

Abstract
Transforming growth factor beta 1 (TGF-β1)-induced epithelial-mesenchymal transition (EMT) in kidney epithelial cells plays a key role in renal tubulointerstitial fibrosis in chronic kidney diseases. As hypoxia-inducible factor (HIF)-1α is found to mediate TGF-β1-induced signaling pathway, we tested the hypothesis that HIF-1α and its upstream regulator prolyl hydroxylase domain-containing proteins (PHDs) are involved in TGF-β1-induced EMT using cultured renal tubular cells. Our results showed that TGF-β1 stimulated EMT in renal tubular cells as indicated by the significant decrease in epithelial marker P-cadherin, and the increase in mesenchymal markers α-smooth muscle actin (α-SMA) and fibroblast-specific protein 1 (FSP-1). Meanwhile, we found that TGF-β1 time-dependently increased HIF-1α and that HIF-1α siRNA significantly inhibited TGF-β1-induced EMT, suggesting that HIF-1α mediated TGF-β1 induced-EMT. Real-time PCR showed that PHD1 and PHD2, rather than PHD3, could be detected, with PHD2 as the predominant form of PHDs (PHD1:PHD2 = 0.21:1.0). Importantly, PHD2 mRNA and protein, but not PHD1, was decreased by TGF-β1. Furthermore, over-expression of PHD2 transgene almost fully prevented TGF-β1-induced HIF-1α accumulation and EMT marker changes, indicating that PHD2 is involved in TGF-β1-induced EMT. Finally, Smad2/3 inhibitor SB431542 prevented TGF-β1-induced PHD2 decrease, suggesting that Smad2/3 may mediate TGF-β1-induced EMT through PHD2/HIF-1α pathway. It is concluded that TGF-β1 decreased PHD2 expression via a Smad-dependent signaling pathway, thereby leading to HIF-1α accumulation and then EMT in renal tubular cells. The present study suggests that PHD2/HIF-1α is a novel signaling pathway mediating the fibrogenic effect of TGF-β1, and may be a new therapeutic target in chronic kidney diseases.
Keywords: Prolyl hydroxylase domain-containing proteins, Smad signaling pathway, renal fibrosis
INTRODUCTION
Renal tubulointerstitial fibrosis, characterized by accumulation of extracellular matrix, is the key underlying pathology in the progression of chronic kidney diseases and is the final common pathway for End-Stage Renal Disease [1–2]. Epithelial-to-mesenchymal transition (EMT) is a process by which epithelial cells lose their epithelial specific markers, undergo cytoskeletal remodeling, and gain a mesenchymal phenotype. More and more studies show that tubular EMT is an important resource of fibrogenic myofibroblasts and plays a central role in tubulointerstitial fibrosis [3–4], such as in diabetes nephropathy [5–6]. Furthermore, there are overwhelming evidence implicating that transforming growth factor-beta 1 (TGF-β1) may act as the key mediator of tubular EMT [2, 7]. For example, TGF-β1 was found to induce EMT in a rat tubular epithelial cell line (NRK-52E), and streptozotocin-induced diabetic rats exhibited increased gene expression of TGF-β1 in the kidneys and enhanced mesenchymal markers α-smooth muscle actin (α-SMA) and collagen with a concomitant decrease in epithelial marker such as E-cadherin [8]. Yeh et al reported that TGFβ1-induced EMT plays a critical role during chronic tubulointerstitial fibrosis [9]. Moreover, transgenic mice with increased expression of TGF-β1 develop renal fibrosis [10].
Hypoxia-inducible factor 1α (HIF-1α) is a transcriptional factor that has been recently associated with the progression of chronic renal injuries [11–13]. Notably, HIF-1α has been shown to play a critical role in EMT. Higgins et al reported that hypoxia induced significant increase in EMT marker fibroblast-specific protein 1 (FSP-1) and cell migration in murine primary tubular epithelial cells. However, this hypoxia-induced EMT was not observed in HIF-1α-deficient cells [13]. In vivo study showed that genetic ablation of epithelial HIF-1α inhibited the development of tubulointerstitial fibrosis and FSP-1-positive cells in unilateral ureteral obstruction kidney [13]. Hypoxia induced significant EMT in hepatocytes from wild type mouse, but failed to induce EMT in hepatocytes isolated from HIF-1α-deficient mice [14]. Moon et al demonstrated that in bile duct ligations-induced liver fibrosis animal model, liver underwent hypoxic, and HIF-1α was activated. After HIF-1α was deleted, bile duct ligations-induced collagen I and α-SMA expression in the liver were significantly decreased [15]. Another study in alveolar epithelial cells showed that hypoxia-induced EMT was significantly attenuated by HIF-1α shRNA [16]. All these data demonstrated an important role of HIF-1α in EMT.
Interestingly, it has been shown that TGF-β1 stimulates HIF-1α accumulation and that HIF-1α functions as a mediator in many TGF-β1-induced actions [17–19]. For example, in colorectal cancer cells, HIF-1α was found to mediate TGF-β1-induced glutathione peroxidase-1 and protects from H2O2-induced cell death in colorectal cancer cells [17]. It was also reported that HIF-1α mediated TGF-β1-induced fibrogenic protein such as collagen and plasminogen activator inhibitor expression in renal epithelial cells and in alveolar macrophages [18–19]. However, the contribution of HIF-1α to TGF-β1-induced EMT in renal epithelial cells has not been evidenced. In addition, the role of HIF prolyl-hydroxylases, the enzymes that promote the degradation of HIF-1α [20–22], in EMT process has not been investigated. A recent study reported that TGF-β1 inhibited PHD2 level via Smad2/3-dependent mechanism in tumor cells [23], indicating a possible role of HIF prolyl-hydroxylases in TGF-β1-induced EMT.
HIF prolyl-hydroxylases hydroxylate HIF-1α at the specific proline site using oxygen as a cofactor and the prolyl-hydroxylated HIF-1α is then recognized and targeted for degradation by the ubiquitin-proteasome pathway [20–21]. Under hypoxia, prolyl hydroxylase activity is inhibited and HIF-1α becomes stabilized to induce the transcription of its target genes. In addition to oxygen homeostasis-related regulation, PHDs also respond to non-hypoxic stimulations including TGF-β1 and regulate HIF-1α via oxygen-independent mechanisms [23–24]. Three isoforms of HIF prolyl hydroxylase, including prolyl hydroxylase domain-containing proteins (PHDs) 1, 2, and 3, have been identified and PHD2 is the primary PHD in the kidneys [25–28]. Given the fact that (1) TGF-β1 induces HIF1-α accumulation, (2) HIF1-α mediates EMT, and (3) HIF prolyl-hydroxylases are present in the kidneys and regulate HIF-1α levels in renal cells [25–28], the present study tested the hypothesis that PHD2/HIF-1α pathway mediates TGF-β1-induced EMT thereby leading to fibrogenesis in renal tubular cells.
MATERIALS AND METHODS
Cell culture
NRK-52E cells, a rat renal tubular cell line, were purchased from ATCC and maintained in Dulbecco's modified Eagle's medium with 4 mmol/L l-glutamine adjusted to contain DMEM/Ham's F12 (DMEM/F12) medium, supplemented with 10% fetal calf serum (FCS), glutamine (2 mM), penicillin (100 IU/ml), and streptomycin (100 μg/ml). Cells were cultured at 37 °C in a humidified atmosphere of 5% CO2 in air [29]. For EMT experiments, cells were treated with 5 ng/ml TGF-β1 for 48 h.
Transfection of HIF-1α siRNA
Transfection of siRNA was performed using the siLentFect lipid reagent (Bio-Rad, Hercules, CA) according to the manufacturer's instructions as we described previously [30]. For a 10 cm dish, 200 pmoles of siRNA was used. After 6 h incubation in transfection reagent, the cells were then switched to normal medium. The sequence of HIF-1α siRNA was: sense, 5'-GGAAAGAGACUCAUAGAAA-3' antisense, 5'-UUUCUAUGACUCUCUUUCC-3' (Sigma-Aldrich, St Louis, MO). A scrambled small RNA (QIAGEN, Valencia, CA), which was confirmed as non-silencing double-stranded RNA, was used as control for siRNA experiments.
Transfection of plasmids expressing rat PHD2 into the cells
Plasmid transfections were performed using lipids (DOTAP/DOPE; Avanti Polar Lipids, Alabaster, AL) according to the manufacturer's instructions as we described previously [30]. In brief, 5 μg of DNA was mixed with lipid solution in a ratio of 1:10 (DNA/lipid, w/w) in serum-free culture medium (5 ml for a 10 cm dish). Cells were incubated with this transfection medium for 6 h and switched to normal medium for another 16 h. The cells were then ready for experiment. In preliminary experiments, almost all cells were positive after transfection with luciferase plasmids when detected by bioluminescent imaging (IVIS200; Caliper Life Sciences, Hopkinton, MA), demonstrating a high transfection efficiency (data not shown). Plasmids encoding full-length rat PHD2 were generous gifts from Dr Frank S. Lee (University of Pennsylvania). The expression and function of rat PHD2 protein by this plasmid has been validated by Dr Lee [31–32] and in our previous study [28, 33–34]. Luciferase plasmids (Promega, Madison, WI) were used as control for PHD2 expression vector transfection experiments.
RNA extraction and quantitative RT-PCR analysis
Total RNA was extracted using TRIzol solution (Life Technologies, Inc. Rockville MD) and then reverse-transcribed (RT) (cDNA Synthesis Kit, Bio-Rad, Hercules, CA). The RT products were amplified using a TaqMan Gene Expression Assays kit (Applied Biosystems). A kit for detecting the levels of 18S ribosomal RNA was used as an endogenous control. The relative gene expressions were calculated in accordance with the ΔΔCt method. Relative mRNA levels were expressed by the values of 2−ΔΔCt.
Western blot analysis
Cytosolic protein and nuclear protein preparation, as well as western blotting, were performed as we described previously [28, 30, 35]. Briefly, after boiling for 5 min at 95°C in a 5× loading buffer, cytosolic protein and nuclear protein were subjected to SDS-PAGE, transferred onto a PVDF membrane and blocked by solution with dry milk respectively. For cytosolic protein, the membrane was probed with primary antibodies of anti-P-cadherin (1: 500, R&D System), anti-α-SMA (1:5000, R&D System), anti-FSP-1 (1:500, Abcam) and anti-PHD2 (1:500, Novus) overnight at 4 °C followed by incubation with horseradish peroxidase-labeled secondary antibody (1:5000); β-actin was detected by using horseradish peroxidase-labeled anti-β-actin antibody (1:5000, Santa Cruz Biotechnology) as a loading control. For nuclear protein, HIF-1α was detected using anti-HIF-1α antibody (1:500, GeneTex) followed by incubation with horseradish peroxidase-labeled secondary antibody (1:3000). Transcription factor II D (TFIID) was detected using anti-TFIID antibody (1:100, Santa Cruz Biotechnology) followed by incubation with horseradish peroxidase-labeled secondary antibody (1:3000) as a loading control for nuclear protein [36]. The immunoreactive bands were detected by chemiluminescence methods and visualized on Kodak Omat X-ray films. The densitometry analyses of the blots were performed using Image J software (free download from National Institutes of Health http://rsbweb.nih.gov/ij/download.html). To calculate the relative values of blot intensities, band intensities in control group were averaged and then all the band intensities were normalized to the mean value of control group. The normalized values in different groups were averaged and expressed as fold change with the mean value of control group as 1.
Immunofluorescent microscopy
Immunofluorescent staining was performed using cultured renal tubular cells on cover slips. After fixation, the cells were incubated with goat anti-FSP-1 (1:50 dilution) (Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA), goat anti-P-cadherin (1:25 dilution), or mouse anti-α-SMA (1:300 dilution) (R&D system, Minneapolis, MN, USA) antibodies, respectively, at 4°C overnight. After washing, the slides were incubated with corresponding Alex-555-labeled secondary antibodies and then mounted and subjected to examinations using a confocal laser scanning microscope (Fluoview FV1000, Olympus, Japan). These experiments were performed to observe the changes of EMT markers in renal tubular cells. Integrated optical intensity (IOD) was calculated by using image-pro plus v6.0 software (Media Cybernetics, Silver Spring, MD). The IOD values in control group were averaged, and all the IOD values normalized to the mean value of the control group. The normalized values in different groups were averaged and expressed as fold change with the mean value of control group as 1.
Statistics
Data are presented as means ± s.e.m. Significant differences between and within multiple groups were examined using ANOVA for repeated measures, followed by Duncan's multiple-range test. Student t test was used to evaluate the significance of differences between two groups of experiments. A value of P < 0.05 was considered statistically significant.
RESULTS
HIF-1α siRNA blocked TGF-β1-induced EMT
We first evaluated whether TGF-β1 would have any effect on HIF-1α protein levels in renal tubular cells. As shown in Figure 1 A&B, TGF-β1 increased HIF-1α protein level when cells were treated for 16 h, and HIF-1α reached the highest level after 24 h and 48 h treatment. Interestingly, epithelial marker P-Cadherin was decreased when cells were treated with TGF-β1 for 16 h, and the protein level reached the lowest level after 24 and 48 h treatment. In contrast, mesenchymal marker α-SMA was increased when cells were treated with TGF-β1 for 16 h, 24 h and 48 h (Figure 1 C&D).
Fig. 1. Effect of TGF-β1 on HIF-1α and EMT marker protein content.
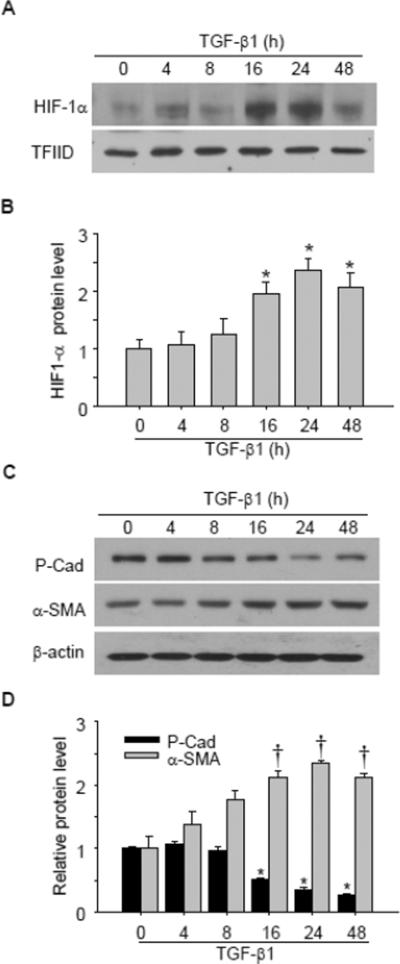
Representative gel documents (A) and summarized data (B) showing the effect of TGF-β1 on HIF-1α protein level. Transcription factor II D (TFIID) was used as a loading control for nuclear protein. Representative gel documents (C) and summarized data (D) showing the effect of TGF-β1 on epithelial marker P-Cadherin and mesenchymal marker α-SMA protein level. All band densities were normalized to the mean value of control group and the normalized values were then calculated and presented in figure. P-cad: P-cadherin. n=4 batches of cells. *P<0.05 vs. other groups in B, or other groups for P-cadherin in D; †P<0.05 vs. other groups for α-SMA in D.
Previous studies showed that HIF-1α was increased in response to hypoxia, and the increased HIF-1α was involved in hypoxia-induced EMT. We then determined whether the increased HIF-1α in response to TGF-β1 stimulation mediated TGF-β1-induced EMT. As shown in Figure 2, TGF-β1 significantly decreased epithelial marker P-cadherin, and increased mesenchymal markers including cytoskeletal protein α-SMA and signal transduction protein FSP-1, indicating that EMT occurred in response to TGF-β1 stimulation. In cells pretreated with HIF-1α siRNA, TGF-β1-induced EMT was significantly inhibited as indicated by the increase in epithelial marker P-cadherin, and decrease in mesenchymal markers α-SMA and FSP-1 compared with TGF-β1-treated group. These results indicate that HIF-1α mediates TGF-β1-induced EMT in renal tubular cells.
Fig. 2. HIF-1α siRNA blocked TGF-β1-induced changes in P-cad, α-SMA and FSP-1.

Representative gel documents (A) and summarized data (B) showing the effect of HIF-1α siRNA on TGF-β1-induced decrease in epithelial marker P-cadherin, and the increase in mesenchymal markers α-SMA and FSP-1. Veh: Vehicle; Scram: Scrambled RNA; P-cad: P-cadherin. The values were normalized to control. n=6 batches of cells, *P<0.05 vs. other groups.
TGF-β1 decreased PHD2 mRNA and protein level
It has been shown that prolyl hydroxylase is the main regulator of HIF-1α in cells. We then determined the expression and abundance of PHD subtypes in renal tubular cells by real-time PCR. Among three different PHDs (PHD1, PHD2, PHD3), only PHD1 and PHD2 were detected. The ΔCt value of PHD1 and PHD2 were 15.6 and 13.4 respectively, and the calculated ratio of PHD1 to PHD2 was 0.21:1.0 (n=6), indicating that PHD2 is the predominant form of PHDs in renal tubular cells.
Next, we evaluated whether TGF-β1 had any effect on mRNA/protein levels of PHD1 and PHD2. As presented in Figure 3, TGF-β1 had no significant effect on PHD1 mRNA (Figure 3 A) and protein level (Figure 3 B&C). In contrast, TGF-β1 dramatically decreased PHD2 mRNA levels with sustained effect from 16 h (Figure 4A). Similarly, TGF-β1 time-dependently decreased PHD2 protein level, which reached its maximum value from 1.0 ± 0.08 to 0.26 ± 0.08 (P < 0.05) at 24 h as shown in Western blot assay (Figure 4 B&C).
Fig. 3. Effect of TGF-β1 on PHD1 mRNA and protein level.
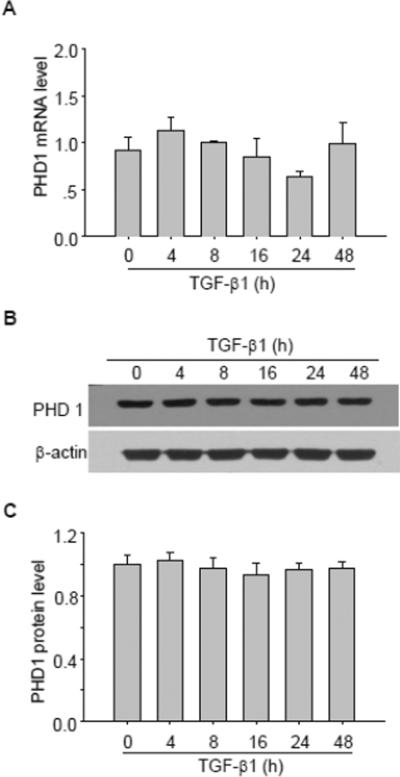
A, Summarized data showing the effect of TGF-β1 on PHD1 mRNA level. Representative gel documents (B) and summarized data (C) showing the effect of TGF-β1 on PHD1 protein level. The values were normalized to control. n=4–6 batches of cells, *P<0.05 vs. other groups.
Fig. 4. TGF-β1 decreased PHD2 mRNA and protein level.

A, Summarized data showing the effect of TGF-β1 on PHD2 mRNA level. Representative gel documents (B) and summarized data (C) showing the effect of TGF-β1 on PHD2 protein level. The values were normalized to control. n=4–5 batches of cells, *P<0.05 vs. other groups.
PHD2 transgene prevented TGF-β1-induced HIF-1α increase and EMT
To determine whether the decreased PHD2 contributes to the HIF-1α increase and consequent EMT after TGF-β1 stimulation, PHD2 overexpression plasmid was transfected into renal tubular cells, and then the effect on TGF-β1-induced changes in PHD2 and HIF-1α, as well as EMT, was evaluated. The gene transfection efficiency was validated by western blot showing that TGF-β1 treatment decreased PHD2 protein level, and this decrease was reversed when cells were transfected with PHD2 overexpression plasmid (Figure 5 A&B). As shown in Figure 5 C&D, PHD2 transgene effectively prevented TGF-β1-induced HIF-1α increase. In Figure 6, TGF-β1 induced significant EMT, as indicted by the decrease in epithelial marker P-cadherin, and the increase in mesenchymal markers α-SMA and FSP-1. When the cells were pretreated with PHD2 overexpression plasmid, TGF-β1-induced EMT was significantly inhibited, as shown by the increase in epithelial marker P-cadherin, and decrease in mesenchymal markers α-SMA and FSP-1 compared with TGF-β1-treated group. These results indicate that PHD2 mediates TGF-β1-induced HIF-1α increase and EMT.
Fig. 5. PHD2 transgene blocked TGF-β1-induced HIF-1α changes.
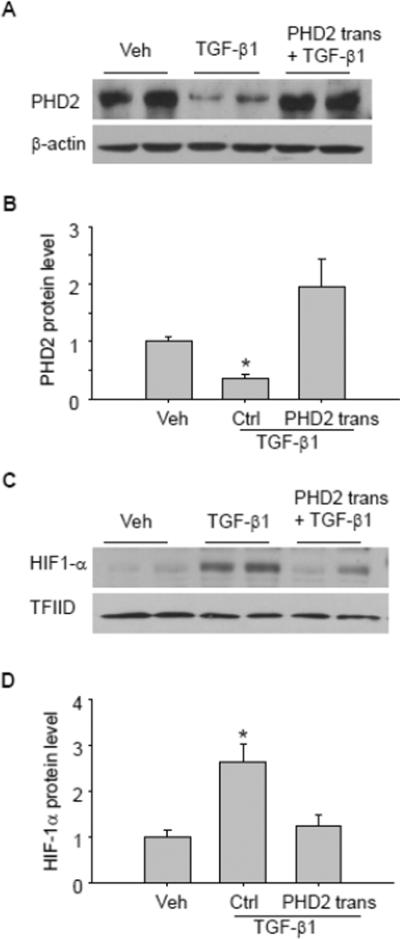
Representative gel documents (A) and summarized data (B) showing the effect of PHD2 transgene on TGF-β1-induced PHD2 decrease. Representative gel documents (C) and summarized data (D) showing the effect of PHD2 transgene on TGF-β1-induced HIF-1α increase. Veh: Vehicle; Ctrl: Control plasmid; trans: transgene. TFIID was used as a loading control for nuclear protein. The values were normalized to control. n=4–6 batches of cells, *P<0.05 vs. other groups.
Fig. 6. PHD2 transgene blocked TGF-β1-induced changes in EMT markers.

Representative gel documents (A) and summarized data (B) showing the effect of PHD2 transgene on TGF-β1-induced decrease in epithelial marker P-cadherin, as well as the increase in mesenchymal markers α-SMA and FSP-1. Veh: Vehicle; Ctrl: Control plasmid; trans: transgene; P-cad: P-cadherin. The values were normalized to control. n=4–6 batches of cells, *P<0.05 vs. other groups.
HIF-1α siRNA and PHD2 transgene prevented TGF-β1-induced EMT as detected by fluorescence microscopy
The involvement of HIF-1α and PHD2 in TGF-β1-induced EMT was further investigated by fluorescence confocal assay. Immunostaining analysis of EMT markers was performed in cells with or without stimulation of TGF-β1. As shown in Figure 7, under basal condition renal tubular cells were enriched with epithelial marker P-cadherin, and it is mainly located on the plasma membrane. When these renal tubular cells were treated with TGF-β1, the expression of P-cadherin was markedly reduced as shown in decreased red fluorescence in TGF-β1-treated cells, and P-cadherin was delocalized from plasma membrane (Figure 7). When cells were pre-treated with HIF-1α siRNA or transfected with PHD2 overexpression plasmid, TGF-β1-induced P-cadherin delocalization were blocked.
Fig. 7. HIF-1α siRNA and PHD2 transgene blocked TGF-β1-induced changes in P-cad, α-SMA and FSP-1 by immunofluorescent microscopy assay.

A. Representative images showing the inhibitory effect of HIF-1α siRNA and PHD2 transgene on TGF-β1-induced decrease in epithelial marker P-cadherin, and increases in mesenchymal markers α-SMA and FSP-1. B. Summarized data show the inhibitory effect of HIF-1α siRNA and PHD2 transgene on integrated optical intensity of α-SMA and FSP-1. Veh: Vehicle; Ctrl: Scrambled RNA as control for HIF-1α siRNA treatment, and control plasmid for PHD2 transgene treatment; trans: transgene; P-cad: P-cadherin. Integrated optical intensity (IOD) was calculated by using image-pro plus v6.0 software. All the IOD values were normalized to the mean value of control group and then the normalized values were calculated and presented. n=5 batches of cells, *P<0.05 vs. other groups.
In contrast, the abundance of two mesenchymal markers, α-SMA and FSP-1 were very low in renal tubular cells under control condition as indicated by the weak fluorescence in confocal images. When these cells were stimulated by TGF-β1, the expression of both α-SMA and FSP-1 was remarkably increased as shown by the increased fluorescence in confocal images. When renal tubular cells were pretreated with HIF-1α siRNA or transfected with PHD2 overexpression plasmid, TGF-β1 failed to increase α-SMA and FSP-1 expression (Figure 7). These results further confirmed that HIF-1α and PHD2 mediate TGF-β1-induced EMT.
HIF-1α siRNA and PHD2 transgene prevented TGF-induced collagen I expression
It has been shown that collagen expression is increased in cells undergoing EMT, which is pathologically related to fibrosis. We then evaluated whether PHD2/HIF-1α mediated TGF-β1-induced collagen expression upon EMT. Figure 8 shows that TGF-β1 induced significant increase in collagen I expression in renal tubular cells. After cells were pretreated with HIF-1α siRNA or PHD2 overexpression plasmid, TGF-β1-induced collagen increase was almost fully inhibited, indicating that HIF-1α/PHD2 mediates TGF-β1-induced collagen expression, and this change may be caused by HIF-1α/PHD2-mediated EMT.
Fig. 8. HIF-1α siRNA and PHD2 transgene blocked TGF-β1-induced collagen I increase.

Representative gel documents (A) and summarized data (B) showing the effect of HIF-1α siRNA and PHD2 transgene on TGF-β1-induced collagen I changes in renal tubular cells. Veh: Vehicle; Ctrl: Scrambled RNA as control for HIF-1α siRNA treatment, and control plasmid for PHD2 transgene treatment; trans: transgene. The values were normalized to control. n=4–6 batches of cells, *P<0.05 vs. other groups.
Smad pathway mediates TGF-β1-induced PHD2 expression
Finally, we investigate whether TGF-β1-induced PHD2 change is mediated by Smads signaling pathway, since previous studies show that TGF-β1 decreased PHD2 level in a Smad-dependent manner. As shown in Figure 9, TGF-β1 induced significant PHD2 decrease in PT cells, this effect was abolished in the presence of Smad2/3 inhibitor SB431542, indicating that Smad signaling pathway was involve in TGF-β1-induced PHD2 decrease.
Fig. 9. Smad inhibition blocked TGF-β1-induced PHD2 decreases.

Representative gel documents (A) and summarized data (B) showing the effect of Smad inhibitor SB431542 on TGF-β1-induced PHD2 decrease. Veh: Vehicle; Ctrl: Control. The values were normalized to control. n=6 batches of cells, *P<0.05 vs. other groups.
DISCUSSION
The present study demonstrated that TGF-β1 decreased PHD2 expression, thereby leading to HIF-1α accumulation. It was found that this PHD2/HIF-1α signaling pathway mediates TGF-β1-induced EMT, since both HIF-1α siRNA and PHD2 overexpression blocked TGF-β1-induced lose of epithelial marker P-cadherin and gain of mesenchymal markers α-SMA and FSP-1. In addition, Smad2/3 inhibitor SB431542 prevented TGF-β1-induced PHD2 decrease. These data support a direct role for PHD2/HIF-1α pathway as a crucial mediator in TGF-β1-induced EMT via a Smad-dependent mechanism in renal tubular cells.
As epithelial cells transdifferentiate into mesenchymal cells during EMT, levels of cytoskeletal proteins (e.g. α-SMA) [37–38], and signal transduction proteins (e.g. FSP-1) are increased [37–38], and the expression of epithelial genes, including P-cadherin is repressed [39–40]. Morphologically, the epithelial marker cadherin protein is delocalized from cell membrane during EMT [41]. Therefore, the changes in α-SMA, FSP-1 and P-cadherin have been widely used as indicators for EMT [40, 42]. In the present study, exposure of proximal tubular cells to TGF-β1 stimulated the expression of α-SMA and FSP-1, and inhibited P-cadherin expression, demonstrating that TGF-β1 stimulated EMT in tubular cells, which is consistent with previous reports [43–44]. It has been shown that TGF-β1 induces HIF-1α accumulation under normoxic conditions in different cells such as HT1080 and vascular smooth muscle cells and that HIF-1α accumulation is involved in TGF-β1-induced various effects, such as fibrosis, apoptosis and tumor angiogenesis [17, 19, 23, 45–46]. In the present study, we demonstrated that TGF-β1 increased HIF-1α protein level in renal tubular cells, and provided novel data by showing that this HIF-1α accumulation mediated EMT in response to a non-hypoxic stimulator TGF-β1 [13–14, 16].
The role of PHDs in EMT was also evaluated in the present study since it represents the most important signaling pathway in regulating HIF-1α. The present study found that PHD2 is the major form of PHDs in renal tubular cells, which is consistent with previous studies showing that PHDs are present in the kidneys, with PHD2 as the predominant isoform of PHDs, although PHD1 and PHD3 can also be detected [26–28, 47–48]. Furthermore, we found that PHD2, but not PHD1, was decreased in response to TGF-β1 stimulation, and that this PHD2 decrease was responsible for the increased HIF-1α accumulation, suggesting divergences in their regulation [23, 49–51]. It has been well documented that PHDs are emerging as an important regulator of HIF-1a in response to both hypoxic and non-hypoxic stimulation, and are involved in many physiological and pathological processes such as collagen expression, cell death, tumor suppression, and blood pressure regulation [17–19, 52]. By using both Western blot and confocal staining, the present study shows novel finding by demonstrating that PHD2, as an upstream regulator of HIF-1α, mediates non-hypoxic agonist TGF-β1-induced EMT in renal tubular cells.
It has been shown that myofibroblasts are activated fibroblasts and the main source for extracellular matrix, thus contributing to kidney fibrosis [53–54]. In particular, it has been demonstrated that the tubular epithelial cells undergoing EMT contribute to one-third of myofibroblast during kidney fibrosis [4, 55]. Consistently, the present study shows that PHD2/HIF-1α mediated TGF-β1-induced collagen I expression, which is even considered as a EMT marker in previous studies [53]. Considering the critical roles of TGF-β1 and EMT in tubulointerstitial fibrosis, the findings in the present study suggest that PHD2/HIF-1α may be an important signaling pathway in mediating this pathological change.
Smad signaling pathway, especially Smad2 and Smad3, plays a central role in TGF-β1-induced effects including EMT in various cell types, such as human bronchial epithelial cells [56–57], murine cloned corneal progenitor cells [58], and panc1 cells [59]. Furthermore, Smad2/3 signaling pathway has been reported to mediate EMT in response to hypoxia in hepatocytes [14]. In animal studies, targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction [60]. It has been suggested that Smad2/3 can regulate the expression of EMT-related genes after interact with other proteins [2, 61]. The present study provided novel mechanisms underlying Smad-mediated EMT by showing that Smad2/3 mediated TGF-β1-induced PHD2 decrease, which then leads to HIF-1α increase and the subsequent EMT.
It should be noted that although EMT has been well acknowledged, there are debates about the involvement of EMT in renal fibrosis in vivo [54, 62–65]. Evidence supporting EMT in vivo includes the loss of epithelial markers, acquisition of mesenchymal markers and collagen synthesis by epithelial cells in diseased kidneys from both human and animal studies, as well as effective strategies to treat experimental fibrosis based on EMT mechanism. However, studies using lineage-tracing techniques to detect tubular epithelial cell-derived fibroblasts show controversial results. An early experiment showed that labeled tubular cells gained EMT markers and migrated into the peritubular interstitium, while several recent similar fate-tracing studies did not detect the EMT markers in labeled tubular cells and failed to find labeled cells in peritubular interstitium. One of the arguments for the conflicting results from these in vivo cell lineage-tracking studies is that the technical differences may account for the disparity. One opinion is that detection of intermediate stages of EMT in injured kidney is straightforward and the current gold standard, and to observe EMT process as well as cell migration in real time in vivo is not feasible with current technology. Nevertheless, our current study revealed a novel mechanism for EMT in renal tubular cells.
In summary, the present study demonstrated that TGF-β1 decreased PHD2 expression via a Smad-dependent mechanism, thereby leading to HIF-1α accumulation, and that this PHD2/HIF-1α signaling pathway mediated TGF-β1-induced EMT in renal tubular cells. This novel signaling pathway in renal tubular cells may contribute to renal tubulointerstitial fibrosis in chronic kidney diseases.
Highlights
-
▶
HIF-1α siRNA blocked TGF-1β-induced EMT in renal proximal tubular cells.
-
▶
The mRNA and protein level of PHD2, an enzyme that promotes the HIF-1α degradation, was decreased by TGF-β1.
-
▶
Over-expression of PHD2 transgene reduced HIF-1α levels and also blocked TGF-β1-induced EMT
-
▶
Smad2/3 inhibitor prevented TGF-β1-induced PHD2 reduction.
-
▶
TGF-β1-induced EMT is mediated by PHD2/HIF-1α through smad2/3 pathway in renal proximal tubular cells
Acknowledgment
This study was supported by grants from the National Institutes of Health (HL89563 and HL106042).
ABBREVIATIONS
- α-SMA
α-smooth muscle actin
- EMT
epithelial-to-mesenchymal transition
- FSP-1
fibroblast-specific protein 1
- HIF-1α
hypoxia-inducible factor 1α
- PHD
prolyl hydroxylase domain
- TFIID
Transcription factor II D
- TGF-β1
transforming growth factor beta 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures None.
REFERENCES
- [1].Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7:684–696. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hills CE, Squires PE. The role of TGF-beta and epithelial-to mesenchymal transition in diabetic nephropathy. Cytokine Growth Factor Rev. 2011;22:131–139. doi: 10.1016/j.cytogfr.2011.06.002. [DOI] [PubMed] [Google Scholar]
- [3].Hills CE, Squires PE. TGF-beta1-induced epithelial-to-mesenchymal transition and therapeutic intervention in diabetic nephropathy. Am J Nephrol. 2010;31:68–74. doi: 10.1159/000256659. [DOI] [PubMed] [Google Scholar]
- [4].Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kanwar YS, Wada J, Sun L, Xie P, Wallner EI, Chen S, Chugh S, Danesh FR. Diabetic nephropathy: mechanisms of renal disease progression. Exp Biol Med (Maywood) 2008;233:4–11. doi: 10.3181/0705-MR-134. [DOI] [PubMed] [Google Scholar]
- [6].Sharma K, Ziyadeh FN. Hyperglycemia and diabetic kidney disease. The case for transforming growth factor-beta as a key mediator. Diabetes. 1995;44:1139–1146. doi: 10.2337/diab.44.10.1139. [DOI] [PubMed] [Google Scholar]
- [7].Carew RM, Wang B, Kantharidis P. The role of EMT in renal fibrosis. Cell Tissue Res. 2012;347:103–116. doi: 10.1007/s00441-011-1227-1. [DOI] [PubMed] [Google Scholar]
- [8].Burns WC, Twigg SM, Forbes JM, Pete J, Tikellis C, Thallas-Bonke V, Thomas MC, Cooper ME, Kantharidis P. Connective tissue growth factor plays an important role in advanced glycation end product-induced tubular epithelial-to-mesenchymal transition: implications for diabetic renal disease. J Am Soc Nephrol. 2006;17:2484–2494. doi: 10.1681/ASN.2006050525. [DOI] [PubMed] [Google Scholar]
- [9].Yeh YC, Wei WC, Wang YK, Lin SC, Sung JM, Tang MJ. Transforming growth factor-{beta}1 induces Smad3-dependent {beta}1 integrin gene expression in epithelial-to-mesenchymal transition during chronic tubulointerstitial fibrosis. Am J Pathol. 2010;177:1743–1754. doi: 10.2353/ajpath.2010.091183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schnaper HW, Hayashida T, Hubchak SC, Poncelet AC. TGF-beta signal transduction and mesangial cell fibrogenesis. Am J Physiol Renal Physiol. 2003;284:F243–252. doi: 10.1152/ajprenal.00300.2002. [DOI] [PubMed] [Google Scholar]
- [11].Nangaku M, Fujita T. Activation of the renin-angiotensin system and chronic hypoxia of the kidney. Hypertens Res. 2008;31:175–184. doi: 10.1291/hypres.31.175. [DOI] [PubMed] [Google Scholar]
- [12].Nangaku M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J Am Soc Nephrol. 2006;17:17–25. doi: 10.1681/ASN.2005070757. [DOI] [PubMed] [Google Scholar]
- [13].Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Copple BL. Hypoxia stimulates hepatocyte epithelial to mesenchymal transition by hypoxia-inducible factor and transforming growth factor-beta-dependent mechanisms. Liver Int. 2010;30:669–682. doi: 10.1111/j.1478-3231.2010.02205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Moon JO, Welch TP, Gonzalez FJ, Copple BL. Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G582–592. doi: 10.1152/ajpgi.90368.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhou G, Dada LA, Wu M, Kelly A, Trejo H, Zhou Q, Varga J, Sznajder JI. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1120–1130. doi: 10.1152/ajplung.00007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Huang Y, Fang W, Wang Y, Yang W, Xiong B. Transforming growth factor-beta1 induces glutathione peroxidase-1 and protects from H2O2-induced cell death in colon cancer cells via the Smad2/ERK1/2/HIF-1alpha pathway. Int J Mol Med. 2012;29:906–912. doi: 10.3892/ijmm.2012.901. [DOI] [PubMed] [Google Scholar]
- [18].Basu RK, Hubchak S, Hayashida T, Runyan CE, Schumacker PT, Schnaper HW. Interdependence of HIF-1alpha and TGF-beta/Smad3 signaling in normoxic and hypoxic renal epithelial cell collagen expression. Am J Physiol Renal Physiol. 2011;300:F898–905. doi: 10.1152/ajprenal.00335.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ueno M, Maeno T, Nomura M, Aoyagi-Ikeda K, Matsui H, Hara K, Tanaka T, Iso T, Suga T, Kurabayashi M. Hypoxia-inducible factor-1alpha mediates TGF-beta-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L740–752. doi: 10.1152/ajplung.00146.2010. [DOI] [PubMed] [Google Scholar]
- [20].Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- [21].Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- [22].Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- [23].McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem. 2006;281:24171–24181. doi: 10.1074/jbc.M604507200. [DOI] [PubMed] [Google Scholar]
- [24].Tug S, Delos Reyes B, Fandrey J, Berchner-Pfannschmidt U. Non-hypoxic activation of the negative regulatory feedback loop of prolyl-hydroxylase oxygen sensors. Biochem Biophys Res Commun. 2009;384:519–523. doi: 10.1016/j.bbrc.2009.05.016. [DOI] [PubMed] [Google Scholar]
- [25].Takeda K, Cowan A, Fong G-H. Essential Role for Prolyl Hydroxylase Domain Protein 2 in Oxygen Homeostasis of the Adult Vascular System. Circulation. 2007;116:774–781. doi: 10.1161/CIRCULATIONAHA.107.701516. [DOI] [PubMed] [Google Scholar]
- [26].Rosenberger C, Rosen S, Shina A, Frei U, Eckardt KU, Flippin LA, Arend M, Klaus SJ, Heyman SN. Activation of hypoxia-inducible factors ameliorates hypoxic distal tubular injury in the isolated perfused rat kidney. Nephrol Dial Transplant. 2008;23:3472–3478. doi: 10.1093/ndt/gfn276. [DOI] [PubMed] [Google Scholar]
- [27].Takeda K, Aguila HL, Parikh NS, Li X, Lamothe K, Duan LJ, Takeda H, Lee FS, Fong GH. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood. 2008;111:3229–3235. doi: 10.1182/blood-2007-09-114561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li N, Yi F, Sundy CM, Chen L, Hilliker ML, Donley DK, Muldoon DB, Li PL. Expression and actions of HIF prolyl-4-hydroxylase in the rat kidneys. Am J Physiol Renal Physiol. 2007;292:F207–216. doi: 10.1152/ajprenal.00457.2005. [DOI] [PubMed] [Google Scholar]
- [29].Hills CE, Al-Rasheed N, Al-Rasheed N, Willars GB, Brunskill NJ. C-peptide reverses TGF-beta1-induced changes in renal proximal tubular cells: implications for treatment of diabetic nephropathy. Am J Physiol Renal Physiol. 2009;296:F614–621. doi: 10.1152/ajprenal.90500.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang Z, Tang L, Zhu Q, Yi F, Zhang F, Li PL, Li N. Hypoxia-inducible factor-1alpha contributes to the profibrotic action of angiotensin II in renal medullary interstitial cells. Kidney Int. 2011;79:300–310. doi: 10.1038/ki.2010.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Huang J, Zhao Q, Mooney SM, Lee FS. Sequence determinants in hypoxia-inducible factor-1alpha for hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J Biol Chem. 2002;277:39792–39800. doi: 10.1074/jbc.M206955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Percy MJ, Zhao Q, Flores A, Harrison C, Lappin TR, Maxwell PH, McMullin MF, Lee FS. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci U S A. 2006;103:654–659. doi: 10.1073/pnas.0508423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhu Q, Liu M, Han W, Li PL, Wang Z, Li N. Overexpression of HIF Prolyl-Hydoxylase-2 transgene in the renal medulla induced a salt-sensitive hypertension. J Cell Mol Med. 2012 doi: 10.1111/j.1582-4934.2012.01590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhu Q, Wang Z, Xia M, Li PL, Van Tassell BW, Abbate A, Dhaduk R, Li N. Silencing of hypoxia-inducible factor-1alpha gene attenuated angiotensin II-induced renal injury in Sprague-Dawley rats. Hypertension. 2011;58:657–664. doi: 10.1161/HYPERTENSIONAHA.111.177626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang Z, Zhu Q, Xia M, Li PL, Hinton SJ, Li N. Hypoxia-inducible factor prolyl-hydroxylase 2 senses high-salt intake to increase hypoxia inducible factor 1alpha levels in the renal medulla. Hypertension. 2010;55:1129–1136. doi: 10.1161/HYPERTENSIONAHA.109.145896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tacchini L, Gammella E, De Ponti C, Recalcati S, Cairo G. Role of HIF-1 and NF-kappaB transcription factors in the modulation of transferrin receptor by inflammatory and anti-inflammatory signals. J Biol Chem. 2008;283:20674–20686. doi: 10.1074/jbc.M800365200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- [38].Masszi A, Di Ciano C, Sirokmany G, Arthur WT, Rotstein OD, Wang J, McCulloch CA, Rosivall L, Mucsi I, Kapus A. Central role for Rho in TGF-beta1-induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. Am J Physiol Renal Physiol. 2003;284:F911–924. doi: 10.1152/ajprenal.00183.2002. [DOI] [PubMed] [Google Scholar]
- [39].Robert G, Gaggioli C, Bailet O, Chavey C, Abbe P, Aberdam E, Sabatie E, Cano A, Garcia de Herreros A, Ballotti R, Tartare-Deckert S. SPARC represses E-cadherin and induces mesenchymal transition during melanoma development. Cancer Res. 2006;66:7516–7523. doi: 10.1158/0008-5472.CAN-05-3189. [DOI] [PubMed] [Google Scholar]
- [40].Li CX, Xia M, Han WQ, Li XX, Zhang C, Boini KM, Liu XC, Li PL. Reversal by growth hormone of homocysteine-induced epithelial-to-mesenchymal transition through membrane raft-redox signaling in podocytes. Cell Physiol Biochem. 2011;27:691–702. doi: 10.1159/000330078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3' kinase/AKT pathways. Oncogene. 2005;24:7443–7454. doi: 10.1038/sj.onc.1209091. [DOI] [PubMed] [Google Scholar]
- [42].Tamiya S, Liu L, Kaplan HJ. Epithelial-mesenchymal transition and proliferation of retinal pigment epithelial cells initiated upon loss of cell-cell contact. Invest Ophthalmol Vis Sci. 2010;51:2755–2763. doi: 10.1167/iovs.09-4725. [DOI] [PubMed] [Google Scholar]
- [43].Zhou Q, Zeng R, Xu C, Liu L, Chen L, Kou P, Pei G, Bai S, Zhang Y, Li C, Rong S, Han M, Xu G. Erbin inhibits TGF-beta1-induced EMT in renal tubular epithelial cells through an ERK-dependent pathway. J Mol Med (Berl) 2012;90:563–574. doi: 10.1007/s00109-011-0833-4. [DOI] [PubMed] [Google Scholar]
- [44].Kopp JB. TGF-beta signaling and the renal tubular epithelial cell: too much, too little, and just right. J Am Soc Nephrol. 2010;21:1241–1243. doi: 10.1681/ASN.2010060676. [DOI] [PubMed] [Google Scholar]
- [45].Shih SC, Claffey KP. Role of AP-1 and HIF-1 transcription factors in TGF-beta activation of VEGF expression. Growth Factors. 2001;19:19–34. doi: 10.3109/08977190109001073. [DOI] [PubMed] [Google Scholar]
- [46].Chae KS, Kang MJ, Lee JH, Ryu BK, Lee MG, Her NG, Ha TK, Han J, Kim YK, Chi SG. Opposite functions of HIF-alpha isoforms in VEGF induction by TGF-beta1 under non-hypoxic conditions. Oncogene. 2011;30:1213–1228. doi: 10.1038/onc.2010.498. [DOI] [PubMed] [Google Scholar]
- [47].Takeda K, Cowan A, Fong GH. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation. 2007;116:774–781. doi: 10.1161/CIRCULATIONAHA.107.701516. [DOI] [PubMed] [Google Scholar]
- [48].Schodel J, Klanke B, Weidemann A, Buchholz B, Bernhardt W, Bertog M, Amann K, Korbmacher C, Wiesener M, Warnecke C, Kurtz A, Eckardt KU, Willam C. HIF-prolyl hydroxylases in the rat kidney: physiologic expression patterns and regulation in acute kidney injury. Am J Pathol. 2009;174:1663–1674. doi: 10.2353/ajpath.2009.080687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Aprelikova O, Chandramouli GV, Wood M, Vasselli JR, Riss J, Maranchie JK, Linehan WM, Barrett JC. Regulation of HIF prolyl hydroxylases by hypoxia-inducible factors. J Cell Biochem. 2004;92:491–501. doi: 10.1002/jcb.20067. [DOI] [PubMed] [Google Scholar]
- [50].Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P, Metzen E. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. Biochem J. 2004;381:761–767. doi: 10.1042/BJ20040620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Janssen HL, Haustermans KM, Sprong D, Blommestijn G, Hofland I, Hoebers FJ, Blijweert E, Raleigh JA, Semenza GL, Varia MA, Balm AJ, van Velthuysen ML, Delaere P, Sciot R, Begg AC. HIF-1A, pimonidazole, and iododeoxyuridine to estimate hypoxia and perfusion in human head-and-neck tumors. Int J Radiat Oncol Biol Phys. 2002;54:1537–1549. doi: 10.1016/s0360-3016(02)03935-4. [DOI] [PubMed] [Google Scholar]
- [52].Su Y, Loos M, Giese N, Metzen E, Buchler MW, Friess H, Kornberg A, Buchler P. Prolyl hydroxylase-2 (PHD2) exerts tumor-suppressive activity in pancreatic cancer. Cancer. 2012;118:960–972. doi: 10.1002/cncr.26344. [DOI] [PubMed] [Google Scholar]
- [53].Mackinnon AC, Gibbons MA, Farnworth SL, Leffler H, Nilsson UJ, Delaine T, Simpson AJ, Forbes SJ, Hirani N, Gauldie J, Sethi T. Regulation of transforming growth factor-beta1-driven lung fibrosis by galectin-3. Am J Respir Crit Care Med. 2012;185:537–546. doi: 10.1164/rccm.201106-0965OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zeisberg M, Duffield JS. Resolved: EMT produces fibroblasts in the kidney. J Am Soc Nephrol. 2010;21:1247–1253. doi: 10.1681/ASN.2010060616. [DOI] [PubMed] [Google Scholar]
- [55].Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol. 1995;130:393–405. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Camara J, Jarai G. Epithelial-mesenchymal transition in primary human bronchial epithelial cells is Smad-dependent and enhanced by fibronectin and TNF-alpha. Fibrogenesis Tissue Repair. 2010;3:2. doi: 10.1186/1755-1536-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell. 2005;16:1987–2002. doi: 10.1091/mbc.E04-08-0658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kawakita T, Espana EM, Higa K, Kato N, Li W, Tseng SC. Activation of Smad-mediated TGF-beta signaling triggers epithelial-mesenchymal transitions in murine cloned corneal progenitor cells. J Cell Physiol. 2012 doi: 10.1002/jcp.24126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Brandl M, Seidler B, Haller F, Adamski J, Schmid RM, Saur D, Schneider G. IKK(alpha) controls canonical TGF(ss)-SMAD signaling to regulate genes expressing SNAIL and SLUG during EMT in panc1 cells. J Cell Sci. 2010;123:4231–4239. doi: 10.1242/jcs.071100. [DOI] [PubMed] [Google Scholar]
- [60].Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 2003;112:1486–1494. doi: 10.1172/JCI19270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kriz W, Kaissling B, Le Hir M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J Clin Invest. 2011;121:468–474. doi: 10.1172/JCI44595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Fragiadaki M, Mason RM. Epithelial-mesenchymal transition in renal fibrosis - evidence for and against. Int J Exp Pathol. 2011;92:143–150. doi: 10.1111/j.1365-2613.2011.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zeisberg M, Neilson EG. Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol. 2010;21:1819–1834. doi: 10.1681/ASN.2010080793. [DOI] [PubMed] [Google Scholar]