | siRNA | [77] |
| QD | Confocal laser scanning microscopy, TEM | Electrostatic | 2-vinylpyridine | In vitro | siRNA | [78] |
| QD | Confocal microscopy, Flow cytometry | Electrostatic | L-arginine, β-cyclodextrin | In vitro | siRNA | [79] |
| QD | FRET, Time-lapse confocal microscopy | Electrostatic | Amphiphilic polymer | In vitro | siRNA | [82] |
| QD | FRET, Time-lapse confocal microscopy | Electrostatic | PEI | In vitro | siRNA | [81] |
| QD | FRET, Time-lapse confocal microscopy | Electrostatic | Polycation | In vitro | pDNA | [83] |
Inorganic particles, such as iron oxide, gold, and quantum dots, have gained significant interest in the field of bioimaging due to their inherent optical and magnetic properties, as well as their relative ease of processing and resistance to degradation under physiological conditions compared to organic materials.[10] The optical, magnetic, and electrical properties of inorganic particles can be manipulated by their size, composition, geometry, and structure[10, 11], making them an enticing tunable detection modality. Specifically, inorganic nanoparticles are being widely investigated for bioimaging applications such as sensors to identify pathologic processes, diagnostic agents for early detection, and molecular tracking to monitor therapy efficacy.[12, 13]
Particularly intriguing are inorganic particles in the nanometer diameter regime. Such particle size allows for detection specificity and sensitivity on the molecular scale[14, 15] as well as the ability for intracellular uptake.[15] Nanoparticles also provide a large surface area for various modifications including bioconjugation with targeting modalities and therapeutic agents, or functionalization to enhance solubility, biocompatibility, or reactivity.[10, 15, 16] Bioconjugation using methods such as 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC)- N-hydroxysuccinimide (NHS) chemistry or avidin-biotin interaction [17] has been successful and the use of various targeting modalities including proteins, antibodies, and small molecule ligands has been documented.[18-21] Furthermore, given the toxicity concerns associated with inorganic materials, a minimal effective dosage is desirable. Inorganic nanoparticles offer superb detection properties with a small relative dosage, as well as the potential for multi-modal imaging to further enhance dosage efficiency. Some of the most widely used types of inorganic nanoparticles in theranostic applications are magnetic nanoparticles, gold nanoparticles, and semiconductor nanocrystals.
2. Magnetic Nanoparticles
Magnetic nanoparticles (MNPs) provide unique theranostic advantages for gene delivery. Their use as contrast agents for magnetic resonance imaging (MRI) allows for improved tracking inside the body without the limitations that tend to impede imaging methods that depend on the transmission of visible or near infrared light. The susceptibility of MNPs to magnetic fields also allows for improved targeting and transfection via a method commonly referred to as magnetofection.
2.1 Background and Advantages
MNPs used for gene delivery can be classified as either single domain ferromagnetic or superparamagnetic. Single domain ferromagnetic nanoparticles are in a state where the electron orbital spins are aligned in a single direction, which allows for a directional response to a magnetic field. Superparamagnetism occurs when a single domain particle is small enough (<~10nm) that the thermal energy in the environment is enough to cause a switch in the orbital spins.
Magnetic nanoparticles have proven to be effective contrast agents for MRI. This is advantageous as it allows for precise monitoring of particle delivery.[22] The accumulation of MNP-gene vectors can be analyzed quantitatively by calculating translational relaxation rates, R2 values, and comparing pre and post injection images, as seen in Figure 1.[23] MRI also enables tissue differentiation, allowing the accumulation of MNPs to be compared between different tissue types. This option enables non-invasive localization of MNP with respect to different tissues and organs in the body. However, MRI is limited in that it only provides information about the particles, while the presence of the genetic payload has to be assumed. Another limitation is, due to equipment availability and cost, active imaging is not possible. To overcome this disadvantage, MNPs are imaged both by MRI and another fluorescent moiety.[24] The use of dual imaging techniques allows for both real time tracking and precise imaging of gene loaded MNPs in vivo.
Figure 1.
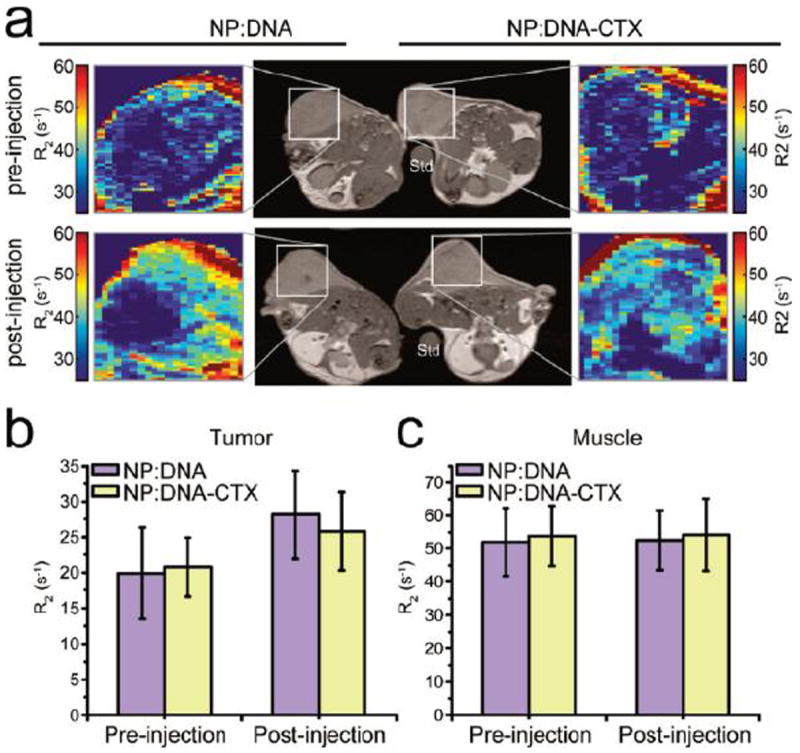
Nanovector delivery to C6 xenograft tumors monitored by MRI. a) T2-weighted images of C6 xenograft tumorbearing mice with an agarose mold standard (Std) and with colorized R2 expanded views of the tumor regions for both NP:DNA (left) and NP:DNA_CTX (right) treatments. Both nontargeted (NP:DNA) and targeted (NP:DNA_CTX) nanovector treatedtumors showed similar enhancement of R2 contrast. (b) Quantitative R2 values for the tumor region in NP:DNA and NP: DNA_CTX treated mice. (c) Quantitative R2 values for muscle in NP:DNA and NP:DNA_CTX treated mice. Reprinted with permission from F.M. Kievit, O. Veiseh, C. Fang, N. Bhattarai, D. Lee, R.G. Ellenbogen, M. Zhang, ACS Nano, 4 (2010). Copyright 2010 American Chemical Society.
MNPs can be made from any number of magnetic materials, such as cobalt, nickel, iron, etc.[25] However, for the purpose of gene delivery, research focuses around the use of iron oxide particles due to their relatively low toxicity and high susceptibility to magnetic fields.[22] There has been a small amount of research into other magnetic materials; however, these materials show no improvement over the transfection efficiency of iron oxide MNPs and can introduce known toxic metals into the body.[26]
The synthesis of magnetic iron oxide nanoparticle (MIONP) is fairly well established, and has been reviewed in depth elsewhere.[27] Briefly, MIONPs can be produced by one of three mechanisms, alkaline co-precipitation, thermal decomposition, or mini-emulsions. For biological functions, alkaline co-precipitation is preferred, as it is simple, scalable, and does not leave the particles coated in a harmful substance. Co-precipitation can create either Fe3O4 or γ-Fe2O3 MNPs depending on which salts are used. [27] The other synthesis methods can be used to impart desired surface coatings. In one instance, thermal decomposition was used to provide an oleic acid coating for further conjugation.[28]
2.2 MNPs applied to oligonucleotide delivery
Naked MNPs are colloidally unstable and incapable of electrostatically binding oligonucleotides on their own, meaning that they require some coating to be effective vectors. The cationic polymer polyethylenimine (PEI) is often used for this purpose. Recent work with PEI coated MNPs has been focused on optimizing gene loading and delivery using these PEI-MNP-gene systems. One study looked into different methodologies of combining these three components.[29] The combinations studied were:
Incubating DNA with PEI coated MNPs (MNP/PEI+DNA)
Coating DNA in PEI and incubating with a bare MNP (MNP+PEI/DNA)
Coating DNA in PEI and incubating with PEI coated MNP (MNP/PEI+PEI/DNA)
Incubating DNA with PEI coated MNPs and then free PEI (MNP/PEI+DNA+PEI)
The eventual intracellular locations of these configurations were studied by fluorescent labeling. DNA was labeled with TOTO3, MNPs with FITC, the nucleus with Hoechst 33342, and lysosomes with Lysotracker blue. The stained systems were incubated with a fixed amount of DNA per well for 1 hour in a magnetic field.
The synthesis method appeared to affect the final destination of the DNA inside the cell. In instances where DNA was coated by PEI, as in the MNP+PEI/DNA and MNP/PEI+DNA/PEI, it managed to localize to the nucleus. When the DNA was attached to the surface of the particle, the complex was entrapped in lysosomes. However, in cases where DNA alone was the outermost layer (MNP/PEI+DNA), the vectors were relatively innocuous, while in configurations where the system was coated with PEI, the particles were significantly cytotoxic. These results indicate that the positively charged PEI, although necessary for lysosomal rupture, can be detrimental to cell health at relatively lower concentrations.[30]
Instead of PEI coatings, another approach is attaching a gene layer directly to the MNP surface by taking advantage of oleic acid coatings from unique synthesis methods. Two methods for attaching genes to MNPs are CLICK chemistry and phosphate linkage to positively charged MNPs. The use of CLICK chemistry requires MNPs functionalized with azides to attach alkyne-modified oligonucleotides. Cellular uptake of oligonucleotide coated MNPs was compared to that of MNPs coated with a negatively charged carboxyl group.[31] MNP uptake was measured via inductively coupled plasma mass spectrometry. This study proved that the gene coating did enhance the uptake compared to the negatively charged counterpart without the use of a transfection agent. However, there is no comparison of this transfection method to commercial standards or other MNP coatings. These gene coated MNPs have limited transfection efficiency because they lack the cationic charge necessary for endosomal escape.
The surface conjugation of MNPs with oligonucleotides has proven an effective method for the delivery of decoy oligonucleotides (dODN). A dODN for the signal transducer and activator of transcription 3 (STAT3) has been successfully adsorbed to the surface of a MNP.[32] The gene coated particles were taken up by the cells, as measured by the response of the cells to the application of magnetic fields, which can be linearly related to the MNP content. MNP uptake was also monitored by the fluorescence of FITC labeled dODNs. This showed that the dODN/MNP complexes were internalized by the cells. It was also shown that the dODNs affected the localization of STAT3. Cells were treated with an anti-STAT3 antibody with red florescence. In control cells STAT3 was localized to the nucleus, while the presence of the dODN/MNP complex localized STAT3 to the cytosol. There is still no evidence that gene coated MNPs alone can effectively deliver genes to the nucleus for transcription, yet it is still a viable option for gene delivery into the cell.
Another common MNP coating is cationic lipid layers. Recent research has looked at coating MNPs with cationic lipids by way of CLICK chemistry. One group attached a twelve-carbon chain liposome to a MNP coated with N,N-bis-(2-aminooxyethyl)-N,N-dimethylammonium iodide.[33] These particles were then loaded with Luciferase pDNA at a variety of MNP:pDNA weight ratios. Transfection efficiency was measured in relative light units of fluorescence produced by the Luciferase gene. Initial results showed a slight increase in transfection over Lipofectamine 2000, however nothing significant. Improved transfection was achieved by incubation with N,N-dimethyl-bis(2-tetradecylideneaminooxy-ethyl) ammonium iodide, which was the free equivalent of the lipid layer coating the MNP. This double lipid layer left the positively charged end as the external coating of the complex with a positive terminal end. These new lipid coated MNPs had transfection efficiencies orders of magnitude over Lipofectamine 2000. These particles also outperformed Lipofectamine 2000 in terms of cytotoxicity; however, there is still an issue of cytotoxicity. At the level of pDNA:MNPs necessary for optimal transfection significant cell death was observed.
Dendrimers have also been investigated in more depth in recent years. Research examined the effect of the number of dendrimer generations on transfection and cytotoxicity. MNPs coated with polyamidoamine (PAMAM) dendrimers of 0-5 generations were used to deliver an antisense survivin oligodeoxynucleotide (asODN) to human breast cancer cells. The asODN was designed to inhibit growth of the cancer cells. This inhibition rate was determined by MTT assays, determining the ratio of viable cells in the test case to that in the control. This study showed that as the number of generations increased from 0-5 so did the inhibition rate. Controls of dendrimer coated MNPs alone did not affect inhibition rate at all, showing even a slight decrease compared to the naked MNPs, yet still showing an increase in cell death compared to control cells.[34] Again, the issue is cytotoxicity as dendrimer coated MNPs are still slightly cytotoxic on their own.
One method proposed for getting around these cytotoxic effects without substantially modifying the surface of the MNPs is MNP clustering. MNP clusters were formed by anchoring branched PEI to MNP surfaces with dihydroxy-L-phenylalanine and then PEGylating the clusters with methoxy PEG succinimidyl-succinate. Gene silencing was performed on GFP overexpressing PC-3 cells by anti-GFP siRNA. Anti-GFP siRNA was conjugated to MNPs and MNP clusters (cMNPs) at varying weight ratios of MNP:siRNA. After incubation, the amount of GFP produced was determined by a spectrofluorometer. The cMNPs showed a significant increase in biocompatibility, having relatively no cytotoxic effects at PEI concentrations that caused almost complete cell death by non-clustered PEG-coated MNPs. cMNPs also proved more effective in terms of gene silencing. Relative GFP expression was reduced to 50% of the control compared to the 85% achieved by the non-clustered MNPs.[35]
2.2.1 Magnetofection
Magnetofection defines therapeutic delivery aided by a permanent magnetic field. It has been proven as an effective method for gene delivery without requiring a transfection agent such as the TAT peptide, particularly in vitro. Recent research has started looking into the effectiveness of magnetofection in more complex systems that can be related to in vivo conditions.
One such study looked at the effect a constant flow has on magnet driven transfection. MNPs were coated with a cationic lipid, Metafectene, and a helper-lipid, dioleolyphosphatidyl-ethanolamine. HeLa cells expressing firefly luciferase and NCI-H441 cells expressing eGFP were seeded onto the surface of a flow channel, individually. The lipid coated MNPs (LCMNP) were loaded with Luci GL3 siRNA and GFP 22 siRNA and used to silence their respective genes. For negative controls, GFP 22 siRNA and a control siRNA (Dharmacon) were used for Luciferase and eGFP expressing cell lines respectively. A reservoir containing cell media and siRNA loaded LCMNPs were attached to the flow channel via a peristaltic pump, and passed over the cultured cells at a velocity of 16cm s-1. A permanent magnet (NbFeB, field strength 1.3 T) was placed under a small section of the flow channel. Images of the cells from over the magnet and random sections throughout the channel were collected with a fluorescence microscope at 24 hour intervals. Cells near the magnet had strong evidence of silencing, with a marked absence of fluorescence, while the cells away from the magnet still had strong fluorescence. The cause of this fluorescent decrease was confirmed to be silencing, as cytotoxicity studies showed no substantial decrease in cell viability when cells were incubated with siRNA containing LCMNPs.[36]
Magnetofection also provides for a potential solution to tissue infiltration. To examine how effectively magnetofection can deliver genes in 3 dimensions, it was performed with a variety of gene cargos into a 3D scaffold.[37] A 3D collagen matrix of NIH 3T3 cells was produced and transferred into a tissue culture plate. A solution of PEI coated MNPs, loaded with either GFP plasmid DNA, FAM-siRNA or a toxic shRNA was added to the collagen matrix then an NdFeB magnet was placed at the bottom of the plate. Z stack images of a profile of the matrix showed that transfection only took place 2.1-2.3mm from the surface. However, when cell matrices were limited to a total depth of 1.9 mm, over 80% of the cells in the matrix possessed fluorescence indicative of GFP expression or FAM-siRNA delivery. The factors affecting this dimensional limitation are yet unknown and more studies are needed. The combined studies into the effect of flow and the addition of a third dimension give good ground to support that magnetofection could be used for gene delivery and targeting.
This has been proven in mouse models, where permanent magnets were used for successful magnetically guided in vivo gene delivery. MNPs with a cationic lipid coating comprised of O,O’-ditetradecanoyl-N-(atrimethylammonioacetyl)diethanolaminechloride and DOPE in a 1:0.4 ratio were used to deliver an Alexa Fluor 488-labled siRNA (AFsiRNA).[38] Initial experiments were done with the delivery of AFsiRNA to compare the targeting effectiveness of magnets implanted inside the tumor (internal), magnets placed on the mice over the tumor site (external), and a control case with no magnet present. The weight ratio of AFsiRNA was measured as a function of time for the tumor tissue and various other organs.
The results from this study showed a marked improvement in the concentration of siRNA at the tumor site in the presence of magnetic targeting. However, there was no significant difference between internal and external magnets. These studies also revealed an accumulation of particles in the lung, spleen and liver, although the mice experienced no detrimental effects of this undesired accumulation. This magnetofection technique was then tested as a delivery mechanism for an anit-EGFR siRNA on cancer disease models. Mice were inoculated with MKN-74 and NUGC-4 gastric adenocarcinomas, and treated with the anti-EGFP loaded lipid coated MNPs. These studies were done with internal, external, and no magnets. Two days after treatment, angiogenesis, proliferation and apoptosis were measured by immunostaining of vWF, Ki-67 and ssDNA respectively to determine the effectiveness of magnetofection. Significant improvement of these three measurements was noted in the presence of the magnet, yet again, there was no difference between the internal and external placement of the magnet. These studies prove that magnetofection can be used to localize treatment and improve transfection efficiency to tumorous tissue without requiring a targeting ligand. However, even though these studies showed no difference between internal and external magnetic sources, these experiments focused on surface based tumors. More work is needed to examine the impact of the depth of the tumor.
Magnetically guided gene delivery does prove an effective method for cancer models, yet there is no proof that it will work for other diseases or cell lines. The enhanced permeability and retention (EPR) effect is likely assisting in the localization of these particles to the tumor, as shown in Figure 2. Studies still need to be done in order to determine if magnetic guided gene delivery is a possibility for non-fenestrated tissue.
Figure 2.
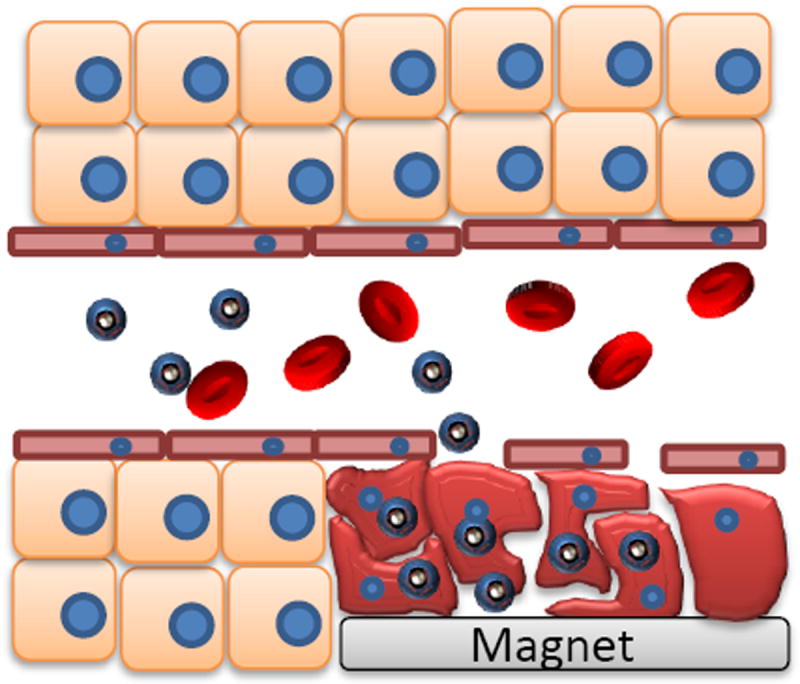
Schematic for nanoparticle accumulation in tumor cells mediated by magnetically guided gene delivery, or magnetofection.
3. Gold Nanoparticles
Gold nanoparticles (AuNPs) are widely researched as theranostic agents due to their notable chemical and optical properties, including well-established and robust synthesis techniques, ease of conjugation, relatively low toxicity, and tunable absorption within the near-infrared spectral range where tissue and bodily fluids are transparent (~700-1000 nm).[11, 13, 39] The useful optical properties result from localized surface plasmon resonance (LSPR) and can be controlled by both the geometry and size of the gold particles; shapes such as spheres, nanoshells and nanorods have different light scattering, absorbance, and light-induced plasmonic heating properties, meaning the particles may be used as imaging and/or photothermal agents.[11, 40]
3.1 Background and Advantages
Synthesis of AuNPs is well established and has been reviewed in detail elsewhere.[41-43] Colloidal gold nanoparticles have been synthesized by reduction of chloroauric acid with sodium citrate in boiling aqueous solution since the 1950s.[44] Particle size can be tuned by varying the type or amount of reducing solution added.[45] Gold nanorods (AuNRs) can be synthesized by a similar seed-mediated method, or by other methods such as a template method or electrochemical methods.[42] Gold nanoshells (AuNS) can be synthesized by functionalizing monodisperse silica nanoparticles with a terminal amine, then covalently bonding small gold colloid particles to the surface via the amine group. These gold-decorated silica particles were then used as seed particles during further reduction of chloroauric acid, resulting in AuNS.[46]
Though colloidal gold is generally accepted as biocompatible, the cytotoxicity of gold nanoparticles has been investigated at concentrations relevant to transfection and cellular uptake; that is, ~1-100 nanoparticles per cell.[47] The toxicity of gold nanoparticles has been reviewed, and results indicate that gold nanoparticles induce little to no toxicity; however, there is correlation between toxicity and the size and shape of the particles as well as the surface charge induced by cationic ligands.[47, 48]
As the toxic effect of AuNPs differs between in vivo and in vitro studies, some researchers have investigated the difference in cytotoxicity of AuNPs between lineage-specific cells types and primary immune cells; the gold nanoparticles (13 ±1 nm) with covalently attached oligonucleotides were found to induce transcriptional activation of the innate immune response in peripheral blood mononuclear cells.[49] This highlights the importance of cytotoxic evaluation using a relevant biological system, as well as the necessity of in vivo studies. Similarly, gold nanoparticles 20 nm in diameter were found to upregulate miRNA expression within lung fibroblasts simply following exposure, without any oligonucleotide cargo.[50] Before AuNPs will be widely accepted as theranostic agents, their effect on cellular function, toxicity, and immune response must be thoroughly studied, particularly in relevant cell models and in vivo.
The ability to absorb within the near-infrared (NIR) spectral region imparts external control over the particles as theranostic agents, since a pulse laser can be used to excite the particles through bodily fluid and into deep tissue.[51] Imaging techniques such as dark-field microscopy, confocal microscopy, and surface-enhanced Raman scattering (SERS) enable spatiotemporal tracking of the particles and their therapeutic payloads.[52, 53] In close proximity, the scattering of the fluorescent dyes and Raman reporters is influenced by the LSPR of the gold particles.[13] SERS is particularly advantageous, as the gold particles enhance the signal of Raman reporter molecules adsorbed to their surface to the limit of single molecule detection without spectral overlap or photobleaching and can be applied in vitro using Raman microscopy or even in deep tissue in vivo.[52, 54]
For example, NIR fluorescent dyes and Raman reporters have been utilized to study the in vivo fate of gold nanorods (AuNRs) in the deep tissues of mice.[55] The AuNRs were coated with the Raman reporter 3,3’-diethylthiatricarbocyanine iodide (DTTC) as well as PEG via Au-S interaction. DTTC alone gave no Raman signal at 785 nm, while the PEG-DTTC-AuNRs gave intense Raman signal due to the SERS effect of the gold.
Additionally, cell viability remained high 24 hours post-treatment with PEG-DTTC-AuNRs; at the maximum dosage, average cell viability was 86%. Nude mice were then injected with the particles and evaluated with a NIR optical imaging system as well as a Raman probe at various time points post-injection, as shown in Figure 3. Strong NIR fluorescence signal was observed in the liver of the mouse in as little as one hour, with lesser signal observed in the tail. SERS spectra confirmed the presence of particles in the liver with a high signal to noise ratio, and showed a decrease in intensity over time. The authors attribute this decrease to the processing and excretion of the PEG-DDTC-AuNRs, but it could also be that the fluorescence and SERS signals of the particles are not stable over time in biological media. Additionally, NIR imaging and SERS were used to confirm the ability of the particles to target a tumor on the mouse due to the EPR effect.
Figure 3.
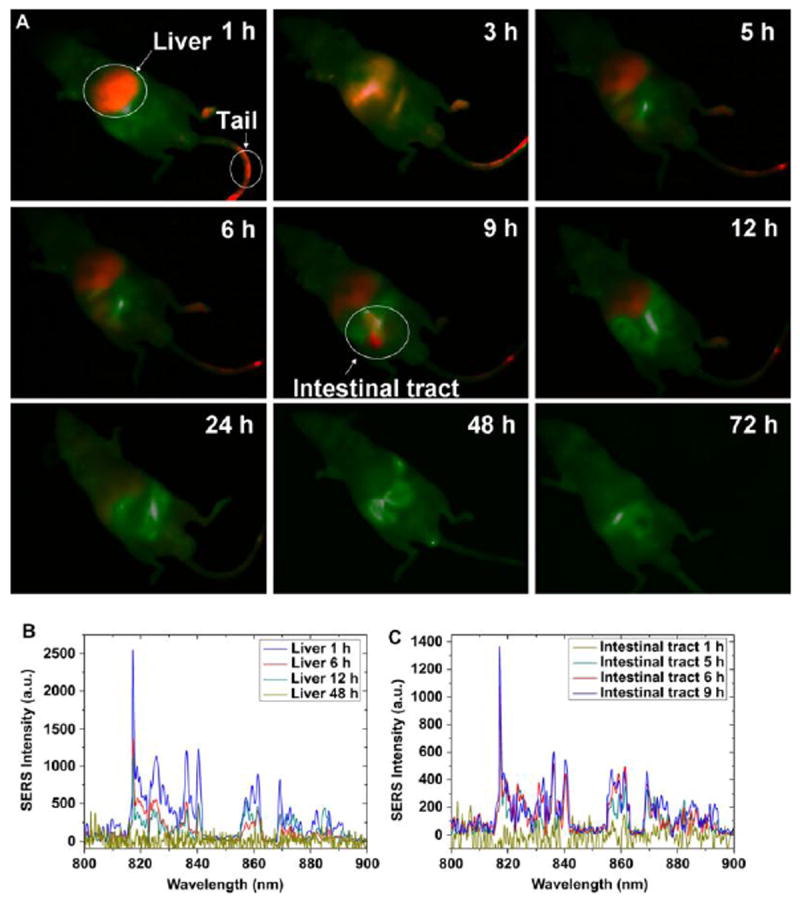
(A) NIR fluorescence imaging over time of the distribution of PEG-DTTC-AuNRs following injection into a nude mouse. NIR fluorescence signal is colored red, autofluorescence is green. (B) NIR SERS spectra from the liver of the mouse at time points post-injection. (C) NIR SERS spectra from the intestine of the same mouse over time.
Figure with permission from J. Qian, L. Jiang, F. Cai, D. Wang, S. He, Biomaterials, 32 (2011) 1601-1610.
3.2 AuNPs applied to oligonucleotide delivery
Dark field microscopy and SERS have been used to track cellular uptake and localization of gold nanoparticles capped with chitosan or branched PEI and loaded with shRNA.[52] Transmission electron microscopy (TEM) showed what appeared to be internalization of the chitosan-AuNPs, with clusters of the particles indicative of entrapment within an endosomal compartment. Dark-field microscopy and SERS were then performed to prove internalization rather than agglomeration of particles on the cell surface. The Raman reporter 4-aminobenzenethiol (ABT) was attached to the surface of the particles, and remained intact throughout the 24-hour incubation period in cell medium as well as internalization within the cell. By obtaining a z-stack of confocal Raman images through the height of the cell, shown in Figure 4, the presence of chitosan/BPEI-AuNPs within the cells was confirmed by the location specific Raman spectra. The particles were found to be located 3-7 μm inside the cell surface, and their location was also correlated an image taken with dark field microscopy. Additionally, both particle types demonstrated some reduction in the target mRNA expression, but toxicity, primarily of the BPEI particles, was a concern. These particles enable a technique which produces convincing evidence of internalization and distribution within the cell, rather than potential misinterpretation of fluorescent microscopy images alone. However, there is no indication that these particles outperform commercial transfection vectors in terms of knockdown efficiency.
Figure 4.
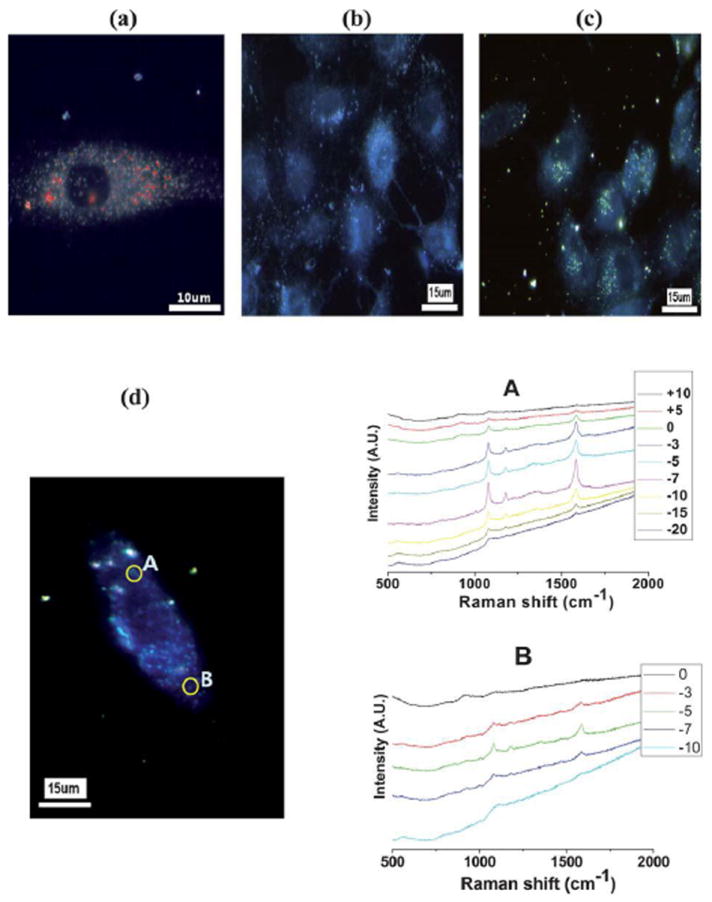
(A) Hyperspectral image of chitosan AuNPs and (B) ABT only (C) chitosan AuNPs only. (D) z-stack confocal Raman spectra of chitosan AuNPs with shRNA. A and B indicate the location of the AuNPs and the corresponding Raman spectra. The insert legends are the relative z-depth positions in micrometers from the surface of the cell.
Figure with permission from S. Jeong, S.Y. Choi, J. Park, J.-H. Seo, J. Park, K. Cho, S.-W. Joo, S.Y. Lee, Journal of Materials Chemistry, 21 (2011) 13853.
The plasmon resonance of gold nanoparticles also endows them with an externally controlled delivery mechanism via laser pulse.[51] Two strategies are generally employed to result in light-induced release of molecules from gold nanoparticles, and are illustrated in Figure 5.[40] In the first method, molecules are attached directly to the surface of the gold particle through an Au-thiol bond, and a femtosecond laser is pulsed to break the covalent bond, shown in Figure 5.A. The drawbacks of this method include reshaping of the nanoparticles with potentially toxic products, and incident energy at a level high enough to potentially induce cell death. The second approach uses and intermediate molecule which is attached covalently to the surface of the gold, and the cargo is then loaded onto this molecule by noncovalent interactions. Upon irradiation, the interaction between the intermediate molecule and cargo is disrupted, releasing the therapeutic cargo, shown in Figure 5.B. This method is appealing because it requires relatively low power densities and short irradiation times.
Figure 5.
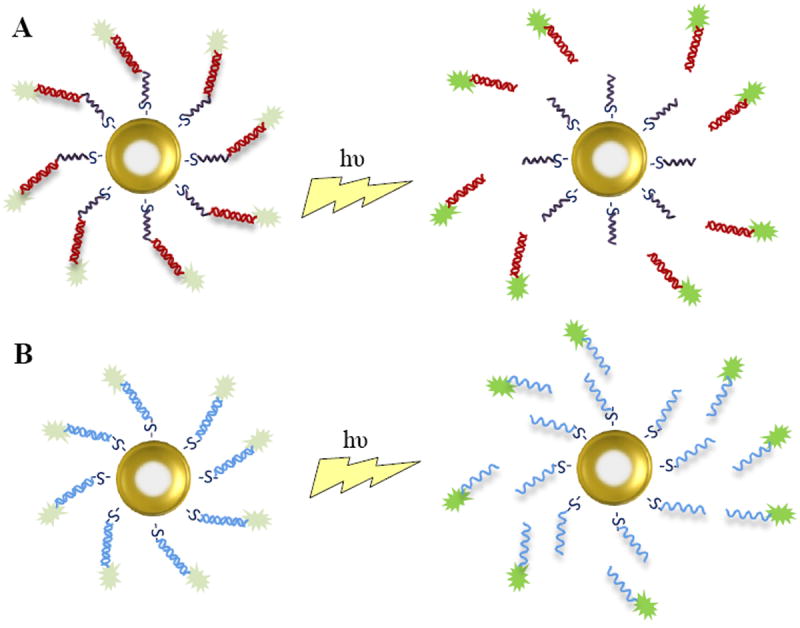
Schematic depicting light-induced release of nucleic acids from AuNS. At close proximity, the fluorescent tag on the oligonucleotide is quenched by the LSRP of the gold particle; after release, the distribution of the oligonucleotide can be tracked by the fluorescent tag. (A) dsRNA is covalently attached to the gold particle via thiol bonds; after a laser pulse, the bond is broken and dsRNA is released. (B) The sense strand of the oligonucleotide is bound to the surface of the gold particle; after a laser pulse, the antisense strand of the oligonucleotide is freed.
Demonstrating the latter approach, dsDNA was attached covalently to gold nanoshells (AuNS) via a thiol bond on one strand of the DNA to act as the intermediate molecule.[56] The fluorescent dye DAPI (4’,6-diamidino-2-phenylindole), which binds reversibly with dsDNA, was then loaded onto the AuNS complex by intercalation with the dsDNA strands. The AuNS-dsDNA-DAPI complexes were incubated in serum containing medium with H1299 lung cancer cells and uptake of the particles was confirmed with dark-field and bright-field microscopy, as the AuNS both absorb and scatter light. Plasmon resonant illumination at a wavelength of 800 nm was then used to trigger the dehybridization and release of the unbound DNA strand and intercalated DAPI. Using fluorescence microscopy, DAPI was observed diffusing throughout the cytoplasm and into the cell nucleus, where its quantum yield and fluorescent intensity increased upon binding with DNA. The nuclei were then isolated and flow cytometry was utilized to quantify the increase in fluorescent intensity after light resonance with the laser; a 33% increase in fluorescent intensity was calculated, indicating externally controlled release of the DNA and DAPI molecules from the AuNS-dsDNA-DAPI complexes. A control comprised of dsDNA-DAPI was tested to show that DAPI release by plasmon resonant illumination does not take place without the incorporation of the AuNS. Following incubation of the AuNS-dsDNA-DAPI complexes for 12 hours, cytotoxicity assays using propidiium iodide showed no significant increase in cell death. The AuNS demonstrated controlled intracellular delivery of DAPI and presumably DNA without significant cytotoxicity, but further studies are needed to confirm DNA delivery to the nucleus as well as demonstrate effective gene therapy.
Following up on a previous study in which gold nanoshells were found to have superior properties to gold nanorods for light-induced release application[40], researchers demonstrated light-induced release of ssDNA and siRNA from silica core/gold nanoshells functionalized with positively charged poly-l-lysine, a schematic of which is shown in Figure 6.[57] Anti-GFP antisense DNA and siRNA were loaded by electrostatic binding onto the surface of the nanoshell-poly-l-lysine (NS-PLL) vector. Upon irradiation at 800 nm, the ssDNA was released from the NS-PLL, possibly due to heating around the nanoshell, photon-induced transfer of electrons that reduced electrostatic interaction, or a combination of the two. It was shown that light-induced release of ssDNA results in efficient release while maintaining relatively low ambient solution temperature, which is important for prevention of cell death due to hyperthermia. It was also demonstrated that release was dependent upon the length of the ssDNA, due to electrostatic interactions. The NS-PLL-ssDNA were then incubated with H1299 lung cancer cells; cellular uptake was confirmed using dark-field microscopy, fluorescent microscopy, and inductively coupled mass spectrometry. Fluorescence microscopy was also used to visualize light-induced release of the ssDNA from the NS-PLL using a fluorescent tag on the ssDNA; prior to laser treatment the fluorescence of the tag was quenched due to the proximity of the AuNS and after laser treatment the green fluorescence on the ssDNA was observed throughout the cell. Gene silencing efficiency of the NS-PLL-anti-GFP was evaluated by measuring knockdown of GFP; 6 hours after laser treatment, GFP was downregulated to ~47% by ssDNA and ~49% by siRNA. Though some downregulation of gene expression was observed prior to irradiation due to the release of loosely bound oligonucleotide, the delivery and gene knockdown was largely controlled by external laser treatment. The loss of oligonucleotide prior to light-induced release may present problems moving forward with the therapy, since it is difficult to quantify and control the amount lost. Since both the NS-PLLs and laser caused minimal cytotoxicity, the system does have therapeutic potential as an externally triggered delivery mechanism, as long as the premature loss of oligonucleotide can be controlled or eliminated. The ability to trigger release is a major advantage over polymeric or liposomal carriers, but it would be beneficial to know how fast the release is in response to the light, and how efficient the release is in an in vivo model.
Figure 6.
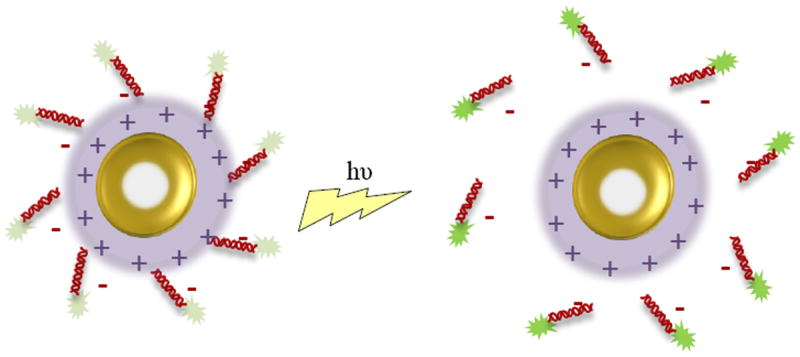
Schematic depicting light-induced release of siRNA by interruption of the electrostatic binding between the siRNA and AuNS surface.
Taking advantage of the ability to control gene delivery by laser-induced cleavage as well as Tat peptide targeting, Cy3-labeled siRNA was conjugated to gold nanoshells and coated in a Tat lipid bilayer via electrostatic interaction.[53] The release of the siRNA following a laser pulse was confirmed by an increase in Cy3 fluorescence, since the gold quenches the fluorescence in close proximity. This technique was used to determine that dosed release can be triggered with a few pulses at high power, or with an increase in pulse rate at low power. As in other studies, reduction in GFP expression was observed only in cells that had been irradiated to release siRNA; however, the cells transfected with NS-siRNA did not show a reduction in GFP expression until 2 days post-transfection, whereas commercially available Lipofectamine showed silencing in as little as 36 hours. While Lipofectamine enabled endosomal escape of the siRNA, the NS-siRNA did not allow sufficient endosomal escape to induce RNA interference (RNAi) without additional laser-induced endosomal rupture. Endosomal rupture also required slightly higher laser power than the cleavage of siRNA, imparting a second degree of control over delivery. The heat caused by the additional laser pulse to the NS did not appear to cause cell death, though the effect of possible thermal degradation of the siRNA is unknown. This study offers a possible alternative to incorporation of potentially cytotoxic cationic materials to induce endosomal rupture. The study also confirmed that decrease in fluorescent quenching by the gold nanoshell can be used to quantify therapeutic release.
The quenching of fluorescent intensity by gold nanoparticles has also been used to study additional aspects of gene delivery such as serum stability and Dicer recognition, essential components of RNAi.[58] Mirkin et al. developed gold nanoparticles functionalized with fluorescein-labeled siRNAs. The fluorescent intensity of the labeled siRNA will increase upon degradation and cleavage of the siRNA from the gold nanoparticle. The siRNA-Au-NP, with various commonly used types of siRNA, were incubated with either Dicer to test specific cleavage or FBS to test nonspecific degradation. A greater increase in fluorescent intensity demonstrated that Dicer has a preference for siRNA with a 3’ overhang, though these siRNA were also 10-15-fold more susceptible to nonspecific degradation. The serum stability of chemically modified siRNAs was then tested, and the results showed that these modifications can decrease nonspecific degradation by ~40%, but Dicer recognition was decreased by ~60%. Finally, the researchers found that serum stability depended upon the orientation of the siRNA on the nanoparticle; siRNA with the more thermally stable base pair distal to the nanoparticle experienced greatly reduced nonspecific degradation while maintaining comparable Dicer recognition. As might be expected, increased serum stability enhanced cellular uptake by 300% and resulted in ~85% GFP knockdown. This shows the versatility of gold nanoparticles as a therapeutic carrier, but also as an analytic agent.
Mirkin et al. have also demonstrated the gene silencing efficacy of gold nanoparticle conjugates with microRNA via thiol bonds.[59] The miRNA-AuNPs downregulated a specific protein by 52%, silenced a reporter gene by 38% relative to a nonspecific miRNA sequence, and even outperformed the commercial transfection agent DharmaFECT. As no change in mRNA levels were observed, it is likely that the miRNA- AuNPs block translation of the mRNA target rather than cleave mRNA strands. The miRNA-AuNPs apparently maintain miRNA-induced apoptosis for least 5 days post-transfection, but it would be interesting to find the maximum duration of therapeutic efficacy. Though these knockdown results were quite promising, maybe it is possible to increase these numbers even more by conjugating a greater number of miRNA to the gold nanoparticle; there is room for optimization of this system.
Research has also been done in the area of targeted intracellular delivery of siRNA using hyaluronic acid (HA) to induce HA-mediated endocytosis.[21] Cysteamine-modified gold nanoparticles (AuCM) were coated with siRNA, PEI, and HA by layer-by-layer assembly. Addition of each layer was confirmed by TEM, atomic force microscopy (AFM), UV-Vis, and zeta-potential measurements. The AuCM/siRNA/PEI/HA complexes were stable in serum-containing media up to 24 hours, presenting no aggregation or precipitation. Cellular uptake of the complexes was visualized by TEM after 24 hours of incubation with B16F1 cells; the complexes were distributed throughout the cytoplasm with no large aggregates present. An MTS cell viability assay showed cell viability of at least 90% for the AuCM/siRNA/PEI/HA complexes as well as the various controls, which is surprisingly given the incorporation of PEI. Anti-luciferase siRNA was used to determine a gene silencing efficiency of ~50% for AuCM/siRNA/PEI/HA complexes in 10 vol% serum. In 50 vol% serum, silencing efficiency of the AuCM/siRNA/PEI/HA complexes improved to 70-80%, which may be a function of the stability of this formulation in serum. This highlights the large effect protein adsorption and serum stability may have on intracellular uptake, and indicates that researchers should investigate this possibility. Anti-VEGF siRNA was also tested, and RT-PCR showed ~70% reduction in gene expression, which outperformed the 20% reduction by siVEGF/Lipofectamine 2000. Dark field microscopy confirmed that cells with HA receptors did uptake AuCM/siRNA/PEI/HA complexes, but the same cells pretreated with HA did not, demonstrating effective targeting of the HA receptors by the complexes. Gene knockdown results corresponded with cellular uptake; gene silencing in the presence of free HA was significantly lower than that without free HA. Finally, the target-specific systemic delivery of the AuCM/siRNA/PEI/HA complex with anti-ApoB siRNA to the liver was tested. Following injection into the tail-vein of Balb/c mice, most of the complexes accumulated in the liver and spleen, as determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES). The ApoB mRNA in the liver was reduced to ~20%, demonstrating effective target-delivery and gene downregulation. This study employed imaging techniques enabled by the optical properties of gold to confirm the intracellular uptake of these particles as well as preliminary biodistribution, but what remains to be determined is the clearance of particles from the RES, which is a challenge for such theranostic systems.
Gold nanorods have also been used to transfect human macrophage cells and deliver siRNA targeted to galectin-1, a gene which is upregulated by methamphetamine and facilitates HIV-1 infection.[60] If effective, the treatment could be used to reduce galectin-1 expression and thus reduce the occurrence of HIV-1 in methamphetamine users. siRNA with a conjugated fluorophore was electrostatically bound to AuNRs coated with polyelectrolytes and used to transfect human monocyte derived macrophages for 72 hours. Intracellular fluorescence was detected at 4 hours post-transfection in the cells incubated with the AuNR/siRNA nanoplexes, as observed with dark-field microscopy. The nanoplexes were able to significantly decrease galectin-1 expression compared to the control at 24, 48, and 72 hours post-transfection. Methamphetamine potentiation of galectin-1 was also reversed by the nanoplexes. The study did not provide cytotoxicity data, which could account for part of the decrease in galectin-1 expression, though a scrambled siRNA sequence did not significantly reduce expression indicating that the coated AuNRs themselves are not cytotoxic.
In summary, gold nanoparticles have been demonstrated as an effective theranostic agent for gene therapy. Especially important is their absorbance and scattering within the NIR region where tissue is transparent, potentiating in vivo imaging of the particles and their nucleic acid cargo, though efficient real-time in vivo imaging has not yet come to fruition. The LSPR of AuNPs allows the delivery of tagged oligonucleotides to be monitored by SERS, dark-field, and confocal microscopy. Biocompatibility of AuNPs is generally accepted as high, though studies in primary cell models at relevant cellular concentrations as well as in vivo would solidify this notion. Additionally, the polycationic coatings often used with AuNP to facilitate endosomal escape present cytotoxicity concerns. The light-induced cytosol delivery of oligonucleotide cargo from AuNS or AuNR may eventually circumvent the need to incorporate inherently toxic polycationic coatings, thereby eliminating the cytotoxic component.
4. Quantum Dots
Semiconductor nanocrystals, also known as quantum dots (QDs), possess fascinating size and shape dependent electronic and optical properties that in recent years have found useful application in imaging and diagnostics. Current research interests include surface modification, intracellular delivery of therapeutic agents, spatiotemporal imaging, and evaluation of toxicity. They have optical properties superior to that of organic dyes and luminescent proteins, including up to 100 times greater brightness and 1000 times better stability against photobleaching.[18] Additionally, QDs are poised to make an impact as an experimental tool, since fluorescent imaging is a widely accessible technique to evaluate cellular and animal studies.
4.1 Background and Advantages
QDs are unique imaging modalities because their fluorescent emission can be tuned by the size of the particle, which is known as the quantum size effect.[61] Additionally, QDs of a small polydispersity possess a wide absorbance range and narrow, highly symmetric emission spectra. In terms of application to bioimaging, these qualities are important because they enable excitation of variously sized QDs with a single wavelength, resulting in distinct emission spectra with little overlapping.[15, 62] This greatly reduces cross-talk between channels and allows improved multicolor detection, thereby enhancing detection efficiency and capability.
The method developed by Bawendi et al. in 1993[63] and slight variations thereof are still used widely today to synthesize QDs. Some made changes to the cadmium species and coordinating ligand[64], and reports of successful synthesis of monodisperse QD in an aqueous phase using milder reaction conditions have started to become more common.[65] It is also worthwhile to note that while size is the predominant mechanism used to tune optical properties, core composition of the crystal will also affect the emission wavelength.[66, 67] For example, CdSe QDs may emit in the 450-650 nm range, while CdTe QDs may emit in the slightly higher range of 500-750 nm.[18]
A QD obtains a “core-shell” structure, shown in Figure 7, following the addition of a passivating layer that serves to preserve the photoluminescent (PL) quantum yield.[18] Typical passivating compounds include CdS, CdSe, and ZnS, with ZnS being the most common passivating compound.[68],[69] The ZnS layer is added to the QD core in a manner similar to QD synthesis, and has been shown to increase the PL quantum yield to 30-50% from 5-15% for unmodified dots.[68] ZnS shell growth is improved by the addition of a small amount of Cd to promote even coating across the QD surface, achieving particles with a quantum yield of up to 95%.[70] A ZnS shell also prevents oxidation and enhances colloidal stability of the particles.[18]
Figure 7.
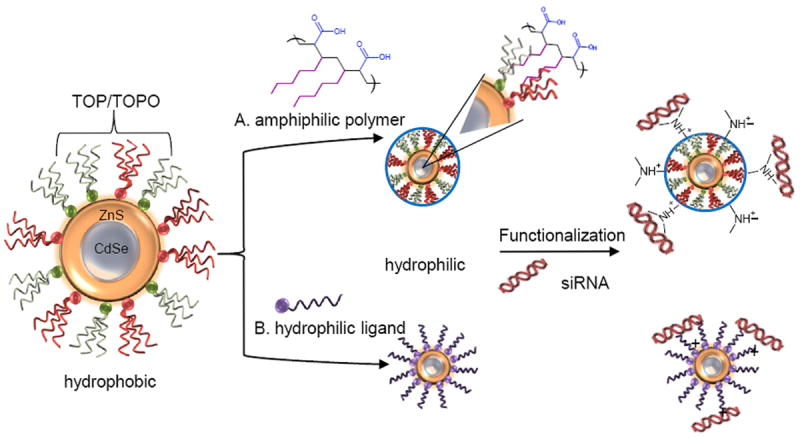
Schematic depicting the composition of a “core-shell” QD and two common strategies for phase transfer into aqueous solution, functionalization, and electrostatic loading of siRNA. Typical reagents reported in current literature are shown. A. Coating of the QD with an amphiphilic polymer. B. Exchange of the TOP/TOPO coordinating ligands with a hydrophilic ligand. QDs are then modified to have a positive surface charge so negatively charged siRNA may be electrostatically bound.
Post synthesis and passivation, QDs may be dried and reconstituted in organic, nonpolar solvents such as hexane, toluene, or chloroform, but they are not inherently water-soluble. This is a fundamental problem, as QDs must be water-soluble for use in in vivo or in vitro bioimaging applications. To resolve this issue QDs can either be encapsulated in a hydrophilic material, often polymeric, or ligand exchange can be used to replace the hydrophobic ligands with water-soluble counterparts. Mattoussi et al. have compiled a comprehensive list of surface capping strategies reported in literature.[69]
4.2 QDs applied to oligonucleotide delivery
Quantum dots have enabled imaging of individual molecules within the cell environment, which can be particularly useful in gene silencing therapies. QDs have been considered as potential gene delivery devices due to their convenient size and inherent stability against photobleaching while imaging, making them an effective theranostic agent. One of the earliest reports using QDs to monitor siRNA transfection appeared in 2005.[71] A conventional transfection agent, Lipofectamine 2000 (Invitrogen), was loaded with both CdSe-core ZnS-shell QDs and siRNA targeting the Lamin A/C gene. Fibroblast cells were then transfected with the loaded liposomes for 5 hours. Following transfection, siRNA uptake and silencing efficacy was measured by flow cytometry, western blotting, and immunofluorescence staining. Cells transfected with QDs and siRNA showed a strong correlation between gene silencing and fluorescence. Additionally, cells transfected with QDs and siRNA underwent ~90% gene knockdown, while cells transfected with siRNA alone experienced only ~20-30% gene knockdown.[71] This study not only demonstrated that QDs are a suitable probe to track siRNA delivery, but also indicated that they may increase gene silencing efficiency, though the authors did not offer an explanation. This study became a springboard for further investigation of QD-mediated siRNA delivery.
Expanding upon their findings, the same researchers sought to reduce the size of the particles and employ targeting moieties with the ultimate goal of improving systemic delivery of siRNA-decorated QDs.[72] To do this, a targeting peptide, F3, was conjugated to the surface of NIR emitting QD cores to enhance internalization of the particles and siRNA was conjugated to the QDs using a disulfide crosslinker which can be cleaved in the reductive environment inside the cell, thereby releasing the siRNA to induce RNAi. Cellular uptake was measured using flow cytometry. The number of F3 peptides and siRNA that could be conjugated was limited by the number of functional groups present on the surface of the QD; consequently, the ratio of F3:siRNA on the QD surface could optimized to maximize uptake and gene knockdown. Moving forward with a formulation of 20 F3 peptides and a single double stranded siRNA per QD, cellular internalization took place but gene knockdown was still not realized 48 hours after transfection.[72] However, upon adding the traditional cationic transfection agent Lipofectamine 2000 to aid in endosomal escape, gene knockdown was observed. The study not only used QDs to image cellular uptake, but also showed that the incorporation of targeting moieties on the QDs could improve gene knockdown by siRNA. Additionally, the study demonstrated that a mechanism for endosomal escape must supplement the QDs to promote effective delivery to the cytoplasm.
In a separate study, thiol-modifed RGD and HIV-Tat peptides were conjugated to CdSe/CdS/ZnS QDs with the goal of inducing brain tumor-specific targeting.[73] Attached to the surface of the QDs via a disulfide linkage, siRNA was selected to specifically suppress epidermal growth factor receptor variant III (EGFRvIII), which is expressed exclusively by tumor cells. Optimal internalization within genetically modified human glioblastoma cells was obtained at a ratio of siRNA/RGD/HIV-Tat of 1:10:10.[73] It was shown that uptake of the targeted siRNA-QDs was higher in tumor cells overexpressing EGFR than in cell lines with less expression of the integrin receptors, confirming effective targeting of the tumor cells. Additionally, significant cell death was observed only in cells treated with siRNA-QDs. This demonstrates that the QDs alone are not significantly cytotoxic and the cell death is a result of siRNA-mediated therapeutic gene knockdown, which was confirmed by Western immunoblotting of downstream proteins. Like the aforementioned QD-mediated siRNA delivery scheme, though, cationic lipid carriers were required to obtain significant gene knockdown. It would be more desirable to incorporate a mechanism for endosomal escape into the structure of the QDs itself, reducing the complexity of the system.
QDs have also been conjugated to ASON to determine cellular uptake mechanisms and the site of ASON activity within the cell.[74] Anti-survivin ASON, a 20-mer single stranded DNA, was conjugated to CdTe QDs via an amide bond. A 9-mer tether was included at one end of the ASON to limit any effect from the QD, and the reaction resulted in about 18 ASON per particle. HeLa cells were transfected with the conjugated QDs at 4°C and 37°C to determine uptake mechanism. It was shown that uptake was greatly reduced at 4°C, indicating uptake by an endocytic pathway. Further studies revealed that these conjugated particles, with an overall anionic charge, were likely endocytosed via macropinocytosis. At a transfection concentration of 50 nM, the CdTe-ASON29 were able to downregulate the surviving mRNA by approximately 70% while inducing moderate cellular toxicity from the CdTe core alone, as demonstrated by the controls. Time-lapse imaging of the QD-conjugates revealed cellular uptake starting at 20 minutes after transfection, with CdTe-ASON29 localizing within the cytoplasm around the nucleus around 40 minutes post-transfection. While cellular uptake of the negatively charged particles and resultant reduction in mRNA expression was demonstrated, the moderate levels of cell toxicity at the QD concentrations is cause for concern when evaluating these particles as a gene delivery vehicle, though they were used successfully to characterize cellular uptake mechanisms.
Other research groups have terminated QDs with a positively charged molecule or coating such as chitosan[75] or other cationic polymers[16, 76] in an effort to achieve effective cellular transfection and gene knockdown with low cytotoxic effect from the particles. One report utilized the endosome-disrupting polymer polyethylenimine (PEI) (Mn= 10 kDa), grafted with polyethylene glycol (PEG) to mitigate the cytotoxic effects associated with PEI.[76] (PEI-g-PEG) was attached to CdSe/CdS/ZnS QDs by a ligand exchange reaction, resulting in a hydrodynamic size of 21-22 nm. It was noted that the PEG grafted nanoparticles were quite stable in acidic conditions as well as biological media, which is often a concern with QDs and is especially relevant to intracellular delivery applications. Confocal microscopy was used to image the modified QDs, and it was observed that cellular uptake via endocytosis or macropinocytosis began as early as 1-2 hours into incubation. It was also noted that the amount of grafted PEG greatly influenced cellular uptake and intracellular distribution. QDs with four grafted PEG chains per PEI chain were apparently trapped in organelles, while QDs with only two grafted PEG chains per PEI chain had apparently escaped endosomal compartments and were released into the cytoplasm, as is necessary for effective siRNA delivery. However, QDs with four PEG chains per PEI molecule displayed less cytotoxicity, as determined by a standard MTT assay. The ability to tune endosomal escape and cytotoxicity by grafted PEG chains gives a greater degree of control over the properties of the QD nanoparticle, but an optimal formulation for intracellular delivery was not reported. Additionally, increasing the number of grafted PEG chains would likely decrease the amount of nucleic acid able to complex with the PEI, resulting in lower therapeutic efficacy, though this has not yet been studied with these particles.
Exploiting of the proven biological advantages of PEG incorporation, CdSe/ZnS QDs were conjugated to a PEG amine, giving the particles a net positive surface charge.[77] siRNAs targeting BACE1 gene were then electrostatically bound to the surface of the QD-PEG. One hour post-transfection, over 90% of the cells showed signal in the emission channel of the QD-PEG as determined by flow cytometry, indicating effective localization on the cell membrane or within the cell. Fluorescent imaging was used to track the QD-PEG and siRNA up to 10 days post-transfection. The QD-PEG/siRNAs achieved about 50% knockdown of the BACE1 gene while maintaining high biocompatibility relative to the controls. However, the performance of the QD-PEG/siRNAs was not compared to traditional transfection agents such as Lipofectamine 2000. It is possible, though, that the amount of PEG amine conjugated to the QDs could be optimized to improve transfection, while maintaining high biocompatibility.
Another approach used silicon QDs functionalized with 2-vinylpyridine, giving the surface a net positive charge to which negatively charged siRNA targeting P-glycoprotein (P-gp) could be electrostatically bound.[78] The functionalized QDs resulted in intracellular delivery of the siRNA, gene knockdown, and reduced efficiency of the P-gp mediated transport pathway across the cell membrane. However, the QDs did not outperform the cationic lipid Lipofectamine 2000, which reduced the protein expression by 71% while siRNA-QDs reduced protein expression by only 50%.[78]
In a more recent report, l-arginine (l-Arg)-modified CdSe/ZnSe quantum dots, with or without β-cyclodextrin (β-CD), were used as siRNA delivery devices to silence the gene HPV18 E6.[79] l-Arg provided a positive surface charge onto which negatively charged siRNA could be electrostatically bound, and β-CD had been shown to induce greater biocompatibility and lower cytotoxicity. HeLa cells were transfected with the QD/siRNA complexes for 24 hours, and cell viability remained >70% for QD concentrations less than 70 μg/mL over this period of time. Confocal microscopy was used to track the QD/siRNA location in real time, shown in Figure 8. Fluorescence intensity measured by flow cytometry was used to quantify cellular uptake. A gene silencing efficiency of nearly 80%, as well as 80% protein suppression was achieved using the QD/siRNA complexes, as determined by RT-PCR and western blotting, respectively. These values were greater than that of commercial transfection agents also tested.[79] This study highlights the utility of QDs to track and quantify cellular uptake in real time, with the additional potential to outperform traditional transfection agents.
Figure 8.
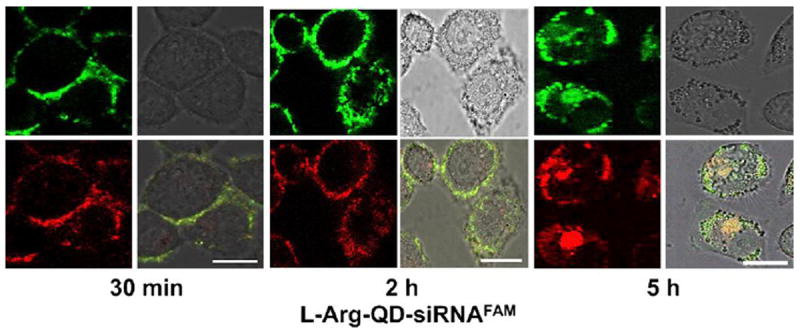
Real-time confocal microscopy images of HeLa cells transfected with CdSe/ZnSe QD/siRNA complexes. Soon after transfection, the QD/siRNA was observed on the cell membrane. At 5 h, QD/siRNA had migrated into the cell cytoplasm. Green fluorescence represents siRNA and red fluorescence represents QDs.
Figure with permission from Li, J.-M., M.-X. Zhao, H. Su, Y.-Y. Wang, C.-P. Tan, L.-N. Ji, and Z.-W. Mao, Multifunctional quantum-dot-based siRNA delivery for hpv18 e6 gene silence and intracellular imaging. Biomaterials, 2011. 32(31): p. 7978-7987.
Further expanding the dexterity and significance of quantum dots, fluorescent resonance energy transfer (FRET) may be utilized to gather information regarding spatial conformation of QDs and siRNA within the cell. FRET is the transfer of energy from a donor fluorophore to an acceptor fluorophore across nanometer-scale distances, resulting in a lower fluorescence intensity for the donor and a higher fluorescence intensity for the acceptor.[80] FRET has been employed with QDs to observe the intracellular delivery of siRNA[81, 82] and DNA[83-85], though most studies utilize QDs as an imaging modality combined with a polymeric or lipid-based transfection agent[86, 87].
In one study, electrostatic binding of FITC-labeled siRNA to amphiphilic polymer-encapsulated QDs was perceived by the quenching of the FITC signal by excited QDs.[82] Time-lapse confocal microscopy was used to monitor the QD-siRNA intracellular interaction; at approximately 1.5 hours after transfection, the particles were present inside the cell and decomplexation between the QD and siRNA was indicated by the appearance of a signal in the FITC channel. At 5 hours post-transfection, siRNA was dispersed throughout the cell. Increased death in cells transfected with HER2 siRNA compared to those transfected with random or no siRNA was indicative of endosomal escape and gene knockdown by RNAi, but was not conclusive. The amphiphilic polymer coating outperformed a conventional polymer, PEI, and was comparable to the commercial transfection agent Lipofectamine in terms of gene silencing efficiency. The nanocomplex carrier has the added benefit of less cytotoxic effect than these other carriers.[82] The authors consider interaction between the QDs and FITC to be FRET, but it could simply be quenching of the signal rather than resonance transfer. However, the quenching was shown to be a function of proximity between the QDs and FITC-siRNA, making this a valid evaluation of siRNA release. Similarly, Lee et al. employed FRET to track the release of cyanine-labeled siRNAs electrostatically bound to the QD-PEI complexes.[81] The QD-PEI complexes displayed knockdown of the VEGF gene comparable to that of commercially available Lipofactamine.
Recently, FRET was used to not only to track delivery of plasmid DNA (pDNA) into the nucleus, but to actually determine the sub-region of the nucleus in which the pDNA was delivered and decondensed.[83] QD-labeled pDNA served as the FRET donor, while rhodamine-labeled polycation was the acceptor onto which the pDNA was condensed. The condensed pDNA complex was delivered via a lipid vector, and the cells were imaged at 3 hours post-transfection. As the pDNA were decondensed, the emission wavelength shifted from that of rhodamine to that of the QD. It was noted that pDNA in the cytoplasm existed in the condensed form, while both condensed and decondensed pDNA were present in the nucleus, as expected due to the protection of the lipid vector outside the nucleus. Two separate regions of the nucleus were identified by an additional fluorescent stain, and the condensation/decondensation of the pDNA in each region of the nucleus was correlated to the number of cationic moieties per molecule of the polycation. Such knowledge is useful in designing DNA therapies targeting a region of the nucleus for higher gene knockdown efficiency. The use of QDs and FRET can produce meaningful insights on particle and therapeutic localization within the cell, but it certainly necessitates careful orchestration of fluorophores that may complicate experimentation.
4.3 Challenges facing QDs as theranostic agents
The circulation, clearance, and accumulation of QDs have been observed using small animal fluorescent imaging in more studies than can be cited here[88-91], and the quantitative biodistribution of CdSe[92] and InAs[93] QDs in various organs of mice has also been reported. The results indicated that there was rapid uptake of a large amount of particles by the liver and spleen, and all QD formulations were cleared from circulation within 10-20 minutes of injection. The studies show that PEGylation and peptide coating both slow RES uptake of QDs, and that the smaller InAs QDs are able to be renally excreted to some extent[93], which is an encouraging finding since other studies suggest that In-containing QDs may be less toxic than Cd-containing QDs.[94] Looking forward, mechanisms for extending the half-life and improving renal clearance while maintaining therapeutic efficacy of the particles are challenges facing in vivo drug delivery and imaging using QDs.
Since QDs have been shown to accumulate and reside in the liver and spleen of living mice, the long-term toxicity of QDs is another major concern for their use as theranostic agents. The greatest risk of toxic effect is due to the release of Cd2+ ions from the nanocrystals, which can be somewhat controlled by encapsulation of the QD in a passivating ZnS shell or crosslinked polymer coating.[94, 95] A report was recently published on the short-term toxicological effects of phospholipid-coated CdSe/CdS/ZnS QDs intravenously injected into monkeys.[96] Levels of metal ions in the blood and various tissues were measured over time, as were biomarkers indicative of organ function and inflammation. It was found that the QDs likely accumulate and degrade over time in the liver and spleen, and the free metal ions accumulate mostly in the kidneys. The biomarkers were all found to be within the normal range. The study concluded that the QD formulation tested had little to no toxic effect in the primates for up to one year, but recommended studies of longer duration. Such studies are especially important to evaluate the effect of repeated doses, as would be necessary for therapeutic effect.
However, adequate long-term in vivo studies have not yet been performed and toxicity of traditional QD formulations remains a concern. Consequently, there has been movement toward Cd-free NIR QDs composed of III-V or I-III-VI2 semiconducting metals such as CuInS2[94] and CuInSe[97]. Both QDs have promising physical characteristics and biocompatibility, but have not yet been used as theranostic agents.
Preliminary studies indicate favorable toxicity over traditional cadmium QDs, but synthesis, quantum yield, and stability of cadmium-free QDs could use improvement. To that end, work has recently been done to develop a high-throughput, facile synthesis method for ZnxS/AgyIn1-yS2 QDs.[67] The sonochemical decomposition of precursors at ambient reaction conditions resulted in a library of QDs with uniform shape, narrow size distribution, high purity, and tunable PL properties relative to composition. Additionally, absorption and PL remained unchanged over two months of storage at ambient conditions. The ZnxS/AgyIn1-yS2 QDs showed marked improvement in cytotoxicity over CdSe/ZnS QDs; cell viability remained greater than 90% up to a concentration of 100 μg/mL. The QDs were then coated with PEI and conjugated to siRNA and used to transfect tumor cells against EGFP; confocal microscopy showed internalization of QDs as well as a decrease in green fluorescence over 3 days, proving their potential to create a theranostic agent with low toxicity as well as therapeutic efficacy in vitro.
Quantum dots present a powerful and versatile theranostic tool. Particularly advantageous is the ability to conjugate QDs with biological and therapeutic agents such as oligonucleotides and targeting ligands to produce a single particle with optical and biological dual-functionality. Various modification and functionalization techniques have produced QDs with characteristics that make them promising instruments for intracellular oligonucleotide delivery and tracking, especially the ability to use QDs with FRET to image spatiotemporal gene delivery. However, biodistribution of current QD compositions show inadequate clearance from live mice, and the long-term effects of QDs are largely unknown. The development of QDs for biological applications has come a long way in a little over a decade, but techniques for better long-term stability in biological buffers, higher quantum yield following bioconjugation, and reduced toxicity and improved clearance from the body are needed for the potential of quantum dots to be fully realized in the field of siRNA delivery and imaging.
5. Conclusions and Future Perspectives
Theranostic inorganic nanoparticles are effective vectors for gene delivery, even outperforming standard transfection vectors in some instances, while providing mechanisms for targeting, imaging and tracking of gene delivery. In recent years, gold, magnetic, and quantum dot nanoparticles have proven themselves superior in their ability to execute controlled and targeted delivery. Advances in imaging technology have established the capability to image spatiotemporal gene transfection at the single-molecule level with the aid of these theranostic particles. These small successes have opened up new doors for the development of gene therapy options, yet the goal for highly efficient, specific in vivo delivery and tracking of oligonucleotides has still not been fully recognized.
Recent research has focused on increasing transfection efficiency in vitro while minimizing cytotoxicity. In order to successfully administer genetic therapies, the coatings that were successful in vitro need to be combined with common in vivo targeting mechanisms. Some work has looked to combining these areas of research; however, the success has been limited to unique disease models, such as cancer. Disease models without unique anatomies need to be pursed in order to test the limits of nanoparticle-mediated gene delivery.
The success of nanoparticle theranostics will depend on the target diseases and the genes used to treat them. The correct combination of nanoparticle, coating, and targeting mechanism will need to be tailored to each disease/gene combination. Substantial progress has been made toward improving cytotoxicity and transfection efficiency, and future work needs to focus on treatment of specific disease models in vivo, as well as the development of real-time in vivo imaging capability. Further, specific targeting mechanisms and minimum dosage levels must be determined in order for theranostic gene delivery to become a treatment standard.
Highlights.
Inorganic nanoparticles are effective theranostic agents for gene transfection.
The response to light or magnetic field imparts noninvasive delivery control and/or spatiotemporal tracking capabilities.
Further development of in vitro and in vivo testing and increased gene therapy efficacy is necessary for clinical success.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health NCI (Center for Oncophysics Grant CTO PSOC U54-CA-143837; and 1 R21 EB012726-01) and by a grant from the National Science Foundation (CBET-1033746). JMK acknowledges support from the National Science Foundation Graduate Research Fellowship Program (DGE- 1110007).
Abbreviations
- ASON
antisense oligonucleotide
- siRNA
small interfering RNA
- miRNA
micro RNA
- shRNA
small hairpin RNA
- EDC
1-ethyl-3-(3-dimethylaminopropyl) carbodiimide)
- NHS
N-hydroxysuccinimide
- MNP
magnetic nanoparticles
- MRI
magnetic resonance imaging
- MIONP
magnetic iron oxide particles
- PEI
polyethylenimine
- PEI max
deacylated polyethylenimine
- PEG
polyethylene glycol
- dODN
decoy oligonucleotide
- STAT 3
signa transducer activator of transcription 3
- PAMAM
polyamidoamine
- asODN
antisense surviving oligodeoxynucleotide
- cMNP
magnetic nanoparticle clusters
- LCMNP
lipid coated magnetic nanoparticle
- AFsiRNA
Alexa Fluor 488-labled siRNA (AFsiRNA)
- EPR
enhanced permeability and retention effect
- AuNP
gold nanoparticle
- LSPR
localized surface plasmon resonance
- AuNR
gold nanorod
- AuNS
gold nanoshell
- NIR
near-infrared
- SERS
surface-enhanced Raman spectroscopy
- DTTC
3,3’-diethylthiatricarbocyanine iodide
- TEM
transmission electron microscopy
- ABT
4-aminobenzenethiol
- BPEI
branched polyethylenimine
- dsDNA
double-stranded DNA
- DAPI
4’,6-diamidino-2-phenylindole
- ssDNA
single-stranded DNA
- GFP
green fluorescent protein
- PLL
poly-l-lysine
- RNAi
RNA interference
- HA
hyaluronic acid
- AFM
atomic force microscopy
- QD
quantum dot
- TOP
trioctylphosphine
- TOPO
trioctylphosphine oxide
- CdSe
cadmium selenide
- CdTe
cadmium telluride
- PL
photoluminescent
- CdS
cadmium sulfide
- ZnS
zinc sulfide
- EGFRvIII
epidermal growth factor receptor variant III
- P-gp
P-glycoprotein
- L-Arg
L-arginine
- β-CD
β-cyclodextrin
- FRET
fluorescence resonance energy transfer
- HER2
human epidermal growth factor 2
- pDNA
plasmid DNA
- RES
reticuloendothelial system
- InAs
indium arsenide
Biographies

Jennifer M. Knipe is a National Science Foundation Graduate Research Fellow and Ph.D. candidate in the Department of Chemical Engineering at The University of Texas at Austin, working under the direction of Dr. Nicholas Peppas. She is developing pH-responsive hydrogel systems for the delivery of siRNA to specific intestinal regions via an oral administration route as a treatment for intestinal diseases. Jennifer received a B.S. in chemical engineering from West Virginia University in May of 2010.

Jonathan T. Peters is currently working towards a Ph.D. in Chemical Engieering at The University of Texas at Austin under the tutelage of Dr. Nicholas Peppas. His research focuses on the inclusion of inorganic nanoparticles in polymeric hydrogels for theranostic approaches to cancer treatment. Jonathan graduated summa cum laude with a B.S. in Chemical and Bimolecular Engineering from North Carolina State University in May of 2011.

Nicholas A. Peppas is the Fletcher S. Pratt Chair in engineering, a chaired professor of Chemical Engineering, Biomedical Engineering, and Pharmacy, and the Director of the Center for Biomaterials, Drug Delivery, Bionanotechnology and Molecular Recognition at The University of Texas at Austin. He is a member of the National Academy of Engineering (2012 Founders Award), the Institute of Medicine of the National Academies, the National Academy of Pharmacy of France, and the Royal Academy of Spain. He received his diploma in engineering (DEng) from the National Technical University of Athens, Greece in 1971 and his ScD from MIT in 1973, both in chemical engineering. He holds honorary doctorates from the Universities of Ghent, Parma, Athens and Ljubljana.
Footnotes
Additional information
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Atkinson H, Chalmers R. Genetica. 2010;138:485–498. doi: 10.1007/s10709-009-9434-3. [DOI] [PubMed] [Google Scholar]
- 2.Robbins PD, Ghivizzani SC. Pharmacol Ther. 1998;80:35–47. [PubMed] [Google Scholar]
- 3.Forbes DC, Peppas NA. J Controlled Release. 2012;162:438–445. doi: 10.1016/j.jconrel.2012.06.037. [DOI] [PubMed] [Google Scholar]
- 4.Verma IM, Weitzman MD. Annu Rev Biochem. 2005;74:711–738. doi: 10.1146/annurev.biochem.74.050304.091637. [DOI] [PubMed] [Google Scholar]
- 5.Schaffert D, Wagner E. Gene Ther. 2008;15:1131–1138. doi: 10.1038/gt.2008.105. [DOI] [PubMed] [Google Scholar]
- 6.Whitehead KA, Langer R, Anderson DG. Nat Rev Drug Discovery. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fattal E, Bochot A. Int J Pharm. 2008;364:237–248. doi: 10.1016/j.ijpharm.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 8.Niidome T, Huang L. Gene Ther. 2002;9:1647–1652. doi: 10.1038/sj.gt.3301923. [DOI] [PubMed] [Google Scholar]
- 9.Caldorera-Moore ME, Liechty WB, Peppas NA. Acc Chem Res. 2011;44:1061–1070. doi: 10.1021/ar2001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho EC, Glaus C, Chen J, Welch MJ, Xia Y. Trends in Mol Med. 2010;16:561–573. doi: 10.1016/j.molmed.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pissuwan D, Niidome T, Cortie MB. J Controlled Release. 2011;149:65–71. doi: 10.1016/j.jconrel.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 12.Cormode DP, Skajaa T, Fayad ZA, Mulder WJM. Arterioscler Thromb Vas Biol. 2008;29:992–1000. doi: 10.1161/ATVBAHA.108.165506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bardhan R, Lal S, Joshi A, Halas NJ. Acc Chem Res. 2011;44:936–946. doi: 10.1021/ar200023x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim CT, Han J, Guck J, Espinosa H. Med Biol Eng Comput. 2010;48:941–943. doi: 10.1007/s11517-010-0677-z. [DOI] [PubMed] [Google Scholar]
- 15.West JL, Halas NJ. Annu Rev Biomed Eng. 2003;5:285–292. doi: 10.1146/annurev.bioeng.5.011303.120723. [DOI] [PubMed] [Google Scholar]
- 16.Yezhelyev MV, Qi L, O’Regan RM, Nie S, Gao X. J Am Chem Soc. 2008;130 doi: 10.1021/ja800086u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenthal SJ, Chang JC, Kovtun O, McBride JR, Tomlinson ID. Chem Biol. 2011;18:10–24. doi: 10.1016/j.chembiol.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith A, Duan H, Mohs A, Nie S. Adv Drug Delivery Rev. 2008;60:1226–1240. doi: 10.1016/j.addr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hlavacek A, Bouchal P, Skládal P. Microchim Acta. 2011 [Google Scholar]
- 20.Jackeray R, Zainul Abid CKV, Singh G, Jain S, Chattopadhyaya S, Sapra S, Shrivastav TG, Singh H. Talanta. 2011;84:952–962. doi: 10.1016/j.talanta.2011.02.052. [DOI] [PubMed] [Google Scholar]
- 21.Lee M-Y, Park S-J, Park K, Kim KS, Lee H, Hahn SK. ACS Nano. 2011;5:6138–6147. doi: 10.1021/nn2017793. [DOI] [PubMed] [Google Scholar]
- 22.Gubin SP. Wiley-VCH Verlag GmbH & Co KGaA; Weinheim, Germany: 2009. p. 451. [Google Scholar]
- 23.Kievit FM, Veiseh O, Fang C, Bhattarai N, Lee D, Ellenbogen RG, Zhang M. ACS Nano. 2010;4:4587–4594. doi: 10.1021/nn1008512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar M, Yigit M, Dai G, Moore A, Medarova Z. Cancer Res. 2010;70:7553–7561. doi: 10.1158/0008-5472.CAN-10-2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hyeon T. Chem Commun. 2003:927–934. doi: 10.1039/b207789b. [DOI] [PubMed] [Google Scholar]
- 26.Bae KH, Lee K, Kim C, Park TG. Biomaterials. 2011;32:176–184. doi: 10.1016/j.biomaterials.2010.09.039. [DOI] [PubMed] [Google Scholar]
- 27.Lu A-H, Salabas EL, Schüth F. Angewandte Chem Int Edit. 2007;46:1222–1244. doi: 10.1002/anie.200602866. [DOI] [PubMed] [Google Scholar]
- 28.Taratula O. Curr Drug Del. 2011;2011:59–69. doi: 10.2174/156720111793663642. [DOI] [PubMed] [Google Scholar]
- 29.Arsianti M, Lim M, Marquis CP, Amal R. Biomacromolecules. 2010;11:2521–2531. doi: 10.1021/bm100748p. [DOI] [PubMed] [Google Scholar]
- 30.Fischer D, Li Y, Ahlemeyer B, Krieglstein J, Kissel T. Biomaterials. 2003;24:1121–1131. doi: 10.1016/s0142-9612(02)00445-3. [DOI] [PubMed] [Google Scholar]
- 31.Cutler JI, Zheng D, Xu X, Giljohann DA, Mirkin CA. Nano Lett. 2010;10:1477–1480. doi: 10.1021/nl100477m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geinguenaud F, Souissi I, Fagard R, Motte L, Lalatonne Y. Nanomed -Nanotechnol. doi: 10.1016/j.nano.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 33.Biswas S, Gordon LE, Clark GJ, Nantz MH. Biomaterials. 2011;32:2683–2688. doi: 10.1016/j.biomaterials.2010.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan B, Cui D, Sheng Y, Ozkan C, Gao F, He R, Li Q, Xu P, Huang T. Cancer Res. 2007;67:8156–8163. doi: 10.1158/0008-5472.CAN-06-4762. [DOI] [PubMed] [Google Scholar]
- 35.Park JS, Na K, Woo DG, Yang HN, Kim JM, Kim JH, Chung H-M, Park K-H. Biomaterials. 2010;31:124–132. doi: 10.1016/j.biomaterials.2009.09.023. [DOI] [PubMed] [Google Scholar]
- 36.del Pino P, Munoz-Javier A, Vlaskou D, Rivera Gil P, Plank C, Parak WJ. Nano Lett. 2010;10:3914–3921. doi: 10.1021/nl102485v. [DOI] [PubMed] [Google Scholar]
- 37.Zhang H, Lee M-Y, Hogg MG, Dordick JS, Sharfstein ST. ACS Nano. 2010;4:4733–4743. doi: 10.1021/nn9018812. [DOI] [PubMed] [Google Scholar]
- 38.Namiki Y, Namiki T, Yoshida H, Ishii Y, Tsubota A, Koido S, Nariai K, Mitsunaga M, Yanagisawa S, Kashiwagi H, Mabashi Y, Yumoto Y, Hoshina S, Fujise K, Tada N. Nat Nano. 2009;4:598–606. doi: 10.1038/nnano.2009.202. [DOI] [PubMed] [Google Scholar]
- 39.Lytton-Jean AKR, Langer R, Anderson DG. Small. 2011;7:1932–1937. doi: 10.1002/smll.201100761. [DOI] [PubMed] [Google Scholar]
- 40.Huschka R, Zuloaga J, Knight MW, Brown LV, Nordlander P, Halas NJ. J Am Chem Soc. 2011;133:12247–12255. doi: 10.1021/ja204578e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen X-J, Sanchez-Gaytan BL, Qian Z, Park S-J. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2012;4:273–290. doi: 10.1002/wnan.1159. [DOI] [PubMed] [Google Scholar]
- 42.Perez-Juste J, Pastorizasantos I, Lizmarzan L, Mulvaney P. Coord Chem Rev. 2005;249:1870–1901. [Google Scholar]
- 43.Daniel M-C, Astruc D. Chem Rev. 2004;104:293–346. doi: 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- 44.Turkevich J, Stevenson PC, Hillier J. Discuss Faraday Soc. 1951;11:55. [Google Scholar]
- 45.Frens G. Nat Phys Sci. 1973;241:20–22. [Google Scholar]
- 46.Oldenburg SJ, Averitt RD, Westcott SL, Halas NJ. Chem Phys Lett. 1998;288:243–247. [Google Scholar]
- 47.Murphy CJ, Gole AM, Stone JW, Sisco PN, Alkilany AM, Goldsmith EC, Baxter SC. Acc Chem Res. 2008;41:1721–1730. doi: 10.1021/ar800035u. [DOI] [PubMed] [Google Scholar]
- 48.Boisselier E, Astruc D. Chem Soc Rev. 2009;38:1759. doi: 10.1039/b806051g. [DOI] [PubMed] [Google Scholar]
- 49.Kim EY, Schulz R, Swantek P, Kunstman K, Malim MH, Wolinsky SM. Gene Ther. 2011;19:347–353. doi: 10.1038/gt.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ng C-T, Dheen ST, Yip W-CG, Ong C-N, Bay B-H, Lanry Yung L-Y. Biomaterials. 2011;32:7609–7615. doi: 10.1016/j.biomaterials.2011.06.038. [DOI] [PubMed] [Google Scholar]
- 51.Barhoumi A, Huschka R, Bardhan R, Knight MW, Halas NJ. Chem Phys Lett. 2009;482:171–179. [Google Scholar]
- 52.Jeong S, Choi SY, Park J, Seo J-H, Park J, Cho K, Joo S-W, Lee SY. J Mater Chem. 2011;21:13853. [Google Scholar]
- 53.Braun GB, Pallaoro A, Wu G, Missirlis D, Zasadzinski JA, Tirrell M, Reigh NO. ACS Nano. 2009;3:2007–2015. doi: 10.1021/nn900469q. [DOI] [PubMed] [Google Scholar]
- 54.Yigit MV, Medarova Z. Am J Nucl Med Mol Imaging. 2012;2:232–241. [PMC free article] [PubMed] [Google Scholar]
- 55.Qian J, Jiang L, Cai F, Wang D, He S. Biomaterials. 2011;32:1601–1610. doi: 10.1016/j.biomaterials.2010.10.058. [DOI] [PubMed] [Google Scholar]
- 56.Huschka R, Neumann O, Barhoumi A, Halas NJ. Nano Lett. 2010;10:4117–4122. doi: 10.1021/nl102293b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huschka R, Barhoumi A, Liu Q, Roth JA, Ji L, Halas NJ. ACS Nano. 2012 doi: 10.1021/nn301135w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patel PC, Hao L, Au Yeung WS, Mirkin CA. Mol Pharm. 2011;8:1285–1291. doi: 10.1021/mp200084y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hao L, Patel PC, Alhasan AH, Giljohann DA, Mirkin CA. Small. 2011;7:3158–3162. doi: 10.1002/smll.201101018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reynolds JL, Law WC, Mahajan SD, Aalinkeel R, Nair B, Sykes DE, Yong K-T, Hui R, Prasad PN, Schwartz SA. J Neuroimmune Pharmacol. 2012;7:673–685. doi: 10.1007/s11481-012-9379-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Resch-Genger U, Grabolle M, Cavaliere-Jaricot S, Nitschke R, Nann T. Nat Methods. 2008;5:763–775. doi: 10.1038/nmeth.1248. [DOI] [PubMed] [Google Scholar]
- 62.Wang D, Rogach AL, Caruso F. Nano Lett. 2002;2:857–861. [Google Scholar]
- 63.Murray CB, Norris DJ, Bawendi MG. J Am Chem Soc. 1993;115:8706–8715. [Google Scholar]
- 64.Qu L, Peng ZA, Peng X. Nano Lett. 2001;1 [Google Scholar]
- 65.Yong K-T, Law W-C, Roy I, Jing Z, Huang H, Swihart MT, Prasad PN. J Biophotonics. 2011;4:9–20. doi: 10.1002/jbio.201000080. [DOI] [PubMed] [Google Scholar]
- 66.Bailey RE, Nie S. J Am Chem Soc. 2003;125:7100–7106. doi: 10.1021/ja035000o. [DOI] [PubMed] [Google Scholar]
- 67.Subramaniam P, Lee SJ, Shah S, Patel S, Starovoytov V, Lee K-B. Adv Mater. 2012;24:4014–4019. doi: 10.1002/adma.201201019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dabbousi BO, Rodriguez-Viejo J, Mikulec FV, Heine JR, Mattoussi H, Ober R, Jensen KF, Bawendi MG. J Phys Chem B. 1997;101:9463–9475. [Google Scholar]
- 69.Mattoussi H, Palui G, Na HB. Adv Drug Delivery Rev. 2011 doi: 10.1016/j.addr.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 70.Mcbride J, Treadway J, Feldman LC, Pennycook SJ, Rosenthal SJ. Nano Lett. 2006;6:1496–1501. doi: 10.1021/nl060993k. [DOI] [PubMed] [Google Scholar]
- 71.Chen AA, Derfus AM, Khetani SR, Bhatia SN. Nucleic Acids Res. 2005;33:e190–e190. doi: 10.1093/nar/gni188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Derfus AM, Chen AA, Min D, Ruoslahti E, Bhatia SN. Bioconjugate Chem. 2007;18:1391–1396. doi: 10.1021/bc060367e. [DOI] [PubMed] [Google Scholar]
- 73.Jung J, Solanki A, Memoli KA, Kamei K-i, Kim H, Drahl MA, Williams LJ, Tseng H-R, Lee K. Angew Chem Int Ed Engl. 2010;49:103–107. doi: 10.1002/anie.200905126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li Y, Duan X, Jing L, Yang C, Qiao R, Gao M. Biomaterials. 2011;32:1923–1931. doi: 10.1016/j.biomaterials.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 75.Tan WB, Jiang S, Zhang Y. Biomaterials. 2007;28:1565–1571. doi: 10.1016/j.biomaterials.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 76.Duan H, Nie S. J Am Chem Soc. 2007;129:3333–3338. doi: 10.1021/ja068158s. [DOI] [PubMed] [Google Scholar]
- 77.Li S, Liu Z, Ji F, Xiao Z, Wang M, Peng Y, Zhang Y, Liu L, Liang Z, Li F. Mol Ther Nucleic Acids. 2012;1:e20. doi: 10.1038/mtna.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klein S, Zolk O, Fromm MF, Schrödl F, Neuhuber W, Kryschi C. Biochem Biophys Res Commun. 2009;387:164–168. doi: 10.1016/j.bbrc.2009.06.144. [DOI] [PubMed] [Google Scholar]
- 79.Li J-M, Zhao M-X, Su H, Wang Y-Y, Tan C-P, Ji L-N, Mao Z-W. Biomaterials. 2011;32:7978–7987. doi: 10.1016/j.biomaterials.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 80.Selvin PR. Nat Struct Mol Biol. 2000;7:730–734. doi: 10.1038/78948. [DOI] [PubMed] [Google Scholar]
- 81.Lee H, Kim I-K, Park TG. Bioconjugate Chem. 2010;21:289–295. doi: 10.1021/bc900342p. [DOI] [PubMed] [Google Scholar]
- 82.Qi L, Gao X. ACS Nano. 2008;2:1403–1410. doi: 10.1021/nn800280r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shaheen SM, Akita H, Yamashita A, Katoono R, Yui N, Biju V, Ishikawa M, Harashima H. Nucleic Acids Res. 2011;39:e48–e48. doi: 10.1093/nar/gkq1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ho Y-P, Chen HH, Leong KW, Wang T-H. J Controlled Release. 2006;116:83–89. doi: 10.1016/j.jconrel.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Biju V, Anas A, Akita H, Shibu ES, Itoh T, Harashima H, Ishikawa M. ACS Nano. 2012;6:3776–3788. doi: 10.1021/nn2048608. [DOI] [PubMed] [Google Scholar]
- 86.Wu Y, Ho Y-P, Mao Y, Wang X, Yu B, Leong KW, Lee LJ. Mol Pharm. 2011;8:1662–1668. doi: 10.1021/mp100466m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang B, Zhang Y, Mallapragada SK, Clapp AR. ACS Nano. 2011;5:129–138. doi: 10.1021/nn1018939. [DOI] [PubMed] [Google Scholar]
- 88.Soo Choi H, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, Bawendi MG, Frangioni JV. Nat Biotechnol. 2007;25:1165–1170. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ballou B, Lagerholm BC, Ernst LA, Bruchez MP, Waggoner AS. Bioconjugate Chem. 2004;15:79–86. doi: 10.1021/bc034153y. [DOI] [PubMed] [Google Scholar]
- 90.Gao X, Cui Y, Levenson RM, Chung LWK, Nie S. Nat Biotechnol. 2004;22:969–976. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 91.Kim S, Lim YT, Soltesz EG, De Grand AM, Lee J, Nakayama A, Parker JA, Mihaljevic T, Laurence RG, Dor DM, Cohn LH, Bawendi MG, Frangioni JV. Nat Biotechnol. 2003;22:93–97. doi: 10.1038/nbt920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schipper ML, Cheng Z, Lee SW, Bentolila LA, Iyer G, Rao J, Chen X, Wu AM, Weiss S, Gambhir SS. J Nucl Med. 2007;48:1511–1518. doi: 10.2967/jnumed.107.040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schipper ML, Iyer G, Koh AL, Cheng Z, Ebenstein Y, Aharoni A, Keren S, Bentolila LA, Li J, Rao J, Chen X, Banin U, Wu AM, Sinclair R, Weiss S, Gambhir SS. Small. 2009;5:126–134. doi: 10.1002/smll.200800003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pons T, Pic E, Lequeux N, Cassette E, Bezdetnaya L, Guillemin F, Marchal F, Dubertret B. ACS Nano. 2010;4:2531–2538. doi: 10.1021/nn901421v. [DOI] [PubMed] [Google Scholar]
- 95.Kim JS, Cho KJ, Tran TH, Nurunnabi M, Moon TH, Hong SM, Lee Y-k. J Colloid Interface Sci. 2011;353:363–371. doi: 10.1016/j.jcis.2010.08.053. [DOI] [PubMed] [Google Scholar]
- 96.Ye L, Yong K-T, Liu L, Roy I, Hu R, Zhu J, Cai H, Law W-C, Liu J, Wang K, Liu J, Liu Y, Hu Y, Zhang X, Swihart MT, Prasad PN. Nat Nanotechnol. 2012;7:453–458. doi: 10.1038/nnano.2012.74. [DOI] [PubMed] [Google Scholar]
- 97.Park J, Dvoracek C, Lee KH, Galloway JF, Bhang H-eC, Pomper MG, Searson PC. Small. 2011;7:3148–3152. doi: 10.1002/smll.201101558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Al-Deen FN, Ho J, Selomulya C, Ma C, Coppel R. Langmuir. 2011;27:3703–3712. doi: 10.1021/la104479c. [DOI] [PubMed] [Google Scholar]
- 99.Kumar A, Jena PK, Behera S, Lockey RF, Mohapatra S, Mohapatra S. Nanomed -Nanotechnol. 2010;6:64–69. doi: 10.1016/j.nano.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Taratula O. Curr Drug Delivery. 2011;2011:59–69. doi: 10.2174/156720111793663642. [DOI] [PubMed] [Google Scholar]
- 101.Pan B, Cui D, Sheng Y, Ozkan C, Gao F, He R, Li Q, Xu P, Huang T. Cancer Res. 2007;67:8156–8163. doi: 10.1158/0008-5472.CAN-06-4762. [DOI] [PubMed] [Google Scholar]
- 102.Cutler JI, Zheng D, Xu X, Giljohann DA, Mirkin CA. Nano Lett. 2010;10:1477–1480. doi: 10.1021/nl100477m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kumar M, Yigit M, Dai G, Moore A, Medarova Z. Cancer Res. 2010;70:7553–7561. doi: 10.1158/0008-5472.CAN-10-2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Namiki Y, Namiki T, Yoshida H, Ishii Y, Tsubota A, Koido S, Nariai K, Mitsunaga M, Yanagisawa S, Kashiwagi H, Mabashi Y, Yumoto Y, Hoshina S, Fujise K, Tada N. Nat Nano. 2009;4:598–606. doi: 10.1038/nnano.2009.202. [DOI] [PubMed] [Google Scholar]
- 105.Pan X, Guan J, Yoo J-W, Epstein AJ, Lee LJ, Lee RJ. Int J Pharm. 2008;358:263–270. doi: 10.1016/j.ijpharm.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kami D, Takeda S, Makino H, Toyoda M, Itakura Y, Gojo S, Kyo S, Umezawa A, Watanabe M. J Artif Organs. 2011;14:215–222. doi: 10.1007/s10047-011-0568-6. [DOI] [PubMed] [Google Scholar]
- 107.Geinguenaud F, Souissi I, Fagard R, Motte L, Lalatonne Y. Nanomed -Nanotechnol. 2012;8:1106–1115. doi: 10.1016/j.nano.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 108.Zhou Y, Tang Z, Shi C, Shi S, Qian Z, Zhou S. J Mater Sci : Mater Med. 2012;23:2697–2708. doi: 10.1007/s10856-012-4720-5. [DOI] [PubMed] [Google Scholar]
- 109.del Pino P, Munoz-Javier A, Vlaskou D, Rivera Gil P, Plank C, Parak WJ. Nano Lett. 2010;10:3914–3921. doi: 10.1021/nl102485v. [DOI] [PubMed] [Google Scholar]
- 110.Liang Z, Liu Y, Li X, Wu Q, Yu J, Luo S, Lai L, Liu S. J Biomed Mater Res Part A. 2011;98A:479–487. doi: 10.1002/jbm.a.33068. [DOI] [PubMed] [Google Scholar]