


Abstract
High-throughput sequencing is becoming a popular research tool but carries with it considerable costs in terms of computation time, data storage and bandwidth. Meanwhile, some research applications focusing on individual genes or pathways do not necessitate processing of a full sequencing dataset. Thus, it is desirable to partition a large dataset into smaller, manageable, but relevant pieces. We present a toolkit for partitioning raw sequencing data that includes a method for extracting reads that are likely to map onto pre-defined regions of interest. We show the method can be used to extract information about genes of interest from DNA or RNA sequencing samples in a fraction of the time and disk space required to process and store a full dataset. We report speedup factors between 2.6 and 96, depending on settings and samples used. The software is available at http://www.sourceforge.net/projects/triagetools/.
INTRODUCTION
High-throughput sequencing (HTS) technology is designed to measure the composition of large and diverse pools of genetic material. The associated data lend itself well to exploratory studies addressing universal issues, such as genetic variation within populations (1,2) or somatic mutation landscapes (3). Despite the important insights gained from such holistic approaches, a significant portion of biology research consists of in-depth analysis of individual genes or pathways. It is therefore relevant to devise effective methods to exploit existing HTS data in these small-scale and in-depth projects without necessitating use of overly specialized computer platforms.
In practice, the raw input to an HTS workflow—regardless of whether it is obtained from an in-house sequencer, service provider or public data repository (4)—typically consists of set of reads in FASTA or FASTQ format. Many workflows begin by mapping the reads onto a reference genome using a fast aligner [e.g. Bowtie (5), GSNAP (6), SOAP (7), SNAP (8), mr(s)Fast (9), RazerS (10,11) and others] and later perform project-specific computations, such as variant detection on the aligned data [SAMtools (12), GATK (13) and others]. Other workflows, for example for detection of fusion genes [deFuse (14) and others], begin their work directly on the raw reads. Due to the large scale associated with HTS, each of the steps can consume many hours of computation time and several gigabytes of storage space for each studied sample.
As the spectrum of data types deposited in HTS repositories is increasing, a researcher studying a small region of a genome (e.g. a gene) may wish to leverage some of this data for her own in-depth investigations. At the same time, she may not be interested, or indeed may not have the infrastructure, to process the entirety of data in the repository. Currently, the researcher can optimize the post-processing steps of the workflows to her needs by extracting relevant data from alignment files using tools, such as SAMtools (12). To do this, however, she must first incur the costs associated with aligning the entire dataset. It would seem more efficient to perform the extraction prior to alignment instead—we call this the problem of targeted extraction. Its implementation would allow the researcher to focus resources onto the intended purpose from the start.
The plausibility of efficient targeted extraction from raw, unaligned data (FASTQ files) can be motivated by recalling how a traditional aligner works. A common strategy utilized in many modern aligners is to split the alignment problem into two parts. In the first, the aligner considers an input read and produces a shortlist of candidate regions of origin. In the second stage, it calculates optimal placement for the read near each candidate site, computes a score for each solution and reports the solution with the best score [some aligners report all solutions up to a specified penalty (15,9,10)]. The second stage, local alignment and scoring, is actually the most time-consuming part of the process, but is not necessary for the purpose of targeted extraction. It is therefore possible to adapt, simplify and optimize the first stage of the alignment process for targeted extraction.
We designed a tool, which we call triage by sequence, specifically to carry out targeted extraction. The tool first scans a file containing a target sequence, for example that of an oncogene. It then scans input file or files with raw FASTQ data. For each read/read pair in the input, the tool determines whether the read can potentially be aligned to the target sequence (the oncogene). The tool does not optimize local alignment of the read against a reference sequence. Instead, it only performs a heuristic test and copies the read to one output file if it is potentially mappable to the target sequence and (optionally) to another output file otherwise. The output files are thus in the same format as the input, but are nonetheless indicative of the region of provenance of the reads within them. The output files can be treated using any existing HTS pipeline and the reduced data load implies that total execution times can be much shorter than in a traditional approach.
Empirically, pre-selection of data with the triage tool followed by alignment can be several times faster than alignment of a full dataset followed by target extraction using SAMtools. For example, starting with an RNA-seq sample with 75 million paired reads, our method makes it possible to extract reads relevant for a gene, align this data and genotype it in less than an hour using a personal workstation or laptop. The traditional approach, in contrast, requires more than 40 h using the same hardware. The ratio of running times for the traditional to the targeted approach, or speedup, is thus above 40.
We incorporated the targeted extraction program into a larger toolkit, TriageTools. The other programs in the kit provide transparent functionality for partitioning raw reads by length, base quality or for identification of (near) duplicate sequences. Together, these programs form a coherent toolkit offering a wide functionality for processing raw data from HTS experiments.
The remainder of this article is structured as follows. In the ‘Materials and Methods’ section, we describe the algorithmic details of our method and explain the factors that determine its performance. In the ‘Results’ section, we describe calibration tests on real RNA-seq data. The ‘Discussion’ section provides a summary of the results, considerations of alternative approaches and perspectives on applications.
MATERIALS AND METHODS
Algorithm
Our method requires three important inputs in addition to a collection of FASTQ reads. The first input is a file(s) with target sequences in FASTA format. The second is an integer, s, which we call the seedlength. The third is another integer, H, which we call the hits threshold. We explain the role of each below.
The algorithm begins by creating an array of 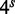 bits in memory, where s is the user-specified seedlength. All the bits in the array are initially set to zero. The algorithm then reads the target sequence, extracts all possible subsequences of length s and calculates a hash code for each one. (Such subsequences are sometimes also referred to in the literature as words, k-grams or n-mers.) In our implementation, the hash code is a two-bit representation of the subsequence, which means that it is a natural index in the boolean array. The bits in the array corresponding to words in the target are set to true. Words in the target sequence that contain non-(ATCG) characters, such as N are given a negative hash code and ignored.
bits in memory, where s is the user-specified seedlength. All the bits in the array are initially set to zero. The algorithm then reads the target sequence, extracts all possible subsequences of length s and calculates a hash code for each one. (Such subsequences are sometimes also referred to in the literature as words, k-grams or n-mers.) In our implementation, the hash code is a two-bit representation of the subsequence, which means that it is a natural index in the boolean array. The bits in the array corresponding to words in the target are set to true. Words in the target sequence that contain non-(ATCG) characters, such as N are given a negative hash code and ignored.
The bit array can be compared with a hash-table/index used in some aligners, such as BLAT (15), GSNAP (6) or SNAP (8), so we will also refer to it as an index. An important difference is that whereas the hash tables in those aligners record the genomic coordinate(s) of each subsequence, this detailed information is missing in the bit array. The bit array is thus more limited but at the same time more light weight. A similar technique of index simplification is behind fast algorithms for clustering of protein sequences, such as Cd-hit (16).
Processing of the input FASTQ files proceeds on a read by read basis. Single-end reads are treated as-is. For paired-end reads, the algorithm first reverse-complements the sequence of the second mate and concatenates it to the first mate with an N character in the middle. The resulting longer sequence is then treated identically as a single-end read.
The algorithm splits a read sequence into all possible words of length s and computes their hash codes. It then looks up the hash codes in the boolean array and counts the number of character hits, h, in the read sequence that are present in the target sequence. For example, consider a read that originates from the target sequences. Initially, the number of hits is set at h = 0. The first word of length s (positions 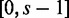 in the sequence) has a hash code that is present in the boolean array, so the hits counter is updated to h = s. The next word of length s (positions
in the sequence) has a hash code that is present in the boolean array, so the hits counter is updated to h = s. The next word of length s (positions 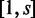 in the sequence) leads to another hit in the boolean array, the character hit counter is incremented from h = s to
in the sequence) leads to another hit in the boolean array, the character hit counter is incremented from h = s to 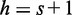 , and so on. The count increment becomes more complex when mismatches arise, but this example conveys the spirit of the calculation.
, and so on. The count increment becomes more complex when mismatches arise, but this example conveys the spirit of the calculation.
A read is classified as potentially originating from a target sequence if the character hit count h reaches or surpasses the user specified threshold H. To speed up the classification, the algorithm looks up seeds starting from both ends of the read and moves inward. This allows the character hit count h to grow quickly initially if the read is indeed from the target region. It also allows the algorithm to abandon the search part way through if too few hits are found to allow h to eventually pass the threshold. If a read fails to pass the threshold, the procedure is repeated using the reverse complement of the read sequence.
We mention that the ratio 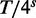 , where T is the length of the target sequence and
, where T is the length of the target sequence and 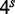 is the size of the boolean array, plays an important role in determining classification performance. This topic is discussed in more detail below, but we note here that if the ratio is close to one, almost any read sequence can accumulate sufficiently many character hits to pass the threshold and thus be classified as ‘potentially mapping to the target sequence’. For this reason, our implementation of targeted extraction is only suitable for relatively short target sequences. Nonetheless, to allow users to target medium-sized regions and maintain good performance, we implemented the possibility of using multiple independent arrays, each specified via a separate target sequence FASTA file. Thus, if a user prepares several files with target sequences, the algorithm creates a distinct bit array for each file and repeats the hits counting and classification calculation using each array. It declares a read as potentially mappable to the target if any one of these calculations gives a positive result.
is the size of the boolean array, plays an important role in determining classification performance. This topic is discussed in more detail below, but we note here that if the ratio is close to one, almost any read sequence can accumulate sufficiently many character hits to pass the threshold and thus be classified as ‘potentially mapping to the target sequence’. For this reason, our implementation of targeted extraction is only suitable for relatively short target sequences. Nonetheless, to allow users to target medium-sized regions and maintain good performance, we implemented the possibility of using multiple independent arrays, each specified via a separate target sequence FASTA file. Thus, if a user prepares several files with target sequences, the algorithm creates a distinct bit array for each file and repeats the hits counting and classification calculation using each array. It declares a read as potentially mappable to the target if any one of these calculations gives a positive result.
The complexity of the algorithm is O(NLI), where N is the number of reads in the full sample, L is the typical read length and I is the number of target files/indexes. Typically, we have I = 1 or I = 2. The algorithm is thus asymptotically optimal and classification is carried out in time directly proportional to the time needed to read the input data.
Classification performance
By construction, reads that match the target sequence are classified as such. However, the approach can also reach some false-negative and false-positive classification decisions.
False negatives can arise as a result of multiple sequencing errors and/or variants in the reads. Consider a read of length L (for paired reads, consider L to be the combined length of the pair). If the read matches the target sequence exactly, the maximum number of hits that it can accumulate is L. If the read contains one single-nucleotide error or variant, the number of hits that it can achieve is smaller than L. The degree of the drop depends on the location of the error/variant in relation to the read sequence. The drop can be a single count if the error is either exactly on one of the edges or far away from the edges. If the error is within a seedlength from one of the edges, the drop can be up to L − s. For n errors, possible values for the drop are in the range 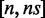 , capped by L. The entire range is possible in principle, but the individual values occur with different probabilities that depend on the properties of error-causing processes. Under a reasonable assumption of uniform error distribution, for example, the probability of the extreme values would be quite small. Irrespective of the details, one can see that the hits threshold H should be set below the read length L to allow for some error tolerance in the classification.
, capped by L. The entire range is possible in principle, but the individual values occur with different probabilities that depend on the properties of error-causing processes. Under a reasonable assumption of uniform error distribution, for example, the probability of the extreme values would be quite small. Irrespective of the details, one can see that the hits threshold H should be set below the read length L to allow for some error tolerance in the classification.
A false positive arises when a read does not actually originate from the target region, but accumulates sufficiently many hits to pass the threshold H. The rate of false positives will again depend on the parameters H and s. Consider a target region of length T and a seedlength s. If T is on the order of 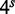 , the boolean array upon which the classification is based will be almost completely full (all bits set to one). In the extreme case of complete filling, all subsequences in all reads, will contribute to the hit count and lead to positive classification. Even if the fill ratio of the boolean array is low, a read may still contain subsequences that correspond to parts of the target purely by chance. As the boolean array does not contain sufficient information to compare the proximity of the various subsequences in the read and in the target region, such cases cannot be eliminated by the classifier. The main conclusion to draw is that low false-positive rates (FPRs) can be achieved by keeping T low, splitting the target into several indexes, setting larger values of H and s or a combination of these.
, the boolean array upon which the classification is based will be almost completely full (all bits set to one). In the extreme case of complete filling, all subsequences in all reads, will contribute to the hit count and lead to positive classification. Even if the fill ratio of the boolean array is low, a read may still contain subsequences that correspond to parts of the target purely by chance. As the boolean array does not contain sufficient information to compare the proximity of the various subsequences in the read and in the target region, such cases cannot be eliminated by the classifier. The main conclusion to draw is that low false-positive rates (FPRs) can be achieved by keeping T low, splitting the target into several indexes, setting larger values of H and s or a combination of these.
It becomes clear that the parameters H and s play important roles in determining the specificity and sensitivity of the targeted extraction classification and must be chosen as to achieve a good compromise. We study this issue in detail in the next section.
False-positive rates
We derive a formula for the expected FPR in the targeted extraction algorithm. The derivation assumes true randomness of sequences and equal representation of nucleotides, which are unrealistic in actual biological genomes or transcriptomes. Nonetheless, the calculation provides some insight into how various factors affect classification performance.
With the usual four-base genetic alphabet, there are 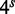 distinct words for a given seedlength s. A target region of length T can contain at most
distinct words for a given seedlength s. A target region of length T can contain at most 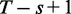 of them; this can be approximated by T as we are typically interested in the case
of them; this can be approximated by T as we are typically interested in the case 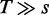 . The fill density of the boolean array, i.e the number of bits set to true divided by the total number of bits, is thus
. The fill density of the boolean array, i.e the number of bits set to true divided by the total number of bits, is thus 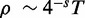 .
.
A read of length R has at most 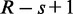 distinct words of lengths s. Even if a read does not originate from the target, some of its words may match to the target by chance. The probability for any word giving a seed hit in the boolean array is equal to the fill density, ρ. In order for the read to pass the character hit threshold, approximately H/s independent (non-overlapping) words must lead to array hits. We estimate that there are R/s independent words in the read. The probability of obtaining a false positive can thus be written as:
distinct words of lengths s. Even if a read does not originate from the target, some of its words may match to the target by chance. The probability for any word giving a seed hit in the boolean array is equal to the fill density, ρ. In order for the read to pass the character hit threshold, approximately H/s independent (non-overlapping) words must lead to array hits. We estimate that there are R/s independent words in the read. The probability of obtaining a false positive can thus be written as:
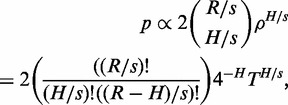 |
where the factor in the parenthesis is a binomial coefficient estimating the number of ways to choose a set of H/s independent words from the read with R/s independent words, and the overall factor of two is due to matching both forward and reverse strand representations. The formula is not exact but we are here only aiming at a rough estimate.
Numerically, this estimate gives the following results. For a one megabase target region 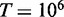 , single-end reads with R = 100, seedlength s = 12 and hits threshold H = 36, we have a fill density
, single-end reads with R = 100, seedlength s = 12 and hits threshold H = 36, we have a fill density 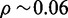 and estimated FPR
and estimated FPR 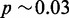 . We would thus expect more than 1% of reads to pass the heuristic test. Changing the required hit count to H = 50, we have
. We would thus expect more than 1% of reads to pass the heuristic test. Changing the required hit count to H = 50, we have 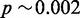 , a more satisfying result. With s = 14 and H = 50, we have
, a more satisfying result. With s = 14 and H = 50, we have 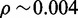 and
and 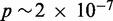 , which is better still.
, which is better still.
In practice, FPRs on truly random data are slightly higher than the above estimates (data not shown). More importantly, observed FPR on real data (‘Results’ section) can be considerably higher. This is due to the fact that actual genomic sequences and reads in HTS samples are not random and contain pronounced similarities/patterns that have been selected during evolution. If a target sequence contains a motif longer than the seedlength that is present in multiple regions of the genome, reads from those alternative regions are picked up by the classification scheme. The estimates, therefore, should be regarded as optimistic and their accuracy can vary greatly depending on the properties and uniqueness of the target sequence in relation to the ensemble of reads. Nonetheless, the formula is indicative of how each parameter affects the classification performance.
RESULTS
To demonstrate and quantify the classification performance of our target extraction method, we performed a number of experiments on publicly available RNA-seq samples from healthy human breast tissue sequenced on an Illumina platform (17). We report details on the experiments on a sample with 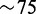 million paired-end reads of 50 bp each.
million paired-end reads of 50 bp each.
As a first step, we produced an alignment of the full RNA-seq sample using Tophat (18) (v2.0.3), which internally employs the fast aligner Bowtie (5) (v2.0.0). We used Tophat’s unsupervised mode, aligned onto the UCSC hg19 reference genome, and left all but one of the parameters at their default values. The exception was that we provided the aligner access to four processor cores. The running time for this procedure was 42 h (2.5 GHz processor; all subsequent experiments were carried out on the same machine). Below, we will refer to the result of this procedure as the ‘full’ alignment.
Single gene selection
For the first set of experiments, we considered a target consisting of a single gene: gene NOTCH1 on chromosome 9. This gene codes for a membrane protein, which is involved in signaling pathways determining cell fate. Mutations in the gene have been linked with human diseases including leukemias [e.g. (19,20)]. Studying the gene using HTS may therefore be of practical use for research and clinical practice.
We began by looking in the full alignment and extracting the ids of the reads that are aligned to the exons of this gene. This yielded 4522 unique id codes. By complementarity, the number of read ids in other regions was ∼75 million. Then, we prepared a FASTA file with the exonic sequence of the gene together with 20 bp adjacent regions. We considered this as the target sequence and ran our triage method to extract those reads from the full sample FASTQ files that potentially originate from this region. We tried all parameters combinations 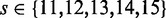 and
and 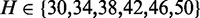 . Post-selection, we applied Tophat using the same settings as for the full sample.
. Post-selection, we applied Tophat using the same settings as for the full sample.
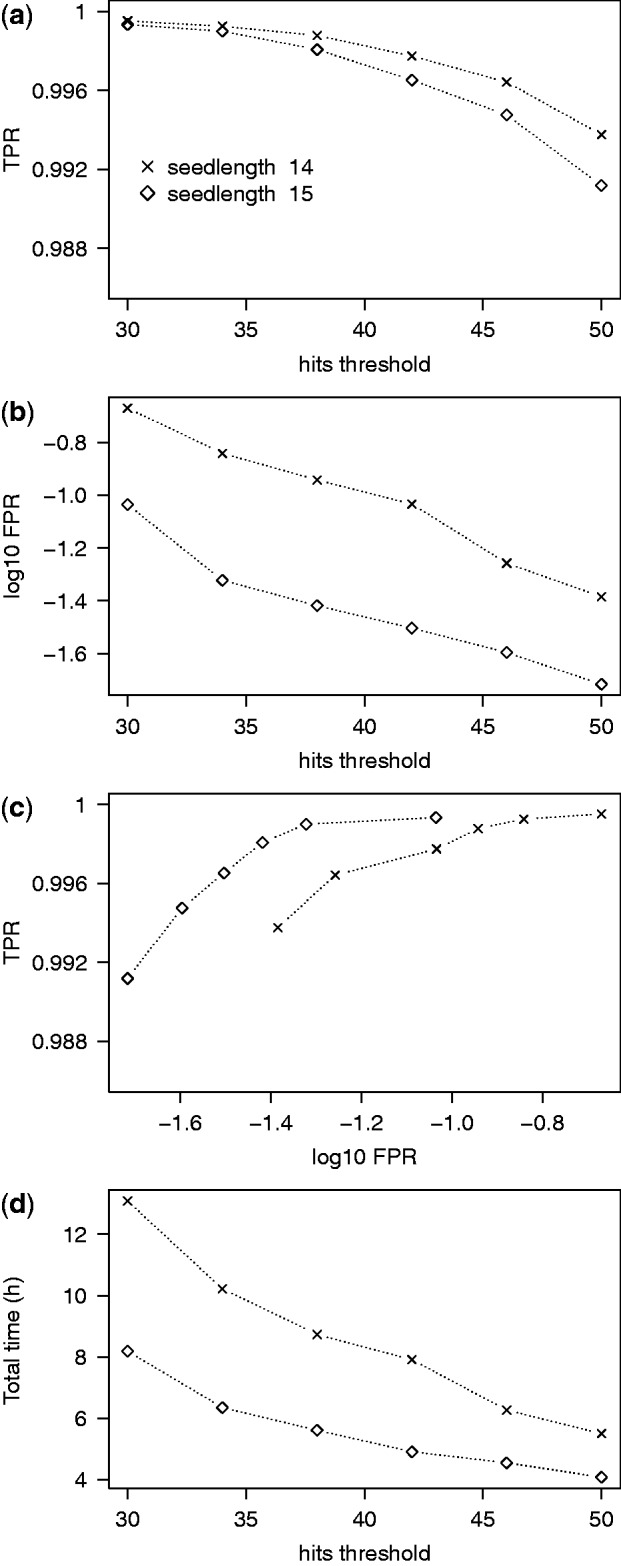
To measure classification performance, we used SAMtools view to extract the names of reads aligning on NOTCH1 exons in the post-triage alignments. We then compared these lists with the one obtained from the full alignment. The results (Figure 1) show several expected trends and trade-offs. Lower seedlengths and hits thresholds give better true-positive rate (TPR) but increase the FPR. For the majority of the combinations we tested, the TPR was above 99%, and the FPR was below 0.1%; in all cases, the respective numbers were 98 and 0.5%. The targeted extraction method is thus validated on data that includes real variants, splicing patterns and sequencing errors.
Figure 1.
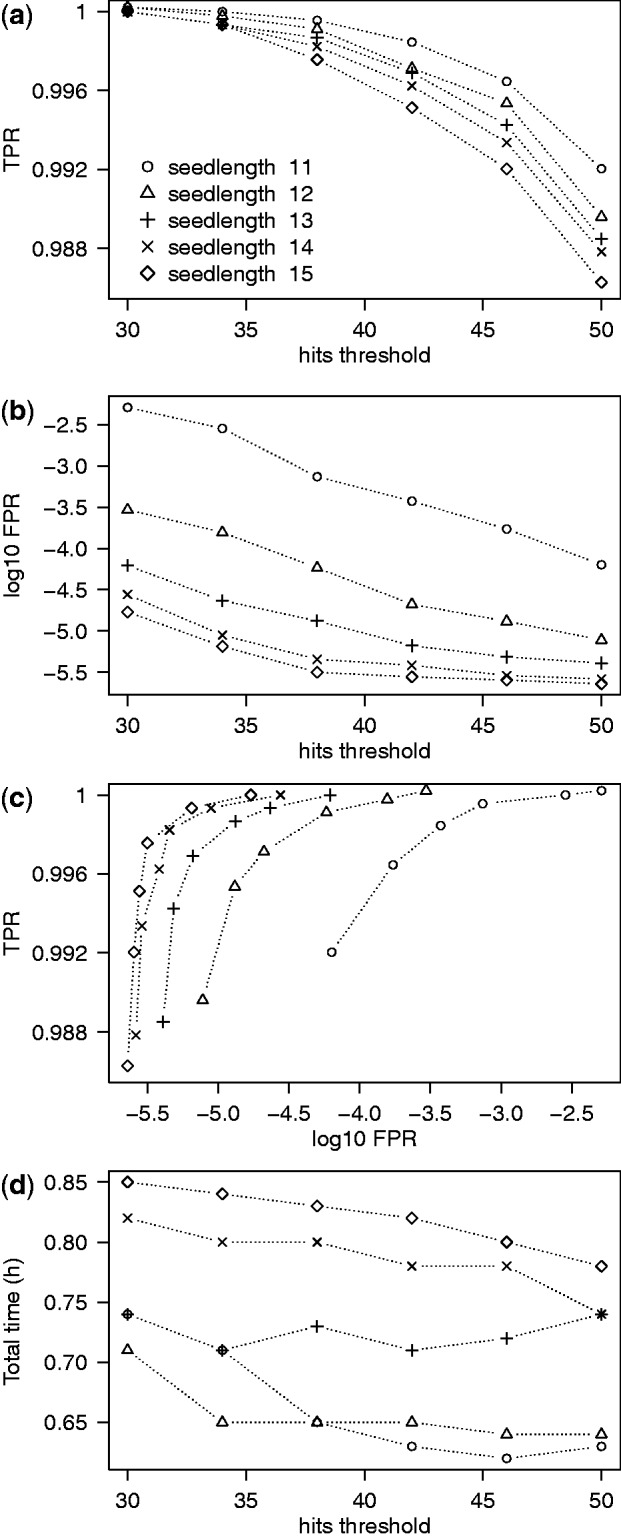
Single-gene classification performance. (a) TPR of classification decreases with the hits parameter. (b) Proportion of reads that are actually off-target but pass through the classification procedure. As expected, this FPR decreases with s and H. (c) A ROC-style representation of the panels (a) and (b). To make the points distinguishable, the vertical scale only shows a small fraction of the full TPR range and the horizontal scale is logarithmic. The series of points in each seedlength group represent different hits thresholds. (d) Running time of classification and mapping. For reference, the running for the full alignment was 42 h.
Since the TPR was sometimes lower than 100%, we investigated the individual reads that were captured by the full alignment but not by the triage procedure. Several of these turned out to be aligned mainly on intronic sequence and which overlapped exons only by a few bases. Their mates were either also aligned on intronic sequence or not aligned at all. We thus conclude that the actual coverage on exons lost by misclassification is minimal. Indeed, running variant calling on the full and post-triage alignments gave equivalent results on the targeted region. In any case, it would be possible to restore much of the lost coverage by extending the flanking regions around exons from 20 to 50 bp.
The total running times (time for targeted extraction using triage plus time for alignment using Tophat/Bowtie) for all parameter combinations were under 1 h (Figure 1d). The times depended only weakly on the hits parameter H or the seedlength s. The fall in times with H can be attributed to the possibility of abandoning scoring off-target reads earlier with higher H. Longer running times for higher s are likely due to the increased size of the index with s, which increases the chance of cache misses during hash code lookups.
Overall, we see that the targeted extraction approach provides data and alignments that are suitable for downstream analysis, such as variant detection. At the same time, the results are obtained with speedups above 40. Disk storage requirements during and after Tophat execution are also significantly reduced, in these cases by factors 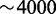 (data not shown).
(data not shown).
Multi-gene selection
In a second case study, we considered a much larger target region to establish versatility for the extraction tool. We again reasoned that a practical application for targeted extraction may be the fast identification of variants or expression levels in genes associated with disease. We thus used a list of 464 cancer-related genes (21) as the target. This list includes the gene NOTCH1 from the previous case study and other well-studied genes, such as TP53, PIK3CA, NRAS, etc.
As the 464-gene region was much larger than before, the classification should be expected to yield many more false positives. Therefore, we split the sequence into two files (see ‘Materials and Methods’ section). We also concentrated on tests with large seedlengths, 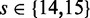 . All measurements of classification performance and running times were carried out like in the previous example.
. All measurements of classification performance and running times were carried out like in the previous example.
The results (Figure 2) are consistent with the previous observations. Of note is that despite the large seedlengths, the TPRs were above 99% in all cases. This implies that the NOTCH1 example was actually a difficult gene to classify and that one can expect better results on average. The number of false positive reads was expectedly much higher than before, which made the total running times longer than before. Nonetheless, the speedups were still between 3 and 6. Disk usage during and after Tophat execution is reduced by similar factors.
Figure 2.
Multiple-gene classification performance. Analogous to Figure 1, but showing results for a target region consisting of 464 cancer genes. In contrast to the case study in Figure 1, the total running is here dominated by alignment rather than extraction. Thus, it is the larger seedlengths that provide higher speedup [panel (d)].
DISCUSSION
We comment on a number of issues related to classification performance.
Optimal choice of parameters
Figures 1 and 2 show that classification performance varies with the seedlength s, the hits threshold H and the size of the target region. It will also vary with the length of the input reads. Therefore, there is no single optimal choice for any of the parameters. Nonetheless, the results above are indicative as to what performance can be expected. As the tool is primarily designed to work with small target regions, we used Figure 1 to set s = 12 and H = 38 as default values to obtain 99.9% TPR on small targets. For other read lengths or for variable-length reads, it is also possible to specify H as a number less than two, in which case the threshold would be determined at run time as a fraction of each processed read.
Generalization to small target regions
The calculation involving 464 cancer genes shows that the classification procedure works well even on a large target region. It also implies that the classification would work effectively were it performed on any subset of the 464 studied genes. It is also reasonable to expect similar performance on genes not part of the cancer set. On such intermediate size targets, the true-positive, false-positive and false-negative rates should be expected to be as good as or better than in Figure 2.
Generalization to large target regions
It is tempting to consider larger target regions as well. While it would be possible in principle to achieve good classification performance on larger regions, we note that this would require splitting the target sequence into several pieces, effectively slowing down the algorithm and increasing its memory footprint. Furthermore, as the extraction would output a larger portion of the input read set, the speedup benefit over traditional alignment would be less pronounced. We therefore do not recommend applying the extraction method on target sequences much larger than the multi-gene set above.
Repetitive regions
Some regions in a genome have high sequence similarity as a result of joint evolutionary history. If such a region is selected as the target (an example is explored in the Supplementary Material), the classification procedure will extract a large number of reads from the similar regions in addition to the ones from the actual locus of interest, effectively increasing the FPR. This is an unavoidable property of our algorithm, which does not keep track of genomic coordinates of the target.
Tolerance to known variants
As explained in the ‘Materials and methods’ section, our algorithm can in rare cases mistakenly discard reads actually originating from the target region if they are short and if they contain several features that distinguish them from the target sequence. Although the achieved TPRs are already consistently above 98% in realistic cases, it is possible to obtain even better results by reducing the negative impact of known variants (SNPs, indels or splice junctions in RNA-seq data). This can be achieved by providing appropriately constructed target sequence files. For SNPs and indels, this would be conceptually similar to SNP-tolerance introduced in SNP-o-matic (22) and used by GSNAP (6) and others (23). For splice junctions, it would mimic the practice of using gene annotations in RNA-seq mapping software, such as Tophat (18) or GSNAP (6).
Stochastic effects
In the single-gene experiments, we observed some reads were placed outside the target region in the full alignment, but inside the target region when running on the collection of reads subset by our triage tool, giving rise to some TPRs above 100%. This can be attributed to a stochastic element in Tophat/Bowtie for handling ambiguous reads. It can lead both to an apparent increase or decrease in the coverage on the target region. We mention this as a curiosity; the effect would not have arisen had we mapped all reads with a fully deterministic aligner.
Other types of HTS data
The detailed experiments above were performed using paired-end RNA-seq data. We also performed some tests on real single-end RNA-seq data (17), as well as synthetic exome (depth 40×) and whole genome (WG) data (depth 8×). For the DNA samples, we compared running times using Bowtie rather than Tophat. The results (Table 1) are not meant to be exhaustive; they can change depending on sample size, aligner, number of processors, etc. Nonetheless, they demonstrate the qualitative performance gains are generalizable to other HTS platforms, read lengths and mapping methods. The speedup values are defined, like before, as time for alignment of full sample divided by the sum of the extraction time plus alignment of the extracted data.
Table 1.
Speedup due to targeted alignment
| NOTCH1 |
Cancer genes |
|||
|---|---|---|---|---|
| Sample | 1 core | 4 cores | 1 core | 4 cores |
| RNA-seq (2 × 50 bp) | 96 | 53 | 6.8 | 6.7 |
| RNA-seq (1 × 75 bp) | 92 | 25 | 4.5 | 4.0 |
| Exome-seq (2 × 100 bp) | 43 | 16 | 3.0 | 3.1 |
| WG-seq (2 × 100 bp) | 66 | 19 | 2.8 | 2.6 |
Speedups computed by comparing the time required to perform the full alignment and the combined time of triage classification plus mapping of the selected reads. For RNA-seq samples, target regions consisted of exons and 20 bp flanking sequences, s = 14 and H = 46. For exome and WG samples, which have longer reads, target regions consisted of exons and 85 bp flanking regions, s = 14 and H = 85. (Speedups are approximate with 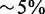 error; results may also vary depending on options used, background load, etc.)
error; results may also vary depending on options used, background load, etc.)
Multiple cores
High-throughput pipelines are sometimes run using multiple cores and sometimes using a single core, the latter option being attractive when treating multiple samples with a series of tools wherein not all are suited for parallel processing. Table 1 shows speedups obtained in both cases. The values for DNA-seq samples are generally lower than for RNA-seq because Bowtie, the aligner used in the former case, makes very efficient use of multiple cores, while Tophat, the program used for mapping spliced reads, spends much time in single-processor mode even when multiple cores are available. The speedups for the small target examples show a marked difference between single- and multi-core settings because the running time of the targeted approach is dominated by the extraction procedure. The speedups for the larger target examples are practically the same for single- and multi-core runs because those running times are dominated by the alignments.
Alternate implementations
Our implementation of targeted extraction is based on a simple index structure and associated algorithm, but it is conceivable to consider alternative approaches. In the Supplementary Material, we explore the following multi-step procedure: create a temporary ‘genome’ with the target sequence and flanking areas, create an index for a traditional aligner using this custom reference, align reads using the custom index and identify relevant reads as those that are mapped successfully. We show that this custom-index approach can be very performant when extracting for large target regions and when processing DNA-based samples. However, our dedicated approach is typically faster on small target regions. Furthermore, it delivers better selection quality in RNA-seq samples as it is capable of properly selecting spliced reads without requiring explicit exon-junction annotations.
CONCLUSION
We presented a method, triage by sequence, designed to extract a subset of reads from raw data from HTS based on sequence similarity with a target region. Its algorithm is closely related to one of the stages performed within a traditional aligner, but we simplified and optimized it for the specific task at hand. We showed that it can handle DNA and RNA sequences with realistic errors, variants and splice patterns. It can achieve very high TPRs, often reaching 99.9%, with FPRs varying with the size of the target region.
Using the triage method, a researcher interested in performing in-depth analysis of a small genomic region can optimize HTS pipelines and achieve significant speedups over the traditional ‘align-then-analyze’ approach. In our practical examples with RNA-seq data, we achieved speedups up to 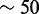 when targeting a single gene and more modest
when targeting a single gene and more modest 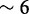 when working with a large collection of genes linked with cancer. We obtained these results running aligners with four processor cores; speedup factors can be greater (we recorded up to
when working with a large collection of genes linked with cancer. We obtained these results running aligners with four processor cores; speedup factors can be greater (we recorded up to 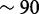 ) for pipelines using fewer cores. In all cases, the targeted selection preserved information relevant for use in variant detection and transcript analysis. These technical improvements should allow researchers to utilize computational resources to efficiently exploit HTS data for their targeted research projects.
) for pipelines using fewer cores. In all cases, the targeted selection preserved information relevant for use in variant detection and transcript analysis. These technical improvements should allow researchers to utilize computational resources to efficiently exploit HTS data for their targeted research projects.
Beside the practical reasoning that motivated this work (‘Introduction’), the targeted extraction procedure has other applications as well. For example, Bhaduri et al. (24) reported an interesting work on detection of viral sequences in human HTS samples. In that work, reads potentially originating from virus species were extracted from a human sample using a multi-stage procedure involving cutting input reads into parts and aligning the pieces onto viral genomes. We note that a similar calculation could be carried out in fewer steps using the sequence-based triage approach.
We also mention that because the quantity of data output by the triage algorithm is much smaller than a full HTS sample, it is conceivable to process this subset using slower but more precise pipelines than those currently in use for HTS data. This should enable researchers interested in small genomic regions to exploit HTS data in ways not currently feasible for genome-wide exploratory studies, even those with access to powerful computer infrastructure. This topic is currently under investigation.
We incorporated the targeted extraction approach into a kit called TriageTools. Apart from the described functionality, the toolkit also provides other means to manipulate, partition and prioritize the analysis of raw HTS data. For example, it includes tools to partition FASTQ data by base quality, or to identify (near) duplicate reads in an alternative way to Picard (12), which can be useful in pipelines acting directly on raw data. Together, these tools should help streamline existing workflows. Details on all the tools, including usage examples, appear in the online software documentation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
TeleVie grant from the Belgian FNRS (to D.F.). Funding for open access charge: IRIBHM.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of IRIBHM for comments and discussions.
REFERENCES
- 1.The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas T, Gilbert J, Meyer F. Metagenomics—a guide from sampling to data analysis. Microb. Inform. Exp. 2012;2:3. doi: 10.1186/2042-5783-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Federhen S, et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2011;39:D38–D51. doi: 10.1093/nar/gkq1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Want J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 8.Zaharia M, Bolosky WJ, Curtis K, Fox A, Patterson D, Shenker S, Stoica I, Karp RM, Sittler T. Faster and more accurate sequence alignment with SNAP. 2011 arXiv: 1111.5572. [Google Scholar]
- 9.Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, Hormozdiari F, Kitzman JO, Baker C, Malig M, Mutlu O, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2009;41:1061–1068. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weese D, Holtgrewe M, Reinert K. RazerS 3: faster, fully sensitive read mapping. Bioinformatics. 2012;28:2592–2599. doi: 10.1093/bioinformatics/bts505. [DOI] [PubMed] [Google Scholar]
- 11.Weese D, Emde AK, Rausch T, Doring A, Reinert K. RazerS-fast read mapping with sensitivity control. Genome Res. 2009;19:1646–1656. doi: 10.1101/gr.088823.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 1000 Genome Project Data Processing Subgroup. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McPherson A, Hormozdiari F, Zayed A, Giuliany R, Ha G, Sun MGF, Griffith M, Moussavi AH, Senz J, Melnyk N, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLOS Comput. Biol. 2011;7:e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kent WJ. BLAT—The BLAST-Like Alignment Tool. Genome Res. 2002;4:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 17. Schroth, G.P. (2011) EMBL-EBI accession no. E-MTAB-513.
- 18.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rossi D, Rasi S, Fabbri G, Spina V, Fangazio M, Forconi F, Marasca R, Laurenti L, Bruscaggin A, Cerri M, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood. 2012;119:521–529. doi: 10.1182/blood-2011-09-379966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mansur MB, Hassan R, Barbosa TC, Splendore A, Jotta PY, Yunes JA, Wiemels JL, Pombo-de-Oliveira MS. Impact of complex NOTCH1 mutations on survival in paediatric T-cell leukemia. BMC Cancer. 2012;12:9. doi: 10.1186/1471-2407-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manske HM, Kwiatkowski DP. SNP-o-matic. Bioinformatics. 2009;25:2434–2435. doi: 10.1093/bioinformatics/btp403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Satya RV, Zavaljevski N, Reifman J. A new strategy to reduce allelic bias in RNA-Seq readmapping. Nucleic Acids Res. 2012;40:e127. doi: 10.1093/nar/gks425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhaduri A, Qu K, Lee CS, Ungewickell A, Khavari PA. Rapid identification of nonhuman sequences in high-throughput sequencing data sets. Bioinformatics. 2012;28:1174–1175. doi: 10.1093/bioinformatics/bts100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.