

Abstract
Multilocus data from the different genomes are essential to understand the phylogeographic history of species, particularly when a species has the male-biased dispersal pattern. Although Black-throated tits (Aegithalos concinnus) are socially monogamous and cooperatively breeding birds, limited observational data suggested that males may have the ability of long-distance dispersal. We have previously detected three highly supported mitochondrial populations within two subspecies of Black-throated tits (A. c. concinnus and A. c. talifuensis). Here, we used several genetic markers with different inheritance patterns to gain insights about their phylogeographic history. Phylogenetic and individual-based Bayesian analysis showed weak geographical structure amongst nuclear sequences (autosomal and Z-chromosomal loci). Coalescent analysis revealed high levels of gene flow among mitochondrial populations, even between allopatric populations. These results strongly suggested that male-biased gene flow was responsible for the discordant cytonuclear phylogeographic patterns. Consistent with expectation on the genetic consequence of long-distance male dispersal, mantel tests revealed a significant pattern of isolation by distance for mitochondrial sequences, but failed to provide a similar pattern for nuclear genes within a continuous population; female Black-throated tits showed a stronger but not significantly different relationship of isolation by distance than males when using mitochondrial DNA alone. We discussed the contribution of male juveniles with delayed dispersal to the non-significantly different IBD patterns between sexes. Our results using multilocus genetic data revealed aspects of the complex evolutionary history of Black-throated tits and the important role of long-distance male dispersal in the population structuring.
Keywords: Aegithalos concinnus, multilocus, cytonuclear discordance, male-biased gene flow, genetic consequence, male dispersal
Introduction
It is generally accepted that lineages are the fundamental units of species (De Queiroz, 2005, 2007) and the distribution pattern of lineages is the consequence of interactions between intrinsic characteristics of species and extrinsic environmental factors (Dawson, 2012). Phylogeographic studies aim to describe the distribution pattern of lineages and the factors driving that pattern by analyzing molecular marker diversity (Avise, 2000). In the past two decades, most avian phylogeographic studies have been conducted on the polymorphism or variation of mitochondrial DNA (mtDNA). Mitochondrial DNA is regarded as a robust indicator of patterns of population history because of a lower effective population size, putative neutrality, higher mutation rate and clock-like evolutionary history (Zink and Barrowclough, 2008). However, mtDNA is only a portion of a species' genome size and the reappraisal of mtDNA inheritance have questioned its supposed biological and evolutionary properties (for example, neutrality, clock-like) (White et al., 2008; Galtier et al., 2009). For example, it only captures a fraction of phylogeographic history and may not be consistent with the evolutionary history of the whole organism. Nuclear genes are needed in phylogeographic studies (Edwards and Bensch, 2009), despite the fact that these genes have been considered as lagging indicators for phylogeographic patterns (Zink and Barrowclough, 2008). Using multiple markers from the different genomes can give a more complete and convincing picture of the phylogeographic history of species (Edwards and Bensch, 2009). For example, recent research using multilocus data has revealed several speciation events and history owing to the Pleistocene glacial cycles in the White-breasted nuthatch (Sitta carolinensis) (Walstrom et al., 2012). A multigene approach has emphasized the important role of male-mediated gene flow in the Blue tit complex (Blue tit Cyanistes caeruleus, Canary blue tit C. teneriffae and Azure tit C. cyanus) where several highly supported mitochondrial lineages had been identified (Illera et al., 2011).
The Black-throated tits (Aegithalos concinnus) are one of two widespread cooperatively breeding long-tailed tits within the genus Aegithalos in East Asia. This species lives in pine forest and open broadleaf forest at mid-range altitudes (600–3200 m). It has six or more subspecies remarkably different in morphology. In the non-breeding season, Black-throated tits live in medium to large stable flocks of more than 20 birds and the ranges of flocks may overlap. As the breeding season approaches, the flock progressively fragments into a number of smaller flocks and pairing occurs between members of the same flock. They breed independently in socially monogamous pairs in and around their wintering range. Males and females share similar roles in parental care from nest building through incubation to feeding. Small flocks and cooperative helpers are frequently observed during the breeding season; however, helping may vary between populations ( Li, 1981; Zhou et al., 2003; Guo et al., 2006).
Recent molecular studies have discovered that mtDNA sequences in Black-throated tits are significantly differentiated between and within subspecies (Eck and Martens, 2006; Dai et al., 2010; Päckert et al., 2010). Based on molecular and morphological findings, Eck and Martens (2006) even speculated that Black-throated tits may contain several cryptic species. However, we believe that convincing conclusions can only be obtained after a fuller understanding of the information regarding the evolutionary processes that led to these genetic differences. In East Asia, several birds have similar divergent patterns to the Black-throated tits in mtDNA (Zou et al., 2007; Saitoh et al., 2010). For example, Grey-cheeked fulvetta (Alcippe morrisonia) subspecies were diverged to the extent of 5%, and a particularly deep division (9%) separates the geographically central clade from the geographically peripheral clade (Zou et al., 2007). Unfortunately, to date, few studies have investigated species' phylogeographic pattern and evaluated its process using multilocus genetic data. Thus, there is little information regarding the divergence process, for example, isolation or isolation with gene flow, in the phylogeographic structuring of East Asian birds. Using recently developed tools that apply coalesent theory to multilocus sequences (for example, isolation with migration), we expect to discern the processes behind those phylogeographic patterns.
Here, we use both mitochondrial and multinuclear loci to evaluate the phylogeography of two subspecies of Black-throated tits, A. c. concinnus and A. c. talifuensis. Our latest work using mitochondrial sequences has found three monophyletic geographical populations within these subspecies (Dai et al., 2011). The genetic distance is 5% between the two highly divergent mitochondrial lineages. Molecular dating and ecological niche modeling were consistent with the Pleistocene glacial cycles having isolated and shaped the mitochondrial phylogeographic pattern. Ecological niche modeling also indicated that the Black-throated tits may have experienced several population decreases and expansions during lengthy lineage divergence. Therefore, it is interesting to know whether the nuclear genes have similar population division to mtDNA and what occurred during mitochondrial lineages divergence. Furthermore, although Black-throated tits are socially monogamous and cooperatively breeding birds, limited observational data have shown that male Black-throated tits may have the ability of long-distance dispersal (Li, 2010). A recent published paper has highlighted the critical role of male-biased dispersal in the population structure conflict between nuclear and mtDNA (Peters et al., 2012). It will be relevant to discern the genetic consequence caused by the long-distance male dispersal in Black-throated tits. This study represents the first step in our long-term aim to completely understand the mechanisms for high Black-throated tit diversity. Samples were hard to collect as the Black-throated tits distributed in several southeast Asian countries such as India, Burma, Nepal, Thailand and Vietnam.
In summary, we used multilocus genetic data to address the following questions:
What are the evolutionary units defined by the multilocus nuclear data and are the phylogeographic structures of nuclear sequences similar to that observed in mtDNA?
What are the reasons for these geographic patterns?
Are mitochondrial lineages diverged in a complete isolation mode or in an isolation with migration mode as suggested by ecological niche modeling?
Does the observed long-distance male dispersal have effect on the population structuring of Black-throated tits?
Materials and methods
Samples and molecular characterization
We have previously analyzed mtDNA sequences of 203 individuals from 32 localities within the breeding range of two subspecies of Black-throated tits and identified two highly divergent (5%) but partially sympatric monophyletic lineages (A and B–C), one of which contained two shallow (0.8%) monophyletic structured lineages (B and C) with a parapatric distribution (Figure 1). Lineage C covers a large area, so we randomly selected two or three individuals from each sampling location. Individuals from the two other monophyletic lineages were all sequenced. These three mitochondrial monophyletic lineages were treated as populations in our analyses. In total we sequenced five nuclear loci (two autosomal and three Z-linked) from 93 individuals.
Figure 1.

Sampling information for Black-throated tits. Colors represent populations identified by mtDNA (Dai et al., 2011) with population A in blue, green and red indicating population B and C, respectively. Locations with letters indicate the sequenced samples in this study.
Total genomic DNA was extracted from blood or tissue samples using the Qiagen DNeasy Tissue kit (Hilden, Germany) following the manufacturer's instructions. The makers, primers and annealing temperatures for PCR are shown in Table 1. PCR was completed in 25 μl reactions under the following parameters: denaturation at 94 °C for 2 min, followed by 35 cycles of 94 °C for 30 s, annealing temperature for 30 s, and 72 °C for 30 s, and then an extension time of 8 min at 72 °C with a 4 °C soak. Reactions contain 12.5 μl PCR master mix, 9.5 μl ddH2O, 1 μl forward and backward primers, respectively, and 1 μl template DNA. The same primers were used for the sequencing reactions. PCR products were sequenced with the corresponding primers in the forward and reverse directions. Complete sequences were assembled using Seqman II (DNASTAR). Sequences were compared visually to the original chromatograms to avoid reading errors. For those sequences that contained more than one heterozygous site, we resolved haplotypes probabilistically using PHASE (Stephens et al., 2001; Stephens and Donnelly, 2003), with files formatted in SeqPHASE (Flot, 2010). However, only individuals with resulting phase probabilities >0.70 were used in subsequent analyses (Harrigan et al., 2008; Carling and Brumfield, 2009). We also used allele-specific priming (Bottema and Sommer, 1993) to determine the gametic phase of alleles that have gap(s). Sampling sizes, localities and number of phased samples for each gene are listed in Supplementary Table S1. All sequences are accessible at GenBank under the accession Nos. JQ339357—JQ339714. Sequences were aligned using ClustalW (Thompson et al., 1994) as implemented in MEGA 4 (Tamura et al., 2007). Gaps were retained in the data set and handled according to the default settings for each program. For example, gaps were treated as the fifth state in the networks but discarded by the Isolation with Migration (IMa) program.
Table 1. Loci, primers and annealing temperatures for polymerase chain reactions used in this study.
| Locus | Annealing temperature | PCR/sequencing primers | Reference |
|---|---|---|---|
| Autosomal | |||
| Recombination activating protein 1 (RAG 1) | 65 | R50: CTGATCTGGTAACCCCAGTGAAATCC | Irestedt et al., 2001 |
| R51: GACCCTCTTTCTGCTATGAGGGGGC | Irestedt et al., 2001 | ||
| RAG1: CTTCTCACTGACATCTCCCATTCCA | This study | ||
| RAG632F: CAGCCTTGGTTTGTGCCT | This study | ||
| RAG632R: CTCCTCCTCCATGTCTTTTAA | This study | ||
| β-fibrinogen gene, intron 5 (FIB 5) | 55 | Fib5: CGCCATACAGAGTATACTGTGACAT | Fuchs et al., 2004 |
| Fib6: CCATCCAACATCTCAGCATGATGAAA | Fuchs et al., 2004 | ||
| Sex-linked | |||
| Chromo-helicase-DNA-binding protein, intron a (CHD a) | 55 | F2550: GTTACTGATTCGTCTACGAGA | Ellegren and Fridolfsson, 1997 |
| R2718: ATTGAAATGATCCAGTGCTTG | Ellegren and Fridolfsson, 1997 | ||
| CHD1F: TATCGTCAGTTTCCTTTTCAGGT | Lee et al., 2010 | ||
| CHD1R: CCTTTTATTGATCCATCAAGCCT | Lee et al., 2010 | ||
| 65 | R1: CCATCAAGCCTCTACAAAGATT | This study | |
| R2: CCATCAAGCCTCTACAAAGATA | This study | ||
| Brahma protein, intron 15 (BRM 15) | 55 | F: AGCACCTTTGAACAGTGGTT | Borge et al., 2005 |
| R: TACTTTATGGAGACGACGGA | Borge et al., 2005 | ||
| Very low density lipoprotein receptor, intron 9 (VLDLR 9) | 55 | F: AAGTGTGAATGTAGCCGTGG | Borge et al., 2005 |
| R: TCGGTTGGTGAAAATCAGAC | Borge et al., 2005 | ||
Genetic diversity and phylogenetic analyses
DNASP version 5.0 (Librado and Rozas, 2009) was used to estimate the number of segregating sites, haplotype diversity and nucleotide diversity of sequences. The statistical test Tajima's D for assessing the neutral mutation hypothesis was also done in this program. Owing to the observed recombination events for nuclear loci (Table 2), networks were used to identify relationships among haplotypes. Networks were constructed by statistical parsimony (Templeton et al., 1992) in TCS version 1.21 (Clement et al., 2000).
Table 2. Summary of genetic diversity for nuclear loci.
| Locus | N | Length | Nps | s | h | Hap | π | D | Rm |
|---|---|---|---|---|---|---|---|---|---|
| RAG 1 | 83 | 905 | 166 | 36 | 61 | 0.945 | 0.0056 | −0.586 | 8 |
| FIB 5 | 43 | 582 | 86 | 36 | 23 | 0.934 | 0.01050 | −0.536 | 5 |
| CHD a | 66 | 530 | 101 | 21 | 21 | 0.901 | 0.00735 | −0.265 | 1 |
| BRM 15 | 58 | 275 | 86 | 26 | 27 | 0.927 | 0.01452 | −0.786 | 5 |
| VLDLR 9 | 64 | 390 | 89 | 17 | 19 | 0.863 | 0.00436 | −1.418 | 1 |
Abbreviation: D, value of Tajima's test; h, number of haplotypes; Hap, haplotype diversity; N, number of phased samples; Nps, number of sequences after phasing; Rm, estimate of the minimum number of recombination events; S, polymorphic sites; π, nucleotide diversity (%).
To examine whether patterns of nuclear DNA (nuDNA) population structure were similar to the patterns in mtDNA observed previously, we used the individual-based Bayesian clustering method in the program STRUCTURE version 2.3.3 (Pritchard et al., 2000) to estimate the number of populations based on nuDNA genotypes and to assign individuals to those populations. We included individuals represented by two or more loci and tested K=1−3 populations (the number of putatively different genetic clusters), using the admixture model with allele frequencies assumed to be correlated and a fixed λ value. Five runs of each value of K were conducted to examine variation in the probability estimates of K. Results are based on 2 000 000 Markov chain Monte Carlo iterations following a burn-in period of 200 000 iterations. The most optimal K was inferred by the posterior probability (Ln P(D)) of the data.
Based on nuDNA genotypes, we performed the analysis of molecular variance using ARLEQUIN 3.5 (Excoffier and Lischer, 2010) to test the segregation of individuals according to the mitochondrial population structure (Dai et al., 2011). Significance levels were determined by conducting nonparametric procedures 1000 times. We also used ARLEQUIN to calculate ΦST values for the nuDNA sequences among populations. Calculation of ΦST was based on Kimura two-parameter distances among haplotypes. As these analyses only depend on the frequencies of individual polymorphisms, we used the full suite of data including sequences with phase probabilities <70% to perform these analyses again.
Coalescent analysis
The isolation-with-migration model can distinguish gene flow from recent divergence with incomplete lineage sorting by estimating the posterior probability of migration since the time of population splitting. The lowest estimated values of migration suggest that the data are consistent with complete isolation and these discordant patterns are consistent with incomplete lineage sorting. In contrast, if posterior distributions do not show the lowest value of migration, then gene flow is necessary to explain phylogenetic patterns (for example, Hey and Nielsen, 2004; Hey, 2005).
We conducted coalescence analyses using the IM model implemented in IMa (Hey, 2005) to determine the level of gene flow among populations. We included all sequences from the mitochondrial, sex-linked and autosomal markers. The IM model involves several simplifying assumptions such as no recombination within each locus, free recombination among all loci, no population structure within each species or populations, no genetic contribution from unsampled populations or species, and selective neutrality (Nielsen and Wakeley, 2001; Hey and Nielsen, 2004). Recent studies have shown that parameter estimates within the IM model are robust to small to moderate violations of IM model assumptions, and to recombination, as long as loci are divided into non-recombining blocks. However, model selection of mutation rate has significant impact on estimates of almost all parameters (Strasburg and Rieseberg, 2010). To avoid these biases, we tested for intralocus recombination using the four-gamete test in DnaSP version 5.0 (Librado and Rozas, 2009). When recombination was detected, we determined the longest region without observed recombination for each locus using the program IMgc (Woerner et al., 2007). Selective neutrality was tested by the Tajima's D statistic. We applied the infinite sites (IS) model of nucleotide substitsution to nuclear loci, and the Hasegawa, Kishino and Yano (HKY) model to mitochondrial loci as suggested by the author. We used locus-specific inheritance scalars to account for autosomal (nuclear loci=1), Z-chromosomal (sex-linked loci=0.75) and maternal (mitochondrial loci=0.25) inheritance.
Several preliminary runs with large parameter intervals were used to determine the starting values of prior distributions for subsequent runs. The final analysis was run with prior bounds (maximum population size=2, maximum migration=10 or 20, maximum time of population splitting=10) and heating parameters (geometric heating model with 10 chains, first and second chain heating parameters=0.8 and 0.95, respectively) to provide good mixing of Markov chains (that is, no autocorrelation and good coverage of the parameter space).
We ran four independent Markov chain Monte Carlo runs (M-mode) with different random seeds for 1 000 000 burn-in plus 2.5 million sampling generations, sampling every 100th generation. Parameter values were inferred from maximum marginal posterior estimates (Nielsen and Wakeley, 2001). Finally, we compared the model that included all six parameters (Θ1, Θ2, ΘA, t, m1 and m2), to a simpler demographic model that did not allow for post population divergence gene flow (m1=m2=0) by a subsequent L-mode run with identical settings to the M-mode. The significance of the difference between models was evaluated by applying likelihood ratio tests as implemented in the software.
Mantel tests for isolation by distance
We compared genetic and geographical distances to test if genetic differentiation was influenced by geographical distance within populations. Restricted by the number of sampled populations and sizes, we only tested the large area covered population C (18 sampled populations). All individuals were used for mtDNA sequences. However, only phased RAG1, BRM15 and VLDLR9 sequences were used for the mantel tests because two other nuclear loci have small sample sizes. We further analyzed the IBD pattern for mtDNA data using individuals that were sequenced for nuDNA. We also compared the pattern of isolation by distance between sexes based on mitochondrial data. Sexes were identified by molecular primers CHD1F/CHD1R (Table 1). We calculated pair-wise Euclidean distances between sampling sites using NCPA (Templeton, 2008). Mantel tests with 10 000 random permutations to test significance were performed using IBDWS (Jensen et al., 2005) between the matrix of pair-wise genetic differentiation (PhiST, K-2P distance) between populations and the matrix of geographical distance.
Results
Sequences characters
The length of the aligned sequence for the nuclear locus ranged from 275–905 bp. The overall level of sequence variation in the five nuclear loci was high. Each locus included several SNPs and a few positions are tri-allelic SNPs. Our alignment of RAG1 gene (905 bp) from 83 individuals included 60 haplotypes based on 36 polymorphic sites. The alignment of FIB5 sequences (582 bp) from 43 individuals identified 23 haplotypes based on 36 polymorphic sites. Twenty-one haplotypes were identified for the CHD a sequences (530 bp) from 66 individuals based on 21 polymorphic sites and 5 indels whereas 27 haplotypes were identified for alignment sequences of BRM 15 (275 bp) from 56 individuals based on 26 polymorphic sites. Sequences of VLDLR 9 (390 bp) from 64 individuals have 19 haplotypes and 17 polymorphic sites respectively. Indels were found in the complete sequences for all loci except RAG1. Values of Tajima's D were negative but not significant for all nuclear loci, which were consistent with selective neutrality of these loci. Recombination events were detected for all nuclear loci with particularly high frequency for the BRM 15 sequences (Table 2).
Phylogenetic analysis
In contrast to the mtDNA analysis in which geographical patterns were observed in haplotype distribution, variations in nuclear loci did not show significant geographic structure. Haplotype analysis and constructed networks indicated that three populations shared many haplotypes at all nuclear loci (Figure 2). We failed to identify distinct population clusters among the broadly sampled Black-throated tits. Based on nuDNA genotypes from 5 loci, the Bayesian analysis of population structure indicated K=1 and K=2 as the more likely results than K=3. However, weak genetic population structuring was confirmed by the fact that very few closely distributed individuals showed a unique affinity to a specific class (Figure 3).
Figure 2.

Constructed networks for nuclear loci by the TCS. Lineages were identified by mtDNA sequences.
Figure 3.

Population structure analyses of Black-throated tits based on nuDNA genotypes. Each individual is represented by a single line, divided in to K colors, where K is the number of clusters estimated.
Analysis of molecular variance analyses did not detect significant genetic differentiation across the three populations and revealed that most of the genetic variance was found within the sampling populations for all nuclear loci (Table 3). However, several values of pairwise ΦST among populations were significant in five nuclear loci (Table 4). These results were also supported when using all the sequences to analyze (Supplementary Tables S2 and S3).
Table 3. Percent of the total genetic variation partitioned among three populations.
| Locus | Between groups (%) | Among populations within group (%) | Within populations (%) |
|---|---|---|---|
| mtDNA | 93.93 | 0.9 | 5.17 |
| RAG 1 | −1.6 | 2.79 | 98.81 |
| FIB 5 | −1.78 | 3.40 | 98.39 |
| CHD a | 2.25 | −1.52 | 99.27 |
| BRM 15 | 0.58 | 3.34 | 96.08 |
| VLDLR 9 | 1.01 | 1.64 | 97.35 |
Table 4. ΦST values for pairwise comparisons among three populations for five nuclear loci.
| Locus | ΦST | ||
|---|---|---|---|
| |
Population A vs B |
Population A vs C |
Population B vs C |
| RAG 1 | 0.0039 | 0.0153a | 0.0281a |
| FIB 5 | −0.0094 | 0.0478a | 0.0284 |
| CHD a | 0.0168 | 0.0011 | −0.0159 |
| BRM 15 | 0.0327a | 0.0513a | 0.0617a |
| VLDLR 9 | 0.0089 | 0.0032 | 0.0069 |
Significance level <0.05.
Coalescent analysis
We also estimated the levels of gene flow between the two allopatric mitochondrial populations. Estimating levels of gene flow between populations in such a distributional pattern would directly indicate whether male-mediated gene flow exists because of absent signal of mitochondrial gene flow. However, analyzing such a pair seems to violate the assumption underlying the IM method as the existence of a more closely related population to the sampled populations. Nevertheless, studies have suggested that the posterior probability of zero gene flow remains a valid test of isolation versus divergence with gene flow (Hey and Nielsen, 2004; Hey, 2005).
The fact that estimated parameters (for example, Θ1, Θ2) from different comparisons were quite similar, suggested that our results from coalescent analysis were robust (Table 5). Our coalescent analysis of the mitochondrial and nuclear sequences produced all parameters in unimodal posterior distributions and clearly rejected the complete isolation model. The marginal likelihood estimations for the migration rates parameter between populations was non-zero. On the contrary, the posterior probability of zero migration between populations is small except for migration from population B to C (Figure 4). The small sampling sizes of population B may lead to the uncertainty of parameter estimating. The effective number of migrants per generation was all higher than two individuals among populations (Table 5). Nested models and log-likelihood ratio tests indicate that the model with gene flow in both directions was significantly better than all models with restricted gene flow in either or both directions (Table 6).
Table 5. MLEs and 90% HPD intervals of demographic parameters.
| Comparisons | Θ1 | Θ2 | ΘA | m1 | m2 | t | 2N1m1 | 2N2m2 |
|---|---|---|---|---|---|---|---|---|
| Lineage A1 versus BC2 | ||||||||
| MLE | 2.75 | 3.73 | 0.009 | 2.79 | 3.27 | 1.49 | 3.84 | 6.1 |
| HPD90Lo | 1.75 | 2.55 | 0.001 | 0.58 | 1.11 | 0.96 | — | — |
| HPD90Hi | 4.24 | 5.28 | 1.551 | 6.91 | 6.88 | 2.3 | — | — |
| Lineage B1 versus C2 | ||||||||
| MLE | 0.91 | 6.62 | 0.05 | 8.64 | 1.31 | 1.15 | 3.93 | 4.3 |
| HPD90Lo | 0.47 | 4.29 | 0.002 | 3.81 | 0.005 | 0.70 | — | — |
| HPD90Hi | 1.83 | 10.90 | 1.35 | 18.79 | 6.91 | 1.87 | — | — |
| Lineage A1 versus C2 | ||||||||
| MLE | 3.51 | 5.79 | 0.01 | 1.46 | 2.11 | 1.80 | 2.56 | 6.1 |
| HPD90Lo | 2.16 | 3.77 | 0.001 | 0.04 | 0.54 | 1.23 | — | — |
| HPD90Hi | 5.47 | 8.58 | 1.61 | 3.89 | 4.43 | 2.51 | — | — |
Θ1: theta for population 1; Θ2: theta for population 2; ΘA: theta for ancestral population; t: time since ancestral population splitting in mutational units; m1: migration rate per mutation from population 2 to population 1; m2: migration rate per mutation from population 1 to population 2; 2N1m1: the effective number of gene migrants per generation from population 2 to population 1; 2N2m2: the effective number of gene migrants per generation from population 1 to population 2.
Figure 4.
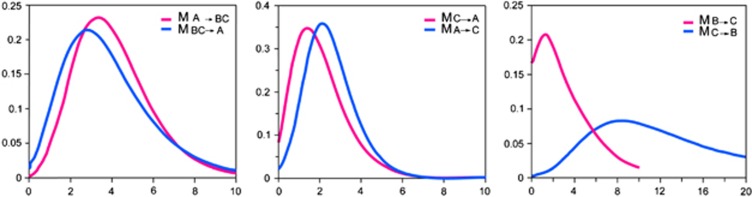
Posterior distributions of scaled migration rate (m1 and m2) estimates for each pairwise comparison among mitochondrial monophyletic populations using the IMa program. Horizontal axes are rates of migration per gene copy per mutation event; vertical axes are marginal posterior probabilities.
Table 6. Summary of likelihood ratio test statistics for the nested models analysis.
| Comparisons | Model | Log(P) | 2LLR | df | P-value |
|---|---|---|---|---|---|
| Lineage A versus BC | ABC0D (m1=0) | −41.8030 | 80.2224 | 1 | <0.001*** |
| ABCD0 (m2=0) | −104.8120 | 206.2405 | 1 | <0.001*** | |
| ABC00 (m1=m2=0) | −460.5170 | 917.6505 | 2 | <0.001*** | |
| ABCDD (m1=m2>0) | −7.1385 | 10.8935 | 1 | <0.001*** | |
| Lineage B versus C | ABC0D (m1=0) | −95.5654 | 186.0926 | 1 | <0.001*** |
| ABCD0 (m2=0) | −112.5921 | 220.1460 | 1 | <0.001*** | |
| ABC00 (m1=m2=0) | −460.5170 | 915.9959 | 2 | <0.001*** | |
| ABCDD (m1=m2>0) | −23.4243 | 41.8103 | 1 | <0.001*** | |
| Lineage A versus C | ABC0D (m1=0) | −102.5104 | 203.9763 | 1 | <0.001*** |
| ABCD0 (m2=0) | −69.5419 | 138.0393 | 1 | <0.001*** | |
| ABC00 (m1=m2=0) | −460.5170 | 919.9896 | 2 | <0.001*** | |
| ABCDD (m1=m2>0) | −17.5071 | 33.9697 | 1 | <0.001*** |
df, degrees of freedom; 2LLR, likelihood ratio statistics. ***P-value, significance level.
Isolation by distance
The molecular sex identification revealed severely male-biased sex ratio in the Black-throated tits. Altogether, we have collected 120 males and 83 females respectively. Within population C, there are 90 males and 65 females respectively. The Mantel test comparing geographical and genetic distances using mtDNA sequences showed significant support for the pattern of isolation by distance (Z=16254.0060, r=0.3556, one-sided P=0.001) (Figure 5a). This significant pattern remained when using only individuals sequenced for nuDNA (Z=12749.6303, r=0.2301, one-sided P=0.012) (Figure 5b). Mitochondrial DNA from males and females both showed significant patterns of isolation by distance, with a stronger but not significant relationship in females (as evidenced by overlapping 95% confidence intervals of slopes between sexes) (females: Z=9321.6943, r=0.4161, one sided P=0.032, slope=0.0004953 (95% CI: 0.0004653–0.0005256); males: Z=7115.4222, r=0.2841, one sided P=0.022, Slope=0.0005371 (95% CI: 0.0005021–0.0005677) (Figures 5c and d). In contrast, tests between geographical and genetic distances using nuDNA sequences failed to yield significant results (RAG1: Z=10649.8380, r=0.0086, one-sided P=0.479; BRM15: Z=26539.2043, r=−0.0493, one-sided P=0.318; VLRLR9: Z=26663.4775, r=−0.0980, one-sided P=0.217) (Figure 6).
Figure 5.

Results of isolation by distance (IBD) analyses based on mtDNA within population C. (a) The pattern for all samples. (b) The pattern for individuals has been sequenced for nuclear data. (c and d) The patterns for females and males respectively.
Figure 6.

Patterns of isolation by distance based on nuclear sequences within population C.
Discussion
Male-mediated gene flow has resulted in cytonuclear discordance
Using mtDNA data to resolve the population genetic structure of Black-throated tits revealed reciprocal monophyletic relationships between regional populations (Dai et al., 2011). Although they had been shown to be narrowly sympatric around Lijiang and Panzhihua (Figure 1), this phylogeographic pattern indicates that regional populations may have diverged to a great extent. Based on the above results for mtDNA, we predict that nuclear sequence loci may exhibit significant differentiation or even reciprocal monophyly, among three populations (for example, three-times rule) (Palumbi et al., 2001). In contrast, the individual-based Bayesian analysis based on nuclear genotypes strongly supported the weak genetic population structuring. Network and analysis of molecular variance analyses also showed that nuclear loci were not significantly differentiated among these mitochondrial monophyletic populations.
Owing to differences in the effective population sizes between mitochondrial and nuclear markers, mtDNA will have a shorter coalescence time than that of nuclear loci (Palumbi et al., 2001). Therefore, the discordant patterns obtained from mitochondrial and nuclear data may have resulted from incomplete lineage sorting at the nuclear loci (Zink and Barrowclough, 2008). Alternatively, ongoing male-biased gene flow also can explain this cytonuclear discrepancy. Several lines of evidence suggest that differences in the rate of lineage sorting between markers cannot fully account for our results.
First, as both mitochondrial and nuclear loci were selective neutral, they should reflect the same history of neutral divergence. If there is no gene flow among populations, the depth of mtDNA divergence between populations should predict similar pattern of differentiation in nuDNA. That is, genetic divergence for nuDNA between populations B and C should be the lowest one as suggested by mtDNA. Quite to the contrary, ΦST values for pairwise comparisons among three populations showed that the lowest divergence was between populations A and B or between populations A and C at five nuclear loci (Supplementary Tables 4 and S2). Combined with the network analysis, this result suggested that ongoing gene flow should occur and result in many shared nuclear polymorphisms among populations. Second, structure result may suggest gene flow among populations. The Bayesian assignment also indicated K=2 as the likely scenario for our nuclear data. When plotting population adherence sorted by Q, the population structure pattern could be interpreted as clinal variation (Supplementary Figure S1). We previously suggested that the two highly divergent populations (between A and B–C) were separated by the Pleistocene climate changes (Dai et al., 2011). This period of allopatry allowed differentiation at nuclear loci but they became admixed when populations came in contact.
Last, and most importantly, our IMa results based on mitochondrial and nuclear data clearly rejected the simple isolation model and strongly supported ongoing male-biased gene flow among populations. The posterior probability of zero migration between two groups was vanishingly small and non-zero gene flow between divergent populations was strongly supported. Furthermore, models incorporating gene flow provided a significantly better fit than nested models with no gene flow. Given detectable gene exchange between populations, these results indicated that the possibility of recent divergence with incomplete lineage sorting at nuclear loci for the discordant phylogeographic pattern is small. Our IMa analysis of two allopatric populations directly indicated the existence of male-biased gene flow in Black-throated tits. Considering the geographical distribution of mitochondrial haplotypes in allopatry, the gene flow detected here cannot be the result of female contribution.
Our coalescent results show that the mitochondrial population structure has diverged with gene flow in Black-throated tits. Gene flow was once perceived as a factor preventing population differentiation and the formation of reproductive isolation (Coyne and Orr, 2004), but a growing numbers of studies have shown that divergence with gene flow is common (Hey, 2005; Nosil, 2008).
Genetic consequence of long-distance male dispersal in Black-throated tits
Li (2010) used the capture-mark-recapture method to access dispersal pattern for Black-throated tits and found that the average dispersal distance of six juvenile females was 500 and 300 m of four juvenile males, whereas a juvenile male ringed two years ago and an adult male dispersed 4800 m away from their natal territory. Although sample sizes were small, he suggested that male Black-throated tits may have the ability of long-distance dispersal. Generally, levels of gene flow prone to decrease with increasing geographic distances in continuous populations (isolation by distance) (Wright, 1943). Therefore, individuals with increasing distances tend to increase in genetic differentiation. Dispersal bias between sexes can lead to the different patterns of isolation by distance for different markers with different inheritance patterns. For example, given that only females contribute to the genetic diversity of maternal markers (mitochondrial) in their offspring but both sexes contribute to biparental markers (nuclear) (Prugnolle and De Meeus, 2002), if female philopatry and male dispersal take place, mitochondrial sequences may demonstrate a stronger pattern of isolation by distance than nuclear loci within a continuous population. Mantel tests showed that mitochondrial sequences have a significant pattern of isolation by distance but no such pattern was found for nuclear sequences. These patterns indicated that mitochondrial gene flow was limited by geographical distance but this was not the case for nuclear gene flow between locations. Besides, we would also expect a weaker isolation by distance relationship for the more dispersing sex (Pearce et al., 2005). If the population structure of Black-throated tits was unaffected by the long-distance male dispersal, we would expect a weaker pattern of isolation by distance for the females. We observed that males demonstrate a weaker but not significant isolation by distance compared with females in mitochondrial data. Therefore, patterns of isolation by distance were consistent with genetic consequence of long-distance male dispersal in the Black-throated tits. We further compared the FST between sexes based on eight sampled populations that have more than 12 individuals in each of population. Females have non-significantly stronger pair-wise FST values than males (data not shown). Sampling collection may be the reason for the non-significant difference in FST and patterns of IBD between sexes. Most of the samples were collected during the late autumn and/or early winter. As the juveniles have delayed dispersal (Li, 1981) and severely male-biased sex ratio in the Black-throated tits, our samples may contain males with delayed dispersal. This hypothesis was supported by the fact that males have many private mtDNA haplotypes when compared with females. When the private male haplotypes were considered as haplotypes of females, IBD patterns between sexes were significantly different (data not shown).
Male-biased dispersal in Black-throated tits?
Two hypotheses could explain observed male-biased gene flow between and among mitochondrial populations: male-biased dispersal and female hybrid sterility or inviability (Haldane, 1922; Illera et al., 2011). Our analyses lack the evidence to reject or support the potential role of female hybrid sterility or inviability in contributing to the discordant genetic population structure. As genetic divergence is 0.8% between two shallow divergent populations, the extent level of divergence to which female hybrid sterility is unlikely occurred; we suggest that the detected gene flow between them was likely due to male-biased dispersal.
Black-throated tits may have a dispersal pattern opposite to the avian norm (Greenwood, 1980; Clarke et al., 1997), whereby males are the dispersing sex and females are more territorial. According to the breeding system hypothesis (Greenwood, 1980), cooperatively breeding birds with biparental care, a socially monogamous and a resource defense mating system should have female-biased dispersal pattern. Several cooperatively breeding birds are yet known to have the male-biased dispersal patterns, such as Brown jays (Cyanocorax morio) (Williams, 2000; Williams and Rabenold, 2005), Seychelles warblers (Acrocephalus sechellensis) (Richardson et al., 2002) and White-throated magpie-jays (Calocitta formosa) (Berg et al., 2009). Interestingly, a close relative to the Black-throated tits, the cooperatively breeding long-tailed tits (Aegithalos caudatus) exhibit female dispersal (Sharp et al., 2011). Although patterns of isolation by distance did not conflict with male-biased dispersal in Black-throated tits, it was unclear that observed dispersing males were under natal dispersal or breeding dispersal. Besides, many studies have showed that even the accidental dispersal event can significantly affect population structure (for example, Alcaide et al., 2009). These findings caution about the case that the observed long-distance male dispersal may just be the accidental dispersal event. The dispersal pattern of Black-throated tits needs to be further evaluated by more effective methods such as identifying fine-scale genetic structure within social groups. As male-biased dispersal is a rare pattern in birds, Black-throated tits may be a good species to evaluate various hypotheses that explain sex-biased dispersal.
Conclusions
This study reveals the complex evolutionary history of Black-throated tits by using multilocus sequence markers with different inheritance patterns (autosomal, Z-chromosomal and mitochondrial markers) and suggests that the Pleistocene climate changes and male-biased gene flow have had important roles in the discordant cytonuclear phylogeographic patterns. The long-distance male dispersal has significantly affected the population structuring whereby patterns of isolation by distance were different between nuclear and mitochondrial markers within a continuous population. Our study adds to the growing evidence that species delimitation (Hebert et al., 2004) and phylogeography based on mtDNA divergence (Zink and Barrowclough, 2008) should be interpreted with caution.
Data archiving
Data have been deposited at GenBank: accession numbers JQ339357–JQ339714.
Acknowledgments
We thank three anonymous reviewers for their constructive suggestions. We also thank Staffan Bensch for the valuable comments on the manuscript and Andrew Bohonak for help using IBDWS. We thank Zuohua Yin, Kaifeng Wang and Kai Chen for the assistance with field sampling and Chunlan Zhang for the statistic help. This work was supported by the National Science Fund for Distinguished Young Scientists (No. 30925008), Major International (Regional) Joint Research Project (No. 31010103901) and NSFC (30870270) to FML.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
Supplementary Material
References
- Alcaide M, Serrano D, Tella JL, Negro JJ. Strong philopatry derived from capture–recapture records does not lead to fine–scale genetic differentiation in lesser kestrels. J Anim Ecol. 2009;78:468–475. doi: 10.1111/j.1365-2656.2008.01493.x. [DOI] [PubMed] [Google Scholar]
- Avise JC. Phylogeography: the History and Formation of Species. Harvard Univ Press: Cambridge, MA; 2000. [Google Scholar]
- Berg EC, Eadie JM, Langen TA, Russell AF. Reverse sex-biased philopatry in a cooperative bird: genetic consequences and a social cause. Mol Ecol. 2009;18:3486–3499. doi: 10.1111/j.1365-294X.2009.04284.x. [DOI] [PubMed] [Google Scholar]
- Borge T, Webster MT, Andersson G, Saetre GP. Contrasting patterns of polymorphism and divergence on the Z chromosome and autosomes in two ficedula flycatcher species. Genetics. 2005;171:1861–1873. doi: 10.1534/genetics.105.045120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottema CDK, Sommer SS. PCR amplification of specific alleles: rapid detection of known mutations and polymorphisms. Mutat Res. 1993;288:93–102. doi: 10.1016/0027-5107(93)90211-w. [DOI] [PubMed] [Google Scholar]
- Carling MD, Brumfield RT. Speciation in Passerina buntings: introgression patterns of sex-linked loci identify a candidate gene region for reproductive isolation. Mol Ecol. 2009;18:834–847. doi: 10.1111/j.1365-294X.2008.04038.x. [DOI] [PubMed] [Google Scholar]
- Clarke AL, Saether B-E, Roskaft E. Sex biases in avian dispersal: a reappraisal. Oikos. 1997;79:429–438. [Google Scholar]
- Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- Coyne JA, Orr HA. Speciation. Sinauer Associates: Sunderland, MA, USA; 2004. [Google Scholar]
- Dai C, Chen K, Zhang R, Yang X, Yin Z, Tian H, et al. Molecular phylogenetic analysis among species of Paridae, Remizidae and Aegithalos based on mtDNA sequences of COI and cyt b. Chinese Birds. 2010;1:112–123. [Google Scholar]
- Dai C, Zhao N, Wang W, Lin C, Gao B, Yang X, et al. Profound climatic effects on two East Asian black-Throated tits (Ave: Aegithalidae), revealed by Ecological Niche Models and phylogeographic analysis. PLoS ONE. 2011;6:e29329. doi: 10.1371/journal.pone.0029329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MN. Parallel phylogeographic structure in ecologically similar sympatric sister taxa. Mol Ecol. 2012;21:987–1004. doi: 10.1111/j.1365-294X.2011.05417.x. [DOI] [PubMed] [Google Scholar]
- De Queiroz K. Ernst Mayr and the modern concept of species. Proc Natl Acad Sci USA. 2005;102:6600–6607. doi: 10.1073/pnas.0502030102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Queiroz K. Species concepts and species delimitation. Syst Biol. 2007;56:879–886. doi: 10.1080/10635150701701083. [DOI] [PubMed] [Google Scholar]
- Eck S, Martens J. Systematic notes on Asian birds. 49. A preliminary review of the Aegithalidae, Remizidae and Paridae. Zool Meded. 2006;80:1–63. [Google Scholar]
- Edwards S, Bensch S. Looking forwards or looking backwards in avian phylogeography? A comment on Zink and Barrowclough 2008. Mol Ecol. 2009;18:2930–2933. doi: 10.1111/j.1365-294X.2009.04270.x. [DOI] [PubMed] [Google Scholar]
- Ellegren H, Fridolfsson AK. Male-driven evolution of DNA sequences in birds. Nat Genet. 1997;17:182–184. doi: 10.1038/ng1097-182. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- Flot J. SeqPHASE: a web tool for interconverting PHASE input/output files and FASTA sequence alignments. Mol Ecol Resour. 2010;10:162–166. doi: 10.1111/j.1755-0998.2009.02732.x. [DOI] [PubMed] [Google Scholar]
- Fuchs J, Bowie RCK, Fjeldså J, Pasquet E. Phylogenetic relationships of the African bush-shrikes and helmet-shrikes (Passeriformes: Malaconotidae) Mol Phylogenet Evol. 2004;33:428–439. doi: 10.1016/j.ympev.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Galtier N, Nabholz B, Glémin S, Hurst GDD. Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Mol Ecol. 2009;18:4541–4550. doi: 10.1111/j.1365-294X.2009.04380.x. [DOI] [PubMed] [Google Scholar]
- Greenwood PJ. Mating systems, philopatry and dispersal in birds and mammals. Anim Behav. 1980;28:1140–1162. [Google Scholar]
- Guo G, Zhou Y, Zhang J, Tang K, Zheng F, Jing X.2006Nest-site selection, breeding ecology and helper behavior of Aegithalos concinnus in northeastern Sichuan, China Chin J Zool 4129–45.(In Chinese). [Google Scholar]
- Haldane JBS. Sex ratio and unisex sterility in hybrid animal. J Genet. 1922;12:101–109. [Google Scholar]
- Harrigan RJ, Mazza ME, Sorenson MD. Computation vs. cloning: evaluation of two methods for haplotype determination. Mol Ecol Resour. 2008;8:1239–1248. doi: 10.1111/j.1755-0998.2008.02241.x. [DOI] [PubMed] [Google Scholar]
- Hebert PDN, Stoeckle MY, Zemlak TS, Francis CM. Identification of birds through DNA barcodes. PLoS Biol. 2004;2:e312. doi: 10.1371/journal.pbio.0020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J. On the number of New World founders: a population genetic portrait of the peopling of the Americas. PLoS Biol. 2005;3:e193. doi: 10.1371/journal.pbio.0030193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004;167:747–760. doi: 10.1534/genetics.103.024182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illera JC, Koivula K, Broggi J, Päckert M, Martens J, Kvist L. A multi-gene approach reveals a complex evolutionary history in the Cyanistes species group. Mol Ecol. 2011;20:4123–4139. doi: 10.1111/j.1365-294X.2011.05259.x. [DOI] [PubMed] [Google Scholar]
- Irestedt M, Johansson US, Parsons TJ, Ericson PGP. Phylogeny of major lineages of suboscines (Passeriformes) analysed by nuclear DNA sequence data. J Avian Biol. 2001;32:15–25. [Google Scholar]
- Jensen J, Bohonak A, Kelley S. Isolation by distance, web service. BMC Genet. 2005;6:13. doi: 10.1186/1471-2156-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JCI, Tsai LC, Hwa PY, Chan CL, Huang A, Chin SC, et al. A novel strategy for avian species and gender identification using the CHD gene. Mol Cell Probes. 2010;24:27–31. doi: 10.1016/j.mcp.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Li G.1981A preliminary study on the breeding ecology of Black-throated tit Aegithalos concinnus Sichuan J Zool 165–70.(In Chinese). [Google Scholar]
- Li JQ. The Breeding Behaviour and Sex-Ratio of the Black-throated Tits (Aegithalos concinnus) and Long-tailed Tits (A. caudatus) Beijing Normal University: Beijing, China; 2010. [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Wakeley J. Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics. 2001;158:885–896. doi: 10.1093/genetics/158.2.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosil P. Speciation with gene flow could be common. Mol Ecol. 2008;17:2103–2106. doi: 10.1111/j.1365-294X.2008.03715.x. [DOI] [PubMed] [Google Scholar]
- Päckert M, Martens J, Sun Y-H. Phylogeny of long-tailed tits and allies inferred from mitochondrial and nuclear markers (Aves: Passeriformes, Aegithalidae) Mol Phylogenet Evol. 2010;55:952–967. doi: 10.1016/j.ympev.2010.01.024. [DOI] [PubMed] [Google Scholar]
- Palumbi SR, Cipriano F, Hare MP. Predicting nuclear gene coalescence from mitochondrial data: the three-times rule. Evolution. 2001;55:859–868. doi: 10.1554/0014-3820(2001)055[0859:pngcfm]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Pearce JM, Talbot SL, Petersen MR, Rearick JR. Limited genetic differentiation among breeding, molting, and wintering groups of the threatened Steller's eider: the role of historic and contemporary factors. Conserv Genet. 2005;6:743–757. [Google Scholar]
- Peters JL, Bolender KA, Pearce JM. Behavioural vs. molecular sources of conflict between nuclear and mitochondrial DNA: the role of male-biased dispersal in a Holarctic sea duck. Mol Ecol. 2012;21:3562–3575. doi: 10.1111/j.1365-294X.2012.05612.x. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prugnolle F, De Meeus T. Inferring sex-biased dispersal from population genetic tools: a review. Heredity. 2002;88:161–165. doi: 10.1038/sj.hdy.6800060. [DOI] [PubMed] [Google Scholar]
- Richardson DS, Burke T, Komdeur J. Direct benefits and the evolution of female-biased cooperative breeding in Seychelles warblers. Evolution. 2002;56:2313–2321. doi: 10.1111/j.0014-3820.2002.tb00154.x. [DOI] [PubMed] [Google Scholar]
- Saitoh T, Alström P, Nishiumi I, Shigeta Y, Williams D, Olsson U, et al. Old divergences in a boreal bird supports long-term survival through the Ice Ages. BMC Evol Biol. 2010;10:35. doi: 10.1186/1471-2148-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp SP, Simeoni M, McGowan A, Nam K-B, Hatchwell BJ. Patterns of recruitment, relatedness and cooperative breeding in two populations of long-tailed tits. Anim Behav. 2011;81:843–849. [Google Scholar]
- Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasburg JL, Rieseberg LH. How robust are ‘Isolation with Migration' analyses to violations of the IM model? A simulation study. Mol Biol Evol. 2010;27:297–310. doi: 10.1093/molbev/msp233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evo. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Templeton AR. Nested clade analysis: an extensively validated method for strong phylogeographic inference. Mol Ecol. 2008;17:1877–1880. doi: 10.1111/j.1365-294X.2008.03731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton AR, Crandall KA, Sing CF. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics. 1992;132:619–633. doi: 10.1093/genetics/132.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J, Higgins D, Gibson T. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walstrom VW, Klicka J, Spellman GM. Speciation in the White-breasted nuthatch (Sitta carolinensis): a multilocus perspective. Mol Ecol. 2012;21:907–920. doi: 10.1111/j.1365-294X.2011.05384.x. [DOI] [PubMed] [Google Scholar]
- White DJ, Wolff JN, Pierson M, Gemmell NJ. Revealing the hidden complexities of mtDNA inheritance. Mol Ecol. 2008;17:4925–4942. doi: 10.1111/j.1365-294X.2008.03982.x. [DOI] [PubMed] [Google Scholar]
- Williams DA. Routes to Fitness in Cooperatively Breeding Brown Jays. Purdue University: West Lafayette, Indiana; 2000. [Google Scholar]
- Williams DA, Rabenold KN. Male-biased dispersal, female philopatry, and routes to fitness in a social corvid. J Anim Ecol. 2005;74:150–159. [Google Scholar]
- Woerner AE, Cox MP, Hammer MF. Recombination-filtered genomic datasets by information maximization. Bioinformatics. 2007;23:1851–1853. doi: 10.1093/bioinformatics/btm253. [DOI] [PubMed] [Google Scholar]
- Wright S. Isolation by distance. Genetics. 1943;28:114–138. doi: 10.1093/genetics/28.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Wang Q, Song Y.2003Breeding ecology of a bird Aegithalos concinnus Chin J Ecol 2224–27.(In Chinese). [Google Scholar]
- Zink R, Barrowclough G. Mitochondrial DNA under siege in avian phylogeography. Mol Ecol. 2008;17:2107–2121. doi: 10.1111/j.1365-294X.2008.03737.x. [DOI] [PubMed] [Google Scholar]
- Zou F, Chuan Lim H, Marks BD, Moyle RG, Sheldon FH. Molecular phylogenetic analysis of the Grey-cheeked Fulvetta (Alcippe morrisonia) of China and Indochina: a case of remarkable genetic divergence in a ‘species'. Mol Phylogenet Evol. 2007;44:165–174. doi: 10.1016/j.ympev.2006.12.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.