
Figure 1.
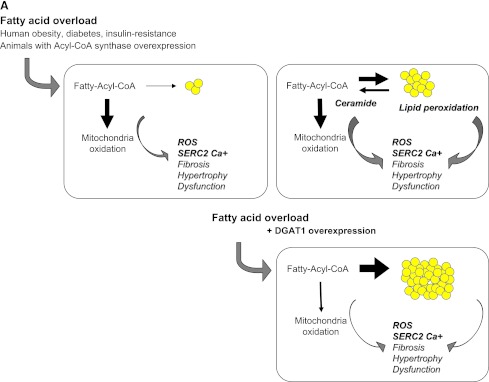
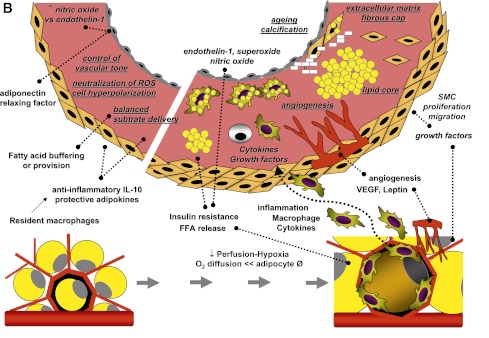
A: Fatty acids entering cardiomyocytes are conjugated with acyl-CoA and transported to the mitochondria to undergo β-oxidation for cellular energy needs. Myocardial fatty acid oxidation is, in fact, increased in human obesity and diabetes and in animal models overexpressing acyl-CoA synthase (top left). Mitochondrial respiration by the electron transport chain and NADPH oxidase are the likely predominant myocardial generators of ROS, resulting in modification of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA2a) as well as cardiac fibrosis and hypertrophy. As oxidation becomes saturated, triglyceride accumulation provides a buffer against the formation of fatty acid intermediate species, but progressive exhaustion of storage capacity provokes the build-up of acyl-CoA and ceramide in the cytoplasm (top right), contributing to lipotoxicity. Amplification of storage capacity by enzymatic overexpression of diacylglycerol acyltransferase 1 (DGAT1) slows the progression of cardiac damage (bottom right), suggesting a defensive role of triglyceride accumulation in fatty acid overload states. B: Adipose tissue surrounding vessels and myocardium may protectively serve as energy source and buffer and promote vascular relaxation (left panel). Its expansion is typical in metabolic and cardiovascular diseases, and leads to a cascade of events (right panel), likely triggered by adipocyte enlargement, hypoxia, consequent macrophage and T-cell recruitment, and inflammation. Changing patterns in the release of adipokines, cytokines, substrates, smooth muscle cell growth factors, and angiogenesis promoters from stromal and fat cells propagate to the subtending tissues, via simple diffusion and through newly formed adventitial vasa vasorum, and may thereby contribute to the progression of cardiac and vascular lipotoxicity, inflammation, and atherosclerosis. FFA, free fatty acid; SMC, smooth muscle cells; VEGF, vascular endothelial growth factor.