

Abstract
Parenteral artesunate (ARS) is the drug of choice for the treatment of severe malaria. Pharmacokinetics data on intramuscular ARS are limited with respect to the main treatment group that carries the highest mortality, namely, critically ill children with severe malaria. A population pharmacokinetic study of ARS and dihydroartemisinin (DHA) was conducted from sparse sampling in 70 Tanzanian children of ages 6 months to 11 years. All the children had been admitted with severe falciparum malaria and were treated with intramuscular ARS (2.4 mg/kg at 0, 12, and 24 h). Venous plasma concentration–time profiles were characterized using nonlinear mixed-effects modeling (NONMEM). A one-compartment disposition model accurately described first-dose population pharmacokinetics of ARS and DHA. Body weight significantly affected clearance and apparent volume of distribution (P < 0.001), resulting in lower ARS and DHA exposure levels in smaller children. An adapted dosing regimen including a practical dosing table per weight band is proposed for young children based on the pharmacokinetic model.
Severe falciparum malaria is one of the main causes of death in African children, who account for >85% of the worldwide malaria-related mortality.1 In children, the course of the disease is more rapid than in adults, with most deaths occurring within the first 24 h after admission despite parenteral antimalarial treatment.2,3
Artesunate (ARS), administered parenterally, is currently the drug of choice for the treatment of severe malaria in all age groups and all malaria-endemic settings.4 A large trial (SEAQUAMAT) carried out in Asia in a study population consisting mostly of adult patients (n = 1,461) with severe falciparum malaria showed a 35% reduction in mortality with ARS treatment as compared with quinine.5 More recently, a larger trial (AQUAMAT) performed in 5,425 African children with severe malaria showed a 22.5% lower mortality in children treated with parenteral ARS as compared with quinine.6
ARS has a broader stage-specificity and more potent parasiticidal effect than quinine.7,8 ARS is water-soluble and can be administered either as an i.v. slow bolus or as an i.m. injection. The latter route is more practical in the majority of African hospital and clinic settings with limited facilities. The absorption of i.m. ARS is rapid and reliable, with peak concentrations occurring within 1 h.9,10 After being injected, ARS is rapidly and almost completely converted into its active metabolite, dihydroartemisinin (DHA).11 The elimination of ARS is very rapid, and antimalarial activity is determined mainly by DHA exposure levels. DHA has a terminal elimination half-life of ~45 min10,12,13 and is ~93% plasma protein–bound in patients with falciparum infection.14 The current dosing recommendation for ARS in the treatment of severe malaria is 2.4 mg/kg on admission, followed by the same dose after 12 h and then a daily dose until the patient is able to take oral antimalarial therapy reliably. This recommendation, for the most part, was derived empirically from studies in adults.15,16
In small children with severe disease, the pharmacokinetic properties of antimalarials may differ from those reported in adults.17,18,19 Only limited data are available on the pharmacokinetic properties of parenteral, rectal, and oral ARS in children with moderate to severe malaria.9,13,20,21,22 The primary aim of this study was to characterize the population pharmacokinetic properties of ARS and its active metabolite DHA in the treatment of severe malaria in African children and to determine a practical dosing regimen.
Results
Clinical details
Seventy patients of ages 7 months to 11 years were included, of whom 59 (84%) were <5 years of age. During the study, nine of the children died (case fatality 13%). Of these, one died within 15 min of admission, and two died within the first 24 h after admission. One patient of age 2.5 years had severe neurologic sequelae 28 days after discharge, comprising spastic hemiparesis, blindness, and hearing impairment. At admission this child had a count of 880,205 parasites/µl and plasma Plasmodium falciparum histidine-rich protein-2 concentration of 1,875 ng/ml, both indicating an extremely heavy parasite burden. At the 3-month follow-up, the neurologic sequelae had resolved completely.
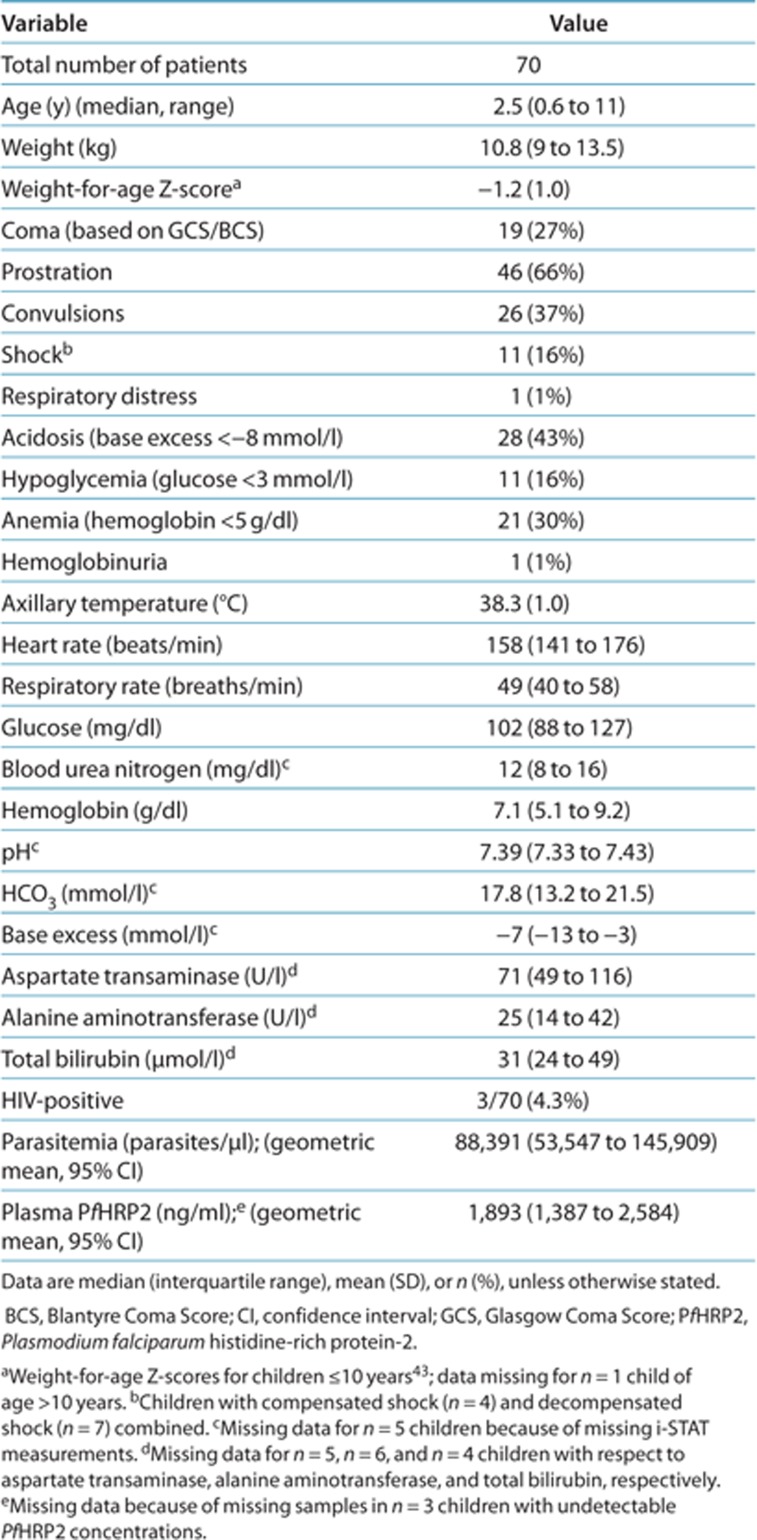
Demographic, clinical, and laboratory characteristics of the study population are described in Table 1. Severe prostration, severe acidosis, convulsions, and severe anemia were the most common severity criteria. Eleven patients (16%) presented with decompensated or compensated shock. In addition, three patients (4.3%) had blood culture–proven septicemia (unspeciated Gram-negative rods, Klebsiella pneumoniae, or Staphylococcus aureus); none of these presented with shock. HIV coinfection was detected in 3/70 (4.3%) of the patients. None of these patients were receiving antiretroviral treatment.
Table 1. Demographic, clinical, and laboratory characteristics of children admitted with severe malaria.
Pretreatment with an oral antimalarial was reported with respect to 31 patients (6 with quinine, 3 with amodiaquine, 13 with sulfadoxine-pyrimethamine, 8 with artemether-lumefantrine, and 1 with amodiaquine followed by artemether-lumefantrine). In addition, 12 patients had received pretreatment with i.m. quinine within the 24 h preceding admission, the median (range) total dose being 16.1 mg/kg (10.1–53.7 mg/kg).
Each of the patients received an i.m. ARS injection of 2.4 mg/kg shortly after admission. Supportive treatments included blood transfusions and fluid resuscitation. Hypoglycemia was corrected with a 10% dextrose bolus at the time of admission and in those who developed hypoglycemia after admission (eight patients). Peripheral blood asexual parasite counts after 24 h were negative in 12/66 (18%) patients (24-h slide was not available for four patients, three of them as a result of death). The geometric mean (95% confidence interval (CI)) parasite count after 24 h was 1,128 (537–2,368) parasites/µl in the rest of the study population (n = 54). The overall geometric mean (95% CI) fractional reduction in parasite counts at 24 h was 96% (94–98%).
Population pharmacokinetic-pharmacodynamic analysis
A total of 274 ARS and DHA postdose samples, randomly distributed over the first 12 h of the study, were included in the model. A one-compartment disposition model for both ARS and DHA was adequate to describe the observed plasma concentration–time data (Table 2). All combinations of two-compartment disposition models displayed significant model misspecification despite significantly lower objective function values. A similar finding has been reported previously for DHA.23 Zero-order distribution/absorption from the injection site(s) to the central compartment provided the best description of the data, but too few samples were collected during the absorption phase and the absorption rate was therefore fixed for an accurate estimation. Interindividual variability could be estimated reliably for ARS and DHA clearance and ARS volume of distribution, showing correlation between ARS clearance and volume.
Table 2. Parameter estimates of the final model describing the population pharmacokinetics of artesunate and dihydroartemisinin in children (n = 70) with severe malaria.
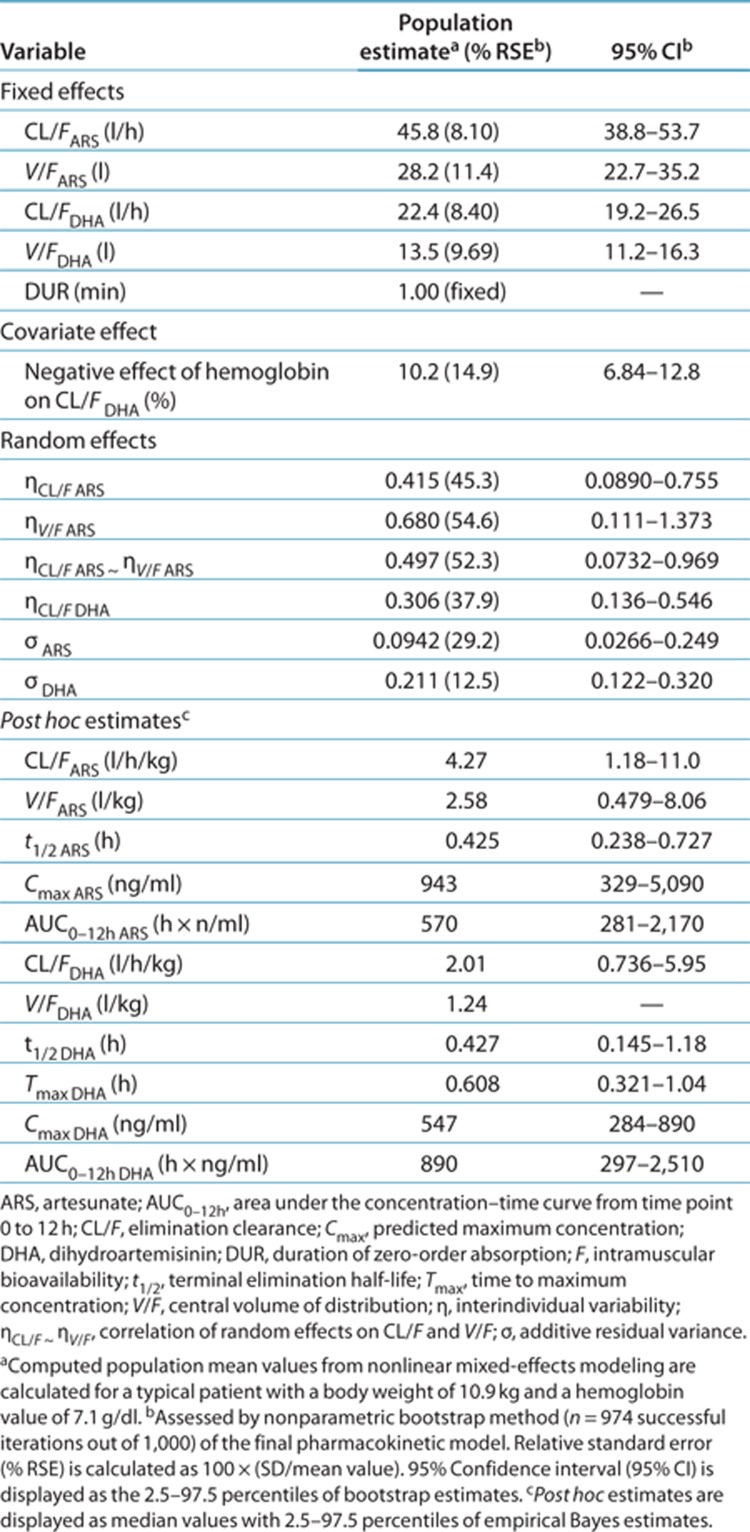
When body weight was used as a fixed allometric function on all elimination clearance (power of 0.75) and apparent volume of distribution (power of 1) parameters, a significantly better model fit was observed (Δobjective function value = −14.6). DHA clearance increased 10.2% per unit (g/dl) of decrease of hemoglobin. Interindividual variability in clearance decreased from 63.6% coefficient of variation to 55.3% coefficient of variation when this covariate was included, suggesting that it accounts for a limited but significant part of the observed variability.
The final model described the observed data well, with adequate goodness-of-fit diagnostics (Figure 1); Eta-shrinkage: elimination clearance for ARS 10.6%, central volume of distribution for ARS 12.2%, elimination clearance for DHA 6.81; Epsilon-shrinkage: 49.4 and 22.5% for ARS and DHA, respectively). A prediction-corrected visual predictive check of the final model resulted in no model misspecification and good simulation properties (Figure 2). The numerical predictive check (n = 2,000) for ARS resulted in 4.35% (95% CI 1.45–9.42) and 5.07% (95% CI 1.45–9.42) of observations above and below the 90% prediction interval, respectively; for DHA, these values were 2.97% (95% CI 1.79–9.52) and 5.95% (95% CI 1.79–8.92), respectively.
Figure 1.

Goodness-of-fit diagnostics of the final population pharmacokinetic model of (a,b,c) artesunate and (d,e,f) dihydroartemisinin in children with severe malaria. The broken line represents a locally weighted least-squares regression; the solid line is the line of identity. The observed concentrations, population predictions, and individual predictions were transformed into their logarithms (base 10).
Figure 2.

Simulation-based diagnostics of the final model describing the population pharmacokinetics of (a,c) artesunate and (b,d) dihydroartemisinin in children with severe malaria. Graphs in a and b display a prediction-corrected visual predictive check with venous plasma concentrations transformed into their logarithms (base 10). Open circles represent observed data points; solid lines represent the 5th, 50th, and 95th percentiles of the observed data; the shaded area represents the 95% confidence interval (CI) of simulated (n = 2,000) 5th, 50th, and 95th percentiles. Graphs in c and d display the observed fraction of data points below the limit of quantification (solid lines) and the 95% CI of the simulated (n = 2,000) fraction below the limit of quantification (shaded area).
ARS and DHA exposures after the standard 2.4 mg/kg dose were simulated at each body weight level (1,000 simulations each at 1-kg intervals from 6 to 25 kg) using a uniform distribution of hemoglobin concentrations within the observed range (2.72–13.6 g/dl) to account for the observed covariate relationship (Figure 3a,b). Children with body weights between 6 and 10 kg showed a mean reduction of 20.4% (P < 0.0001) in DHA exposure as compared with children with body weights between 21 and 25 kg (median (25th to 75th percentile) exposure: 3,380 (2,130–5,470) ng × h/ml in the 6–10 kg patients, 3,780 (2,430–6,060) ng × h/ml in the 11–15 kg patients, 4,100 (2,570–6,590) ng × h/ml in the 16–20 kg patients, and 4,240 (2,700–6,840) ng × h/ml in the 21–25 kg patients).
Figure 3.

Simulated total first-dose exposure levels (AUC0–12h) of (a) ARS and (b) DHA after the standard 2.4 mg/kg dosing in children at different body weights. Simulated total first-dose exposure levels (AUC0–12h) of (c) ARS and (d) DHA after the suggested adjusted dose regimen (Table 3). Open circles represent median values, and bars indicate the 25th to 75th percentiles of simulations (1,000 simulations at each body weight). The broken line represents the median exposure for the largest weight group (i.e., 700 h × ng/ml and 1,230 h × ng/ml for ARS and DHA, respectively). ARS, artesunate; AUC0–12h, area under the concentration–time curve from time point 0 to 12 h; DHA, dihydroartemisinin.
This suggests that smaller children need higher doses of ARS to attain ARS exposures similar to those in children with higher body weights. Using the final model, we evaluated various dosing regimens based on body weight bands. The proposed regimen (Table 3) resulted in similar exposures in all weight bands after the first dose of i.m. ARS (Figure 3c,d). The same simulations were performed assuming a uniform distribution of body weights at different levels of hemoglobin; these resulted in lower DHA exposures with decreasing hemoglobin levels (Figure 4).
Table 3. Proposed body weight–adjusted dosing regimen for i.m. artesunate.
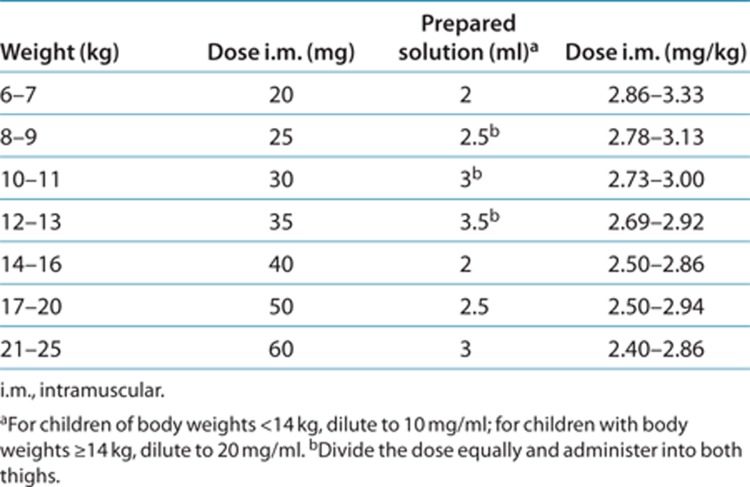
Figure 4.

(a) Simulated total first-dose exposure levels (AUC0–12h) of DHA after the standard 2.4 mg/kg dosing in children at different hemoglobin levels. Open circles represent median values, and bars indicate the 25th to 75th percentiles of simulations (1,000 simulations at each hemoglobin level). (b) Simulated total first-dose exposure (AUC0–12h) of DHA in children at different body weights at very low (open circles: 3 g/dl), low (open squares: 8 g/dl), and normal (open triangles: 13 g/dl) hemoglobin levels. The broken line represents the median exposure for the largest weight group (i.e., 1,230 h × ng/ml). AUC0–12h, area under the concentration–time curve from time point 0 to 12 h; DHA, dihydroartemisinin.
There was no statistical difference between survivors and nonsurvivors with respect to total exposure (ARS P = 0.8060, DHA P = 0.4828) or maximum concentration (ARS P = 0.7655, DHA P = 0.6865) after the first dose. Similarly, an exposure–response relationship could not be established using nonlinear mixed-effects modeling (NONMEM) in a time-to-event approach. However, this might be a consequence of the relatively low number of deaths and the high proportion of pretreatments with different antimalarial drugs, doses, and administration routes.
Discussion
Parenteral ARS is currently the drug of choice for the treatment of severe malaria. Optimal treatment strategies depend on detailed knowledge of the pharmacokinetic properties of drugs in the target population in which the drug is used. Age, disease status, and severity are all factors that may affect drug absorption, distribution, metabolism, and elimination.17 The importance of pharmacokinetics in determining the therapeutic response is illustrated by the study of artemether, the first parenteral artemisinin derivative that was compared with quinine for effectiveness in the treatment of severe malaria in large clinical trials.24,25 In a meta-analysis of randomized trials in severe malaria, artemether significantly reduced mortality in Southeast Asian adults but did not do so in African children.26 Subsequent pharmacokinetic studies showed that the oil-based artemether was released slowly and erratically from the i.m. injection site, and that this likely counterbalanced its pharmacodynamic advantages relative to quinine.10,27 Despite having pharmacodynamic properties similar to those of artemether, ARS is superior to artemether as a treatment because of its more favorable pharmacokinetic properties.28
Dosing regimens for children are often derived from studies in adults, and this practice has led to substantial underdosing of several antimalarials in children. A pharmacokinetic study of sulfadoxine-pyrimethamine in African children with uncomplicated falciparum malaria showed that, with the usual dose of 25/1.25 mg/kg, the area under the concentration–time curve in children 2–5 years of age was half that in adults. This may have caused failure of antimalarial treatment in small children, thereby contributing to the spread of resistance. This information came decades after the introduction of sulfadoxine-pyrimethamine.18 It has also been shown that piperaquine exposure levels are lower in small children if a standard body weight-based dose regimen is followed.19
Only limited data are available on the pharmacokinetics of parenteral, rectal, and oral ARS in children. This is the first population pharmacokinetic study of i.m. ARS in African children with severe malaria. A one-compartment model accurately described the distribution of ARS and DHA. ARS was converted rapidly into DHA, the ARS elimination half-life being ~26 min. This is in agreement with ARS half-life values reported in other pharmacokinetic studies of i.m. ARS9,10,29 but is considerably longer than that associated with the i.v. route.20,30 This is because the elimination rate of ARS is limited by the rate of absorption from the i.m. injection site (i.e., “flip-flop” pharmacokinetics).9 The volume of distribution values, maximum concentration values, and area under the concentration–time curves of ARS and DHA are also comparable with the findings from a previous small and densely sampled pharmacokinetic study of i.m. ARS in children.9 However, the area under the concentration–time curve of DHA in this study was considerably lower than that reported in adult patients and healthy volunteers after i.v. administration of ARS.30,31 Despite the lower predicted maximum concentration value after i.m. injection as compared with i.v. administration, this is still far greater than the in vitro–defined DHA concentration value of 2.28 ng/ml that is required for 99% inhibition (IC99). The excellent bioavailability after i.m. injection (~90%),9,29 fast absorption, and comparable estimates for ARS and DHA exposure support the use of i.m. ARS as a suitable alternative to i.v. ARS.30
Our study had only a relatively small number of patients, thereby limiting its power to detect covariate relationships. A parsimonious approach (P < 0.001 for a covariate to be retained in the model) was applied in order to avoid artifactual covariate relationships. The most significant covariate identified in the present study was body weight. This has also been reported in other population pharmacokinetic studies of oral and rectal ARS in pediatric32,33 and mixed adult–pediatric13 populations. In general, physiological processes do not scale linearly with body weight; consequently children with lower body weights will have higher body weight–normalized elimination clearance values. This has also been reported previously for other antimalarials.18,19 Dosing simulations using an exact dose of 2.4 mg/kg resulted in lower ARS and DHA exposures in small children as compared with those with body weights of 25 kg, suggesting the need for higher dosing in small children. In addition, hemoglobin concentration was also a significant covariate, resulting in lower DHA exposure in more anemic children. Severe anemia is a common presenting feature of severe malaria in young children. In severe malarial anemia there is usually substantial intravascular hemolysis, and the released heme may cause iron-mediated degradation of the artemisinin peroxide bridge. This is also the cause of the degradation of ARS in hemolyzed plasma samples ex vivo.34 Hemoglobin concentration was found to be associated with reduced exposure levels of ARS and DHA, independent of body weight. This supports the idea that young children with severe malaria, who are generally more anemic than older children, should receive adjusted higher doses.35,36
Underdosing in young children with severe malaria may have immediate adverse consequences with respect to outcomes. Parenteral and oral ARS are extremely well tolerated. The only dose-dependent toxicity identified to date is neutropenia. A recent study carried out in Cambodia has shown that oral ARS at a dose of 6 mg/kg/day for 7 days resulted in a reduction in neutrophil counts (to <1.0 × 103/μl) and short-term neutropenia in 19% of the patients.37 Because oral ARS has a bioavailability of ~80%,38,39 this corresponds to a total parenteral dose of 33.6 mg/kg, which would be reached only if parenteral treatment were continued for 14 doses. In the AQUAMAT trial, the median (interquartile range) number of doses of parenteral treatment in surviving children was 3 (2–4) doses.6
ARS is currently the first-choice treatment for severe malaria, and a product produced according to Good Manufacturing Practices, which is WHO prequalified and available. To facilitate implementation of an optimized dosing regimen in the treatment of severe malaria in African children, we defined a simplified weight-band–based dosing regimen based on the current population pharmacokinetics model, taking into account accuracy and practicality issues. We considered 0.5 ml to be the minimum volume of prepared ARS solution that can be measured accurately and administered with commonly available types of syringes. Because the weight bands are smaller for children with body weights <14 kg, we propose an incremental ARS dose increase of 5 instead of 10 mg and a dilution of 10 mg/ml for i.m. administration in this group, similar to the dosage regimen in the current study. In children with body weights of <14 kg, doses should be split and administered in both thighs if injection volumes exceed 2 ml. In children of body weights ≥14 kg, larger injection volumes can be avoided by using a dilution of 20 mg/ml. Binning of weight bands was also chosen in accordance with the currently available vial, which contains a dose of 60 mg, demarcating the upper limits of weight bands at 25 kg.
The dosing recommendations we propose do not extend beyond the weight ranges of the children included in this study. No children with body weight <6.5 kg were included; therefore more studies of ARS population pharmacokinetics are needed to evaluate dosing in these very small children and to support the current dosing recommendations. More extensive sampling in the first 15 min and during the first 12 h after the dose could give more information than the current study provided.
In conclusion, ARS and DHA exposures were lower in small children after i.m. administration of ARS in severe malaria, warranting dose adaptation in this group. Independently, hemolytic anemia may aggravate the lower exposures in young children. We propose a body weight–adjusted and convenient dosing regimen for i.m. ARS in children with body weights between 6 and 25 kg.
Methods
Study design. This pharmacokinetic assessment of ARS was part of the AQUAMAT trial (registration number ISRCTN 50258054), a large multinational trial for which the results have been published elsewhere.6 This substudy was conducted at Teule Hospital in Muheza, Tanzania, from May 2009 to July 2010. Except for the additional blood sampling, the procedures for this substudy were part of the AQUAMAT study protocol.6 Ethical approval was obtained from the Tanzania Medical Research Coordinating Committee and the Oxford Tropical Research Ethics Committee. A total of 18 patients were co-enrolled in the “FEAST” trial evaluating fluid bolus therapy in children with compensated shock.40
Children ≤14 years with a clinical diagnosis of severe malaria confirmed by Plasmodium lactate dehydrogenase (pLDH)-based rapid diagnostic test (OptiMAL, Diamed, Cressier, Switzerland) were recruited, and written informed consent was obtained from the respective parents or caregivers. Severe malaria was defined as the presence of at least one of the following: coma (Glasgow Coma Score ≤10 or Blantyre Coma Score ≤2 in preverbal children), convulsions (duration >30 min or ≥2 episodes in the 24 h preceding admission), respiratory distress (nasal alar flaring, costal indrawing/recession or use of accessory muscles, severe tachypnea) or acidotic breathing (“deep” breathing), shock (capillary refill time ≥3 sec and/or temperature gradient and/or systolic blood pressure <70 mm Hg), severe symptomatic anemia (<5 g/dl with respiratory distress), hypoglycemia (<3 mmol/l), hemoglobinuria, severe jaundice, or, in older children, a convincing history of anuria or oliguria. Patients who had received full treatment with parenteral quinine or a parenteral artemisinin derivative >24 h before admission were excluded.
Physical examination was carried out at admission, and a venous blood sample was taken for peripheral blood parasite count, quantitative assessment of plasma Plasmodium falciparum histidine-rich protein-2 (a measure of total body parasite burden),41 HIV serology (SD Bio-Line HIV 1/2 3.0; Standard Diagnostics, Kyonggi-do, Korea/Determine HIV-1/2, Abbott Laboratories, IL), blood culture, liver function tests (aspartate aminotransferase, alanine transaminase, γ-glutamyltransferase, total bilirubin, creatinine, and urea, by Reflotron, Roche Diagnostics, Basel, Switzerland), hematocrit, biochemistry, and acid–base parameters (EC8+ cartridge for i-STAT handheld blood analyzer, Abbott Laboratories, Abbott Park, IL). Hematocrit was reported from the i-STAT reading or, when not available, measured by HemoCue (HemoCue AB, Ängelholm, Sweden) (n = 5). A neurologic examination was conducted at discharge, and repeated at day 28 in children who had not made a full neurologic recovery at discharge.
Antimalarial treatment. ARS (Guilin Pharmaceutical Factory, Guangxi, China) was administered as an i.m. injection (2.4 mg/kg) shortly after admission, again at 12 h and 24 h, and daily thereafter. The contents of each 60 mg vial of ARS were dissolved in 1 ml 5% sodium bicarbonate (provided with the drug) and further diluted with 5 ml 5% dextrose (final concentration of 10 mg/ml) before administration as a deep i.m. injection into the anterolateral thigh. Dosing was based on the measured body weight of the patient, and injection volumes of >2–3 ml were split and divided into two injections, one in each thigh. When the patient was well enough to take oral medication, but after a minimum of 24 h (two doses of i.m. ARS), a full 3-day course of oral artemether-lumefantrine (Co-artem; Novartis, Basel, Switzerland) was given to complete the treatment.
Patient management. Vital signs and glucose were monitored at least every 6 h and also at any sign of deterioration in clinical condition. A majority of the patients (i.e., other than those who were able to be orally fed) received an infusion with dextrose 5%. Hypoglycemia (defined here as blood glucose <3 mmol/l) was treated with an i.v. bolus of 5 ml/kg of 10% dextrose. Blood transfusion (20 ml/kg) was given to children with hemoglobin concentrations of <5 g/dl. Fluid bolus was given to children with signs of shock.40 All the children were treated empirically with i.v. antibiotics. Convulsions were treated with i.v. diazepam or phenobarbitone if persisting. Peripheral blood smears were repeated after 24 h.
Blood sampling. Blood samples (1.5 ml) were drawn from an indwelling catheter into prechilled fluoride oxalate tubes42 for ARS and DHA quantification before the first dose (at baseline). Four subsequent samples were taken from each patient at preset random time points within the following time windows: 0–1, 1–4, 4–12, and 12–24 h after the first dose. Randomization of sampling times was done by computer-generated randomization (STATA version 12 (Stata, College Station, TX).
Immediately after blood collection, the blood samples for drug measurements were centrifuged at 4 °C at 2,000g for 7 min. Plasma samples (0.5 ml) were stored at −80 °C and shipped on dry ice to the MORU Department of Clinical Pharmacology, Bangkok, Thailand, for drug quantification. ARS drug content and quality were checked in vials taken randomly from the purchase lots (see Supplementary Data online).6
Drug analysis. ARS and DHA plasma concentrations were measured using liquid chromatography–tandem mass-spectrometry.43 Quality control samples at low, middle, and high concentrations were analyzed in triplicate within each analytical batch to ensure accuracy and precision during the analysis. The total coefficients of variation were <8% for all quality control samples. The lower limit of quantification was set at 1.2 ng/ml for ARS and 2.0 ng/ml for DHA.
Pharmacokinetic modeling. Venous plasma concentrations were transformed into molar units and modeled as natural logarithms, using NONMEM v.7 (ICON Development Solutions, Ellicott City, MD). ARS and DHA were modeled simultaneously, using a drug–metabolite model with complete in vivo conversion of ARS into DHA (for details, see Supplementary Data online). The first-order conditional estimation method with interaction was used throughout the modeling. Model selection was based on the objective function values computed by NONMEM, goodness-of-fit graphical analysis, and physiological plausibility. Potential covariates were investigated using a stepwise forward addition and backward elimination approach. A P value of 0.05 was used in the forward step and a P value of 0.001 in the backward step to compensate for the relatively small population studied. Simulation-based diagnostics (visual and numerical predictive checks) and bootstrap diagnostics were used to evaluate the performance of the final model.44
Monte Carlo simulations using the final model with the observed variability were performed for different body weights to obtain representative population estimates of the exposure levels during the first day of dosing (area under the concentration–time curve from time point 0 to 12 h) after prospective dose regimens. No drug exposure target is defined for parenteral ARS; therefore, for the purpose of arriving at a practical parenteral dosing regimen, different body weight “bins” were evaluated to ensure similar target exposures in all weight bands in agreement with the exposure in children of body weight 25 kg. The same simulations were used to evaluate the effect of other significant covariates on drug exposure.
Pharmacodynamics. Peripheral blood smears were taken at admission and after 24 h. Reduction in parasite load over 24 h, survival, and severe neurologic sequelae were evaluated as part of the pharmacodynamic analysis. The effects of ARS and DHA exposures on outcomes were investigated using a time-to-event analysis in NONMEM, with predicted drug concentrations being used to modulate the hazard function in a traditional maximum effect (Emax) relationship. Group comparisons were performed using the nonparametric Mann–Whitney U-test in STATA.
Author Contributions
J.T., I.C.E.H., and A.M.D. wrote the manuscript. J.T., I.C.E.H., N.L., N.P.J.D., L.v.S., N.J.W., and A.M.D. designed research. I.C.E.H., G.M., and A.K. performed research. J.T., I.C.E.H., and N.L. analyzed data. S.G., H.R., and M.M.L. contributed new reagents/analytical tools.
Study Highlights

Acknowledgments
We are grateful to the patients and their caregivers. We thank Ben Amos from Teule Hospital in Muheza for microbiology and laboratory management. Permission to publish this work was given by the director general, National Institute for Medical Research, Tanzania. This work was supported by The Wellcome Trust of Great Britain (grants 076908 and 082541) and was coordinated as part of the Mahidol-Oxford Tropical Medicine Research Programme funded by the Wellcome Trust of Great Britain.
The authors declared no conflict of interest.
Footnotes
SUPPLEMENTARY MATERIAL is linked to the online version of the paper at http://www.nature.com/cpt
Supplementary Material
References
- World Health Organization World Malaria Report 2008. <http://www.who.int/malaria/publications/ atoz/9789241563697/en/index.html> (2008. Accessed 10 October 2011.
- Marsh K., et al. Indicators of life-threatening malaria in African children. N. Engl. J. Med. 1995;332:1399–1404. doi: 10.1056/NEJM199505253322102. [DOI] [PubMed] [Google Scholar]
- Waller D., et al. Clinical features and outcome of severe malaria in Gambian children. Clin. Infect. Dis. 1995;21:577–587. doi: 10.1093/clinids/21.3.577. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Guidelines for the treatment of malaria 2010, second edition-Rev.1. <http://www.who.int/malaria/publications/ atoz/9789241547925/en/index.html> (2011. Accessed 4 May 2011.
- Dondorp A., Nosten F., Stepniewska K., Day N., White N. Artesunate versus quinine for treatment of severe falciparum malaria: a randomised trial. Lancet. 2005;366:717–725. doi: 10.1016/S0140-6736(05)67176-0. [DOI] [PubMed] [Google Scholar]
- Dondorp A.M., et al. Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial. Lancet. 2010;376:1647–1657. doi: 10.1016/S0140-6736(10)61924-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Kuile F., White N.J., Holloway P., Pasvol G., Krishna S. Plasmodium falciparum: in vitro studies of the pharmacodynamic properties of drugs used for the treatment of severe malaria. Exp. Parasitol. 1993;76:85–95. doi: 10.1006/expr.1993.1010. [DOI] [PubMed] [Google Scholar]
- White N.J. Assessment of the pharmacodynamic properties of antimalarial drugs in vivo. Antimicrob. Agents Chemother. 1997;41:1413–1422. doi: 10.1128/aac.41.7.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nealon C., et al. Intramuscular bioavailability and clinical efficacy of artesunate in gabonese children with severe malaria. Antimicrob. Agents Chemother. 2002;46:3933–3939. doi: 10.1128/AAC.46.12.3933-3939.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hien T.T., et al. Comparative pharmacokinetics of intramuscular artesunate and artemether in patients with severe falciparum malaria. Antimicrob. Agents Chemother. 2004;48:4234–4239. doi: 10.1128/AAC.48.11.4234-4239.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navaratnam V., Mansor S.M., Sit N.W., Grace J., Li Q., Olliaro P. Pharmacokinetics of artemisinin-type compounds. Clin. Pharmacokinet. 2000;39:255–270. doi: 10.2165/00003088-200039040-00002. [DOI] [PubMed] [Google Scholar]
- Davis T.M., et al. Pharmacokinetics and pharmacodynamics of intravenous artesunate in severe falciparum malaria. Antimicrob. Agents Chemother. 2001;45:181–186. doi: 10.1128/AAC.45.1.181-186.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J.A., et al. Population pharmacokinetics of artesunate and dihydroartemisinin following intra-rectal dosing of artesunate in malaria patients. PLoS Med. 2006;3:e444. doi: 10.1371/journal.pmed.0030444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batty K.T., Ilett K.F., Davis T.M. Protein binding and alpha : beta anomer ratio of dihydroartemisinin in vivo. Br. J. Clin. Pharmacol. 2004;57:529–533. doi: 10.1046/j.1365-2125.2003.02045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton P.N., et al. The pharmacokinetics of intravenous artesunate in adults with severe falciparum malaria. Eur. J. Clin. Pharmacol. 2006;62:1003–1009. doi: 10.1007/s00228-006-0203-2. [DOI] [PubMed] [Google Scholar]
- Li G.Q., Guo X.B., Jin R., Wang Z.C., Jian H.X., Li Z.Y. Clinical studies on treatment of cerebral malaria with qinghaosu and its derivatives. J. Tradit. Chin. Med. 1982;2:125–130. [PubMed] [Google Scholar]
- Bartelink I.H., Rademaker C.M., Schobben A.F., van den Anker J.N. Guidelines on paediatric dosing on the basis of developmental physiology and pharmacokinetic considerations. Clin. Pharmacokinet. 2006;45:1077–1097. doi: 10.2165/00003088-200645110-00003. [DOI] [PubMed] [Google Scholar]
- Barnes K.I., Little F., Smith P.J., Evans A., Watkins W.M., White N.J. Sulfadoxine-pyrimethamine pharmacokinetics in malaria: pediatric dosing implications. Clin. Pharmacol. Ther. 2006;80:582–596. doi: 10.1016/j.clpt.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Tarning J., et al. Population pharmacokinetics and pharmacodynamics of piperaquine in children with uncomplicated falciparum malaria. Clin. Pharmacol. Ther. 2012;91:497–505. doi: 10.1038/clpt.2011.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremsner P.G., et al. A simplified intravenous artesunate regimen for severe malaria. J. Infect. Dis. 2012;205:312–319. doi: 10.1093/infdis/jir724. [DOI] [PubMed] [Google Scholar]
- Karunajeewa H.A., et al. Artesunate suppositories versus intramuscular artemether for treatment of severe malaria in children in Papua New Guinea. Antimicrob. Agents Chemother. 2006;50:968–974. doi: 10.1128/AAC.50.3.968-974.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethell D.B., et al. Pharmacokinetics of oral artesunate in children with moderately severe Plasmodium falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 1997;91:195–198. doi: 10.1016/s0035-9203(97)90222-4. [DOI] [PubMed] [Google Scholar]
- Tarning J., et al. Population pharmacokinetics of dihydroartemisinin and piperaquine in pregnant and non-pregnant women with uncomplicated malaria. Antimicrob Agents Chemother. 2012;56:1997–2007. doi: 10.1128/AAC.05756-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T.H., et al. A controlled trial of artemether or quinine in Vietnamese adults with severe falciparum malaria. N. Engl. J. Med. 1996;335:76–83. doi: 10.1056/NEJM199607113350202. [DOI] [PubMed] [Google Scholar]
- van Hensbroek M.B., et al. A trial of artemether or quinine in children with cerebral malaria. N. Engl. J. Med. 1996;335:69–75. doi: 10.1056/NEJM199607113350201. [DOI] [PubMed] [Google Scholar]
- Artemether-Quinine Meta-analysis Study Group. A meta-analysis using individual patient data of trials comparing artemether with quinine in the treatment of severe falciparum malaria. Trans R Soc Trop Med Hyg. 2001;95:637–650. doi: 10.1016/s0035-9203(01)90104-x. [DOI] [PubMed] [Google Scholar]
- Murphy S.A., et al. The disposition of intramuscular artemether in children with cerebral malaria; a preliminary study. Trans. R. Soc. Trop. Med. Hyg. 1997;91:331–334. doi: 10.1016/s0035-9203(97)90097-3. [DOI] [PubMed] [Google Scholar]
- Phu N.H., et al. Randomized controlled trial of artesunate or artemether in Vietnamese adults with severe falciparum malaria. Malar. J. 2010;9:97. doi: 10.1186/1475-2875-9-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilett K.F., et al. The pharmacokinetic properties of intramuscular artesunate and rectal dihydroartemisinin in uncomplicated falciparum malaria. Br. J. Clin. Pharmacol. 2002;53:23–30. doi: 10.1046/j.0306-5251.2001.01519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris C.A., Duparc S., Borghini-Fuhrer I., Jung D., Shin C.S., Fleckenstein L. Review of the clinical pharmacokinetics of artesunate and its active metabolite dihydroartemisinin following intravenous, intramuscular, oral or rectal administration. Malar. J. 2011;10:263. doi: 10.1186/1475-2875-10-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byakika-Kibwika P., et al. Pharmacokinetics and pharmacodynamics of intravenous artesunate during severe malaria treatment in Ugandan adults. Malar. J. 2012;11:132. doi: 10.1186/1475-2875-11-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunajeewa H.A., et al. Disposition of artesunate and dihydroartemisinin after administration of artesunate suppositories in children from Papua New Guinea with uncomplicated malaria. Antimicrob. Agents Chemother. 2004;48:2966–2972. doi: 10.1128/AAC.48.8.2966-2972.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepniewska K., et al. Population pharmacokinetics of artesunate and amodiaquine in African children. Malar. J. 2009;8:200. doi: 10.1186/1475-2875-8-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindegardh N., et al. Quantification of dihydroartemisinin, artesunate and artemisinin in human blood: overcoming the technical challenge of protecting the peroxide bridge. Bioanalysis. (2011;3:1613–1624. doi: 10.4155/bio.11.158. [DOI] [PubMed] [Google Scholar]
- Snow R.W., et al. Relation between severe malaria morbidity in children and level of Plasmodium falciparum transmission in Africa. Lancet. 1997;349:1650–1654. doi: 10.1016/S0140-6736(97)02038-2. [DOI] [PubMed] [Google Scholar]
- Billig E.M., O'Meara W.P., Riley E.M., McKenzie F.E. Developmental allometry and paediatric malaria. Malar J. 2012;11:64. doi: 10.1186/1475-2875-11-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethell D., et al. Dose-dependent risk of neutropenia after 7-day courses of artesunate monotherapy in Cambodian patients with acute Plasmodium falciparum malaria. Clin. Infect. Dis. 2010;51:e105–e114. doi: 10.1086/657402. [DOI] [PubMed] [Google Scholar]
- Batty K.T., et al. A pharmacokinetic and pharmacodynamic study of intravenous vs oral artesunate in uncomplicated falciparum malaria. Br. J. Clin. Pharmacol. 1998;45:123–129. doi: 10.1046/j.1365-2125.1998.00655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGready R., et al. Artesunate/dihydroartemisinin pharmacokinetics in acute falciparum malaria in pregnancy: absorption, bioavailability, disposition and disease effects. Br. J. Clin. Pharmacol. 2012;73:467–477. doi: 10.1111/j.1365-2125.2011.04103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitland K., et al. Mortality after fluid bolus in African children with severe infection. N. Engl. J. Med. 2011;364:2483–2495. doi: 10.1056/NEJMoa1101549. [DOI] [PubMed] [Google Scholar]
- Hendriksen I.C., et al. Diagnosing severe falciparum malaria in parasitaemic African children: a prospective evaluation of plasma PfHRP2 measurement. PLoS Med. 2012;9:e1001297. doi: 10.1371/journal.pmed.1001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindegardh N., et al. Major pitfalls in the measurement of artemisinin derivatives in plasma in clinical studies. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008;876:54–60. doi: 10.1016/j.jchromb.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Hanpithakpong W., et al. A liquid chromatographic-tandem mass spectrometric method for determination of artesunate and its metabolite dihydroartemisinin in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008;876:61–68. doi: 10.1016/j.jchromb.2008.10.018. [DOI] [PubMed] [Google Scholar]
- Bergstrand M., Hooker A.C., Wallin J.E., Karlsson M.O. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13:143–151. doi: 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.