

Abstract
Split inteins carry out a naturally occurring process known as protein trans-splicing, where two protein fragments bind to form a catalytically competent enzyme, then catalyze their own excision and the ligation of their flanking sequences. In the past thirteen years since their discovery, chemists and biologists have utilized split inteins in exogenous contexts for a number of biotechnological applications centered around the formation of native peptide bonds. While many protein trans-splicing technologies have emerged and flourished in recent years, several factors still limit their wide-spread practical use. Here, we discuss the development, applications, and limitations of split intein-based technologies and propose that further advancement in this field will require a more fundamental understanding of split intein structure and function.
Keywords: protein splicing, protein semisynthesis
I. Introduction
The convergent chemical synthesis of proteins by peptide fragment condensation has significantly expanded our understanding of protein structure-function relationships. While traditional biochemical techniques have relied on the production of proteins recombinantly, typically limiting their scope to natural amino acid mutagenesis, chemical synthesis has facilitated the introduction of unnatural amino acids, biophysical probes, and post-translational modifications into proteins with relative ease.[1] Furthermore, the development of various fragment condensation methods has allowed for the synthesis of larger structures bearing virtually any desired functionality. In the most widely-used of such strategies, native chemical ligation (NCL), two fragments are ligated by the reaction of a peptide bearing a C-terminal thioester and an N-terminal cysteine containing peptide to first form a cysteinyl peptidyl thioester which rapidly undergoes an S- to N-acyl shift, forming an amide bond.[2] Importantly, this reaction can be carried out on unprotected peptides and results in a native peptide bond.
The mechanism of NCL, along with the abundance of thioester chemistry found in nature (e.g. non-ribosomal peptide synthesis[3] and ubiquitination enzyme cascades[4]), raises the question: does Nature use native chemical ligation? In fact, a virtually identical cysteine/thioester chemistry is used to form peptide bonds by a naturally occurring class of proteins known as inteins.[5] These single-turnover enzymes catalyze their own excision from a larger precursor protein with concomitant ligation of N- and C-terminal flanking sequences (N- and C-exteins) through a native peptide bond. A small subset of inteins exist as split protomers (N- and C-inteins/IntN and IntC) that are transcribed and translated as separate polypeptides but rapidly associate to carry out the canonical ligation reaction. This process, known as protein trans-splicing (PTS), is analogous to native chemical ligation and peptide fragment condensation in general (Figure 1, PTS and NCL). Thus, split inteins are Nature's protein ligases.
Figure 1.

The mechanism of protein trans-splicing and native chemical ligation.
II. History and Applications of Protein Trans-Splicing
Protein splicing was first observed in baker's yeast, Saccharomyces cerevisiae, in 1990.[6] Since then, several hundred inteins have been found in microorganisms from every domain of life.[7] Early studies of these peculiar proteins relied on naturally intact inteins, as there was no notion that such a complex enzyme might exist in parts. Nonetheless, it was apparent that a protein that catalyzes both the cleavage and formation of peptide bonds could be a useful tool for protein engineering and biochemistry. Indeed, intact inteins quickly became a staple of the protein chemist's toolbox, providing access to tagless proteins[8] and recombinant C-terminal protein thioesters.[9] Importantly, the latter can be used as a reactive handle for native chemical ligation onto recombinant proteins, a technology known as expressed protein ligation (EPL).
In 1998, several groups reported that when intact inteins are artificially split and isolated as separate protein fragments, these protomers can reassemble and catalyze protein splicing in trans.[10-12] Although early artificially split intein systems required unfolding, mixing, and refolding steps, these enzymatic fragment condensations opened the door to myriad protein chemistry technologies. The first significant application of PTS, segmental isotopic labeling for NMR spectroscopy (Figure 2a), was demonstrated by Yamazaki et al.[12] In this seminal study, an intein from the thermophile Pyrococcus furiosus (PI-PfuI) was artificially split, and its protomers were fused to two fragments of the C-terminal domain of RNA polymerase α-subunit (αC). The fragment fusions were separately expressed with or without 15N isotopic labeling, and full-length αC was generated by PTS. By splicing a labeled fragment with an unlabeled fragment, the authors showed that the resulting protein had a substantially simplified protein NMR spectrum, bearing only resonances corresponding to the labeled αC portion. Importantly, these resonances coincided precisely with those of a sample generated by expression of the globally labeled contiguous gene.
Figure 2.

Applications of protein trans-splicing.
Although this technology was a potentially powerful tool for facilitating NMR spectroscopy of large protein architectures, its practical application was stifled by a number of technical drawbacks. First, as artificially split inteins could not reassemble without denaturation and cooperative refolding, segmental labeling was limited to protein targets that were known to be refoldable. Furthermore, PI-PfuI and other known inteins were stringent with regards to local extein residues. This often required the introduction of non-native residues in the target protein surrounding the splice junction. Due to these limitations, segmental isotopic labeling by PTS saw limited use and awaited more efficient and well-behaved split inteins. In the meantime, segmental labeling techniques were developed using EPL and subsequently more widely applied.[13-17]
In a groundbreaking study, Liu and coworkers reported that the catalytic subunit of DNA polymerase III (DnaE) from the cyanobacterium Synechocystis sp. strain PCC6803 (Ssp) exists as a discontinuous gene containing two fragments fused to sequences homologous to the N- and C-terminal regions of inteins.[18] When co-expressed in E. coli, these split intein fusions catalyzed protein trans-splicing, demonstrating that functional split inteins exist in Nature. The discovery of the naturally split SspDnaE intein effectively ablated one major limitation of PTS-based segmental isotopic labeling: the need for refolding. Furthermore, since this split intein could assemble and splice under native conditions, it allowed for more efficient in vivo methods for the incorporation of NMR probes.[19]
Beyond its contribution to NMR spectroscopy, the SspDnaE intein proved useful for a number of other applications, most significantly the cyclization of proteins and peptides (Figure 2b). Immediately after the discovery of SspDnaE, Benkovic and coworkers developed a method for in vivo split intein-mediated circular ligation of peptides and proteins (SICLOPPS).[20] By inverting the order of intein fragments around a polypeptide of interest, (IntC-target-IntN), they demonstrated that a target sequence could be head-to-tail cyclized. This reaction resulted in the formation of a native amide bond upon excision of the N- and C-inteins, leaving behind a single cysteine residue. The power of this technology was two-fold. First, it allowed for the production of cyclic proteins with enhanced biological properties conferred by their augmented stability.[21, 22] Perhaps more significantly, it provided a method to generate large libraries of genetically encoded cyclic peptides. Using SICLOPPS, researchers have discovered methyltransferase inhibitors,[23] protease inhibitors,[24] modulators of protein-protein interactions,[25] and molecules that reduce the cellular pathology of Parkinson's disease.[26]
Split inteins have also proven useful for protein semi-synthesis.[27] Like other fragment condensation methods, PTS can be used to ligate polypeptides bearing modifications or non-peptidic moieties. Compared with synthetic methods, however, PTS holds two significant technical advantages: one or both fragments can be generated recombinantly, increasing the size-limits of protein semi-synthesis; and reactions can be carried out efficiently at low concentrations. Rates of chemical ligation reactions are strongly concentration dependent, as they rely on the random collision of peptide fragments.[28] PTS by naturally occurring split inteins, however, is facilitated by a tight protein-protein interaction and thus shows low concentration dependence. Indeed, the dissociation constant of the SspDnaE fragments is low-nanomolar, and their association rate approaches the diffusion limit for molecules of that size.[29]
Despite these benefits, protein semi-syntheisis using SspDnaE has one major drawback: the N- and C-intein fragments are 123 and 36 amino acids, respectively, and the split site is conserved in all other naturally occurring split inteins. Thus, the N-intein is beyond the scope of efficient linear peptide synthesis, and while the C-intein is accessible by solid-phase peptide synthesis, the addition of any synthetic cargo as a C-extein puts this fragment nearly out of reach as well. To address this problem, several groups revisited the notion of artificially split inteins, in search of PTS systems with at least one, short, synthetically tractable intein fragment. One particularly interesting result of these studies was the discovery that the naturally intact DnaB intein from Ssp could be split 11 residues from its N-terminus to generate a functional PTS system.[30] Unlike naturally split inteins, which all have a shorter C-intein, the split SspDnaB intein could be used to splice synthetic moities onto the N-termini of proteins.[31] Additional studies have uncovered artificially split inteins with C-fragments as short as 6 and 15 amino acids.[32, 33] Although these new split inteins resolve the issue of synthetic accessibility, they are typically used at high concentrations, presumably due to their diminished binding affinities relative to naturally occurring split inteins.[34]
The most significant advantage of PTS-based protein semi-synthesis over chemical ligation is that it can be readily applied in vivo (Figure 2c). In purified systems, reaction specificity is governed simply by the functional groups present in the molecules of interest, but in living systems, orthogonal protein chemistry becomes increasingly challenging.[35] Split inteins overcome this problem by acting as a ligation auxiliary that engenders virtually absolute specificity to the splicing reaction of interest. Indeed PTS has been used in vivo to label proteins with synthetic probes[36] and to construct a therapeutic gene product from its fragments.[37] In the later study, the authors overcame the size-limitations of adeno-associated viral vectors for gene therapy by splitting the cargo into two fragments and assembling the therapeutic target in vivo by PTS. Importantly, this work demonstrated the potential utility of split inteins as more than just a research tool.
Conditional protein splicing (CPS), the activation or inhibition of protein splicing in the presence of an extrinsic modulator, is perhaps the most intriguing application of split inteins (Figure 2d). CPS systems show promise as a powerful tool to manipulate protein function in vivo. While such modules have been developed with intact inteins,[38] split inteins provide an optimal platform for CPS, given that control over fragment complementation inherently implies control over protein splicing. Indeed split intein CPS systems have been engineered to be responsive to small molecules,[39, 40] light,[41-43] and proteolysis.[41] In an interesting recent study from Mootz and coworkers, an artificially split SspDnaB intein was outfitted with an uncleavable isopeptide bond bearing a peptidyl side-chain at position 1 of the N-intein and a photo-cleavable 4,5-dimethoxy-2- nitrobenzyl (DMNB) group capping the N-terminus in place of an N-extein.[44] This construct was splicing-incompetent, however upon photolysis of the DMNB moiety, it efficiently catalyzed C-extein cleavage, releasing a protein of interest with a free N-terminus. As proof of concept of this technology, the authors demonstrated that they could control a coagulation cascade in human blood plasma by photo-caging the activity of prothrombin. Importantly, this study not only showed the potential power of CPS, but it also demonstrated that PTS side reactions could be harnessed to control protein function.
III. A New Era for Protein Trans-Splicing
Despite the development of numerous technologies based on PTS, until recently, most variations of this ligation technique were limited by the properties of available split inteins. The activities of most tested split inteins, especially the naturally occurring SspDnaE intein, were shown to be strongly dependent on the identity of extein residues.[45] As a result, ligation sites in a target protein were limited to sequences similar to endogenous extein sequences or required mutation of neighboring residues. Furthermore, barring a few artificially split systems,[46] PTS is a slow process, despite its enzymatic nature. In fact, it was generally believed that protein splicing, which requires the cleavage of two peptide bonds and the formation of a third, always occurred on the time-scale of hours. For many inteins, including SspDnaE, this rate could be even slower in the presence of exogenous extein sequences and at temperatures above 30°C.
In 2003, the Pietrokovski group carried out a genomic study of cyanobacterial dnaE genes and systematically identified split DnaE inteins homologous to SspDnaE in several new organisms (Figure 3a).[47] Despite their discovery and sequence analysis of several new naturally split inteins, few attempts were made to characterize or compare their splicing efficiencies in vivo or in vitro.[48] In a landmark paper, Iwai et al demonstrated that PTS catalyzed by the split DnaE intein from the cyanobacterium Nostoc punctiforme (Npu) is substantially more efficient than that of SspDnaE.[49] Furthermore, the authors showed that NpuDnaE was significantly more promiscuous than SspDnaE with regards to C-extein sequences. In a follow-up study, Mootz and coworkers showed that NpuDnaE could catalyze PTS in vitro with a reaction half-time as fast as 1 minute at 37°C, disproving the notion that there is a practical speed limit for protein splicing.[50]
Figure 3.
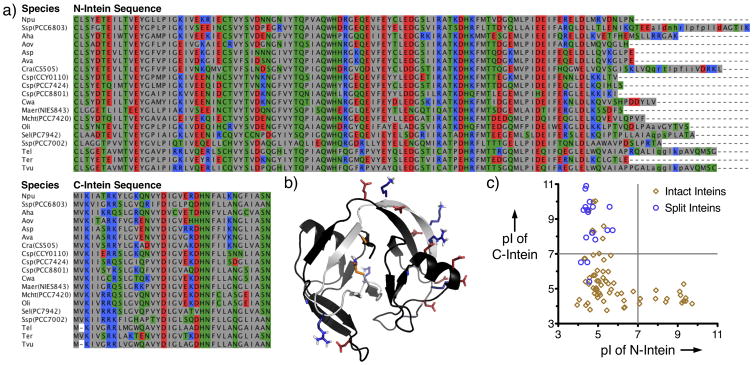
Sequence and structural features of split Dnae inteins. a) Sequence alignment of 19 unique naturally split DnaE inteins. b) NMR structure of NpuDnaE (PDB code: 2KEQ) highlighting intermolecular electrostatic interactions. The N- and C-inteins are shown in black and gray, respectively. Acidic residues are shown in red, basic residues are shown in blue, and terminal catalytic residues are shown in orange. c) Comparison of calculated isoelectric points (pI) for the N- and C-terminal fragments of naturally split inteins and the N- and C-terminal sequences of naturally intact inteins.
Given the superior properties of NpuDnaE compared to other split inteins, it is not surprising that several applications of this intein have emerged in the past few years. In a recent study, Becker and coworkers demonstrated that NpuDnaE could be used to efficiently lipidate the C-termini of proteins for immobilization onto liposomes or lipid-coated nanoparticles.[51] In another semi-synthesis report, NpuDnaE was used to modify trans-membrane and glycosylphosphatidylinositol (GPI) anchored proteins on the surface of living cells.[52] This split intein has also emerged as a useful tool for segmental isotopic labeling of proteins.[53] In an exciting recent study, the wild-type NpuDnaE intein was used in conjunction with a linearly permuted form to carry out tandem three-piece ligation reactions of proteins.[54] This technology was used to isotopically label the central domain of a protein containing three homologous domains with substantial chemical shift overlap. By segmentally labeling each domain of the protein separately, the authors were able to significantly reduce the complexity of its NMR spetra for future structural studies.
IV. A Reductionist Approach to Protein Trans-Splicing
An emerging theme in split intein research is that the limitations of existing PTS systems have fueled the discovery or development of new and improved split inteins. In moving forward, it is apparent that expanding the scope and utility of split inteins will require a deeper understanding of what drives or restricts efficient trans-splicing. Three recent studies highlight this notion. As stated above, the major hindrance in using naturally split inteins for protein semi-synthesis is synthetic accessibility due to size. As the split site is conserved in naturally split inteins (Figure 3a), this hurdle also applies to NpuDnaE. Recognizing this, the Iwai group sought to find a new split site that provided one synthetically tractable intein fragment while still allowing splicing.[55] To achieve this goal, the authors solved the solution structure of a fused form of NpuDnaE by NMR spectroscopy (Figure 3b). Then they employed 15N relaxation measurements to identify flexible regions in the protein structure which might be an appropriate place for a new split site. Using this strategy, they found that a 6 amino acid C-intein could be used in conjunction with its cognate extended N-intein to catalyze PTS. Not only did this study provide a new tool for protein semi-synthesis, but it also provided invaluable structural information for this important intein.
Another aforementioned limitation of PTS is the requirement for specific extein residues beyond the required catalytic cysteine. While NpuDnaE is more promiscuous than SspDnaE and other split inteins in this regard, it still shows some extein dependence. In a study from our group, we demonstrated that in the presence of exogenous C-extein residues, the rate limiting step for PTS by DnaE inteins is succinimide formation and resolution of the branched intermediate.[56] Armed with this information we used an in vivo directed evolution approach to enhance the promiscuity of a Npu/SspDnaE chimera. This study demonstrated that extein dependence is not an unsolvable problem, and it generated the first laboratory selection system for the development of split inteins with enhanced properties. Indeed, in the same study, we used this system to evolve a split DnaE chimera that spliced efficiently at 37°C and showed that this intein could be used in mammalian cells.
The largest known group of naturally split inteins are the cyanobacterial DnaE inteins. Due to their highly homologous nature, N- and C-inteins from various organisms are known to cross-react, prohibiting their simultaneous use for multiple one-pot ligation reactions.[48, 49] In another recent study from our lab, we sought to determine whether this limitation could be overcome by identifying a common sequence feature of split inteins that might promote this cross-reactivity.[57] It had previously been postulated that split intein association is driven by significant charge segregation between the N- and C-intein fragments.[29, 48] A bioinformatic analysis of intein sequences and visual inspection of the NpuDnaE structure showed that this charge segregation is unique to split inteins and manifests as specific intermolecular ion clusters at the intein fragment interface (Figure 3). We demonstrated that swapping charges within several of these ion clusters in NpuDnaE altered split intein binding affinities and splicing kinetics, and we used this strategy to generate a mutant split intein with low cross-reactivity to NpuDnaE. This allowed for the catalysis of two trans-splicing reactions in one pot with high selectivity. Presumably, this new mutant intein is also orthogonal to other split DnaE inteins, given the conserved property that were exploited and perturbed, however this remains to be tested.
V. Summary and Outlook
In the 14 years since its discovery, protein trans-splicing has evolved from a curious but handicapped process to an efficient ligation method with broad technological applications. Split inteins, like their intact counterparts, are now an indispensible member of the protein chemists' toolbox. Despite the persistent limitations of PTS, several technologies based on this technique have thrived. In particular, well established protocols are now in place for the split intein-mediated production of genetically encoded cyclic peptide libraries.[58] Given this, SICLOPPS as emerged as a fruitful strategy for drug discovery, in particular for targeting protein-protein interactions. The application of PTS to structural biology, especially NMR spectroscopy, has also proven to be rewarding.[59] With the development of PTS systems that can facilitate the construction of larger, more complex proteins,[54, 57] the upper size limits for protein analysis by NMR are rapidly being raised. Furthermore, with the discovery of NpuDnaE and the development of non-canonically split inteins, the application of PTS to protein semi-synthesis may soon rival that of chemical ligation techniques.
We now have a variety of highly efficient wild-type and engineered split inteins at our disposal, however some applications of PTS are still in their infancies. Conditional protein splicing with split inteins, for example, is effectively a proof-of-principle application. The promise of this technology both for basic research and therapeutics remains unfulfilled. In large part, this is due to the splicing efficiency of many existing CPS systems, which are based on artificially split inteins. Only two CPS modules have been developed that control the activity of a naturally split intein, SspDnaE, by modulating its reactivity[41] or fragment assembly.[43] This is not surprising, given our lack of understanding of how split inteins bind and fold into the complex intein-fold topology (Figure 3b). Thus, the creation of a truly efficient conditional trans-splicing system would require a thorough understanding of the relationship between fragment assembly and catalysis in a highly active split intein such as NpuDnaE.
As more microbial genomes are sequenced, the number of split inteins is rapidly growing. To date, there are roughly 50 unique sequences of split inteins reported based on genomic data mining, however few of these have been experimentally shown to catalyze PTS.[7, 47, 60, 61] Furthermore, little work has been carried out to rigorously characterize the splicing activities of experimentally validated split inteins in a parallel and systematic fashion,[48, 50] and few studies have measured their fragment binding affinities.[29, 57] The largest established class of split inteins, the cyanobacterial DnaE inteins (Figure 3), come from an ecologically diverse class of organisms.[62] While these microbes live in a wide array of environments, including some with high temperatures or high salinity, protein splicing has yet to be critically examined or utilized under such exotic conditions. Having distinct inteins that could catalyze PTS under unique conditions would be an enormously useful addition to our existing array of protein ligation tools. Thus, it appears as though nature's protein ligases still retain some untapped potential.
Acknowledgments
The authors thank members of the Muir laboratory for valuable discussions. Some of the work discussed in this review was carried out in the authors' laboratory and was supported by grants from the US National Institutes of Health (GM086868 and RC2 CA148354).
Biographies
Neel Shah received his B.S. in Chemistry in 2008 from New York University. At NYU, he worked with Kent Kirshenbaum on developing methods for stabilizing the structures of folded peptidomimetic oligomers through non-covalent interactions. After obtaining his undergraduate degree, he began his graduate studies at The Rockefeller University under the advisement of Tom Muir. N.S. is currently carrying out his doctoral research on the biophysical and biochemical characterization of naturally split inteins as a visiting student at Princeton University.
Tom Muir received his Ph.D. in organic chemistry in 1993 from the University of Edinburgh. After studying bioorganic chemistry at The Scripps Research Institute, he joined The Rockefeller University in 1996 as assistant professor, becoming Richard E. Salomon Family Professor in 2005. He also served as director of the Pels Family Center for Biochemistry and Structural Biology. In 2010 he moved to Princeton University and in 2011 was appointed Van Zandt Williams Jr. Class of 1965 Professor of Chemistry. T.M. has received the Blavatnik Award for Young Scientists, the Vincent du Vigneaud Award, the Irving Sigal Young Investigator Award, the Leonidas-Zervas Award, and a Burroughs Wellcome Fund New Investigator Award. He was deemed an Alfred P. Sloan Research Fellow in 1999 and a Pew Scholar in the Biomedical Sciences in 1997. T.M. is a fellow of the American Association for the Advancement of Science.
References
- 1.Kent SBH. Chem Soc Rev. 2009;38:338. doi: 10.1039/b700141j. [DOI] [PubMed] [Google Scholar]
- 2.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Science. 1994;266:776. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 3.Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 4.Huang DT, Hunt HW, Zhuang M, Ohi MD, Holton JM, Schulman BA. Nature. 2007;445:394. doi: 10.1038/nature05490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mills KV, Perler FB. Protein Pept Lett. 2005;12:751. doi: 10.2174/0929866054864337. [DOI] [PubMed] [Google Scholar]
- 6.Kane PM, Yamashiro CT, Wolczyk DF, Neff N, Goebl M, Stevens TH. Science. 1990;250:651. doi: 10.1126/science.2146742. [DOI] [PubMed] [Google Scholar]
- 7.Perler FB. Nucleic Acids Res. 2002;30:383. doi: 10.1093/nar/30.1.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Southworth M, Amaya K, Evans T, Xu M, Perler F. Biotechniques. 1999;27:110. doi: 10.2144/99271st04. [DOI] [PubMed] [Google Scholar]
- 9.Muir TW, Sondhi D, Cole PA. Proc Natl Acad Sci USA. 1998;95:6705. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Southworth MW, Adam E, Panne D, Byer R, Kautz R, Perler FB. EMBO J. 1998;17:918. doi: 10.1093/emboj/17.4.918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mills KV, Lew BM, Jiang S, Paulus H. Proc Natl Acad Sci USA. 1998;95:3543. doi: 10.1073/pnas.95.7.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamazaki T, Otomo T, Oda N, Kyogoku Y, Uegaki K, Ito N, Ishino Y, Nakamura H. J Am Chem Soc. 1998;120:5591. [Google Scholar]
- 13.Xu R, Ayers B, Cowburn D, Muir TW. Proc Natl Acad Sci USA. 1999;96:388. doi: 10.1073/pnas.96.2.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fushman D, Xu R, Cowburn D. Biochemistry. 1999;38:10225. doi: 10.1021/bi990897g. [DOI] [PubMed] [Google Scholar]
- 15.Camarero JA, Shekhtman A, Campbell EA, Chlenov M, Gruber TM, Bryant DA, Darst SA, Cowburn D, Muir TW. Proc Natl Acad Sci USA. 2002;99:8536. doi: 10.1073/pnas.132033899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao W, Zhang Y, Cui C, Li Q, Wang J. Protein Sci. 2008;17:736. doi: 10.1110/ps.073383708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vitali F, Henning A, Oberstrass FC, Hargous Y, Auweter SD, Erat M, Allain FHT. EMBO J. 2006;25:150. doi: 10.1038/sj.emboj.7600911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu H, Hu Z, Liu XQ. Proc Natl Acad Sci USA. 1998;95:9226. doi: 10.1073/pnas.95.16.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Züger S, Iwai H. Nat Biotechnol. 2005;23:736. doi: 10.1038/nbt1097. [DOI] [PubMed] [Google Scholar]
- 20.Scott CP, Abel-Santos E, Wall M, Wahnon DC, Benkovic SJ. Proc Natl Acad Sci USA. 1999;96:13638. doi: 10.1073/pnas.96.24.13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Camarero J, Muir T. J Am Chem Soc. 1999;121:5597. [Google Scholar]
- 22.Jeffries CM, Graham SC, Stokes PH, Collyer CA, Guss JM, Matthews JM. Protein Sci. 2006;15:2612. doi: 10.1110/ps.062377006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naumann TA, Tavassoli A, Benkovic SJ. Chembiochem. 2008;9:194. doi: 10.1002/cbic.200700561. [DOI] [PubMed] [Google Scholar]
- 24.Young TS, Young DD, Ahmad I, Louis JM, Benkovic SJ, Schultz PG. Proc Natl Acad Sci USA. 2011;108:11052. doi: 10.1073/pnas.1108045108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tavassoli A, Lu Q, Gam J, Pan H, Benkovic SJ, Cohen SN. ACS Chem Biol. 2008;3:757. doi: 10.1021/cb800193n. [DOI] [PubMed] [Google Scholar]
- 26.Kritzer JA, Hamamichi S, McCaffery JM, Santagata S, Naumann TA, Caldwell KA, Caldwell GA, Lindquist S. Nat Chem Biol. 2009;5:655. doi: 10.1038/nchembio.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mootz HD. Chembiochem. 2009;10:2579. doi: 10.1002/cbic.200900370. [DOI] [PubMed] [Google Scholar]
- 28.Dawson P, Churchill M, Ghadiri M, Kent S. J Am Chem Soc. 1997;119:4325. [Google Scholar]
- 29.Shi J, Muir TW. J Am Chem Soc. 2005;127:6198. doi: 10.1021/ja042287w. [DOI] [PubMed] [Google Scholar]
- 30.Sun W, Yang J, Liu XQ. J Biol Chem. 2004;279:35281. doi: 10.1074/jbc.M405491200. [DOI] [PubMed] [Google Scholar]
- 31.Ludwig C, Pfeiff M, Linne U, Mootz HD. Angew Chem Int Ed. 2006;45:5218. doi: 10.1002/anie.200600570. [DOI] [PubMed] [Google Scholar]
- 32.Appleby JH, Zhou K, Volkmann G, Liu XQ. J Biol Chem. 2009;284:6194. doi: 10.1074/jbc.M805474200. [DOI] [PubMed] [Google Scholar]
- 33.Aranko AS, Züger S, Buchinger E, Iwaï H. PLoS ONE. 2009;4:e5185. doi: 10.1371/journal.pone.0005185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ludwig C, Schwarzer D, Mootz HD. J Biol Chem. 2008;283:25264. doi: 10.1074/jbc.M802972200. [DOI] [PubMed] [Google Scholar]
- 35.Prescher JA, Bertozzi CR. Nat Chem Biol. 2005;1:13. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 36.Giriat I, Muir TW. J Am Chem Soc. 2003;125:7180. doi: 10.1021/ja034736i. [DOI] [PubMed] [Google Scholar]
- 37.Li J, Sun W, Wang B, Xiao X, Liu XQ. Hum Gene Ther. 2008;19:958. doi: 10.1089/hum.2008.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buskirk AR, Ong YC, Gartner ZJ, Liu DR. Proc Natl Acad Sci USA. 2004;101:10505. doi: 10.1073/pnas.0402762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mootz HD, Blum ES, Tyszkiewicz AB, Muir TW. J Am Chem Soc. 2003;125:10561. doi: 10.1021/ja0362813. [DOI] [PubMed] [Google Scholar]
- 40.Brenzel S, Mootz HD. J Am Chem Soc. 2005;127:4176. doi: 10.1021/ja043501j. [DOI] [PubMed] [Google Scholar]
- 41.Vila-Perelló M, Hori Y, Ribó M, Muir TW. Angew Chem Int Ed. 2008;47:7764. doi: 10.1002/anie.200802502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tyszkiewicz AB, Muir TW. Nat Methods. 2008;5:303. doi: 10.1038/nmeth.1189. [DOI] [PubMed] [Google Scholar]
- 43.Berrade L, Kwon Y, Camarero JA. Chembiochem. 2010 doi: 10.1002/cbic.201000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Binschik J, Zettler J, Mootz HD. Angew Chem Int Ed. 2011;50:3249. doi: 10.1002/anie.201007078. [DOI] [PubMed] [Google Scholar]
- 45.Amitai G, Callahan BP, Stanger MJ, Belfort G, Belfort M. Proc Natl Acad Sci USA. 2009;106:11005. doi: 10.1073/pnas.0904366106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brenzel S, Kurpiers T, Mootz HD. Biochemistry. 2006;45:1571. doi: 10.1021/bi051697+. [DOI] [PubMed] [Google Scholar]
- 47.Caspi J, Amitai G, Belenkiy O, Pietrokovski S. Mol Microbiol. 2003;50:1569. doi: 10.1046/j.1365-2958.2003.03825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dassa B, Amitai G, Caspi J, Schueler-Furman O, Pietrokovski S. Biochemistry. 2007;46:322. doi: 10.1021/bi0611762. [DOI] [PubMed] [Google Scholar]
- 49.Iwai H, Züger S, Jin J, Tam PH. FEBS Lett. 2006;580:1853. doi: 10.1016/j.febslet.2006.02.045. [DOI] [PubMed] [Google Scholar]
- 50.Zettler J, Schütz V, Mootz HD. FEBS Lett. 2009;583:909. doi: 10.1016/j.febslet.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 51.Chu NK, Olschewski D, Seidel R, Winklhofer KF, Tatzelt J, Engelhard M, Becker CFW. J Pept Sci. 2010;16:582. doi: 10.1002/psc.1227. [DOI] [PubMed] [Google Scholar]
- 52.Dhar T, Mootz HD. Chem Commun. 2011;47:3063. doi: 10.1039/c0cc04172f. [DOI] [PubMed] [Google Scholar]
- 53.Muona M, Aranko AS, Raulinaitis V, Iwaï H. Nat Protoc. 2010;5:574. doi: 10.1038/nprot.2009.240. [DOI] [PubMed] [Google Scholar]
- 54.Busche AEL, Aranko AS, Talebzadeh-Farooji M, Bernhard F, Dötsch V, Iwaï H. Angew Chem Int Ed. 2009;48:6128. doi: 10.1002/anie.200901488. [DOI] [PubMed] [Google Scholar]
- 55.Oeemig JS, Aranko AS, Djupsjöbacka J, Heinämäki K, Iwaï H. FEBS Lett. 2009;583:1451. doi: 10.1016/j.febslet.2009.03.058. [DOI] [PubMed] [Google Scholar]
- 56.Lockless SW, Muir TW. Proc Natl Acad Sci USA. 2009;106:10999. doi: 10.1073/pnas.0902964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shah NH, Vila-Perelló M, Muir TW. Angew Chem Int Ed. 2011;50:6511. doi: 10.1002/anie.201102909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Horswill AR, Benkovic SJ. Curr Protoc Protein Sci. 2006;Chapter 19 doi: 10.1002/0471140864.ps1915s46. Unit 19.15. [DOI] [PubMed] [Google Scholar]
- 59.Volkmann G, Iwaï H. Mol Biosyst. 2010;6:2110. doi: 10.1039/c0mb00034e. [DOI] [PubMed] [Google Scholar]
- 60.Choi JJ, Nam KH, Min B, Kim SJ, Söll D, Kwon ST. J Mol Biol. 2006;356:1093. doi: 10.1016/j.jmb.2005.12.036. [DOI] [PubMed] [Google Scholar]
- 61.Dassa B, London N, Stoddard BL, Schueler-Furman O, Pietrokovski S. Nucleic Acids Res. 2009;37:2560. doi: 10.1093/nar/gkp095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.B. A. Whitton, M. Potts, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2000.