

Abstract
Acute inflammation in response to both exogenous and endogenous danger signals can lead to the assembly of cytoplasmic inflammasomes that stimulate the activation of caspase-1. Subsequently, caspase-1 facilitates the maturation and release of cytokines and also, under some circumstances, the induction of cell death by pyroptosis. Using a mouse line lacking expression of NLRP1, we show that assembly of this inflammasome in cells is triggered by a toxin from Anthrax and that it initiates caspase-1 activation and release of IL-1β. Furthermore, NLRP1 inflammasome activation also leads to cell death, which escalates over three days following exposure to the toxin and culminates in acute lung injury and death of the mice. We show that these events are not dependent on production of IL-1β by the inflammasome but are dependent on caspase-1 expression. In contrast, MDP mediated inflammasome formation is not dependent on NLRP1, but NLRP3. Taken together, our findings show that assembly of the NLRP1 inflammasome is sufficient to initiate pyroptosis, which subsequently leads to a self-amplifying cascade of cell injury within the lung from which the lung cannot recover, eventually resulting in catastrophic consequences for the organism.
Introduction
NLRP1 was first characterized as a member of the CED-4 family of apoptotic proteins, which are required to initiate programmed cell death in developing C. elegans (1–3). Consistent with this relationship, two independent groups showed that over expression of NLRP1/DEFCAP/NAC in mammalian cells leads to cell death through apoptosis (1, 2). Functional homology with CED-4 was further demonstrated by in vitro studies with macrophages that reported that NLRP1 is regulated by the anti-apoptotic proteins Bcl-2 and Bcl-XL. Bcl-2 is homologous to CED-9, an antiapoptotic protein that binds CED-4(4).
A possible mechanism by which NLRP1 could mediate cell death has been proposed based on its detection in a protein complex named the inflammasome, which activates inflammatory caspases (5). Thus, the first inflammasome complex to be described contained the proteins caspase-1, caspase-5, ASC, and NLRP1. In later work using recombinant proteins, caspase-1 activation was initiated by the NLRP1 protein alone in response to a microbial component, muramyl-dipeptide (MDP), and ASC was not required, though it enhanced caspase-1 activity(6). The protease caspase-1 mediates the production of mature Interleukin (IL) -1β and IL-18 cytokines, which stimulate a pro-inflammatory immune response and protect the organism from pathogen-induced death. However, a number of lines of evidence indicate that robust activation of caspase-1 leads to induced cell death under some circumstances. Cell death that is inflammation-associated and caspase-1 dependent has been termed pyroptosis (7). The downstream effects of pyroptosis are largely unknown. Recently, pyroptosis was identified as a mechanism to clear intracellular bacteria. Following Salmonella typhimurium infection, caspase-1 dependent pyroptosis resulted in release of S. typhimurium from infected macrophages, and the bacteria were subsequently killed by neutrophils (8).
NLRP1 is now recognized as one of a number of proteins that can form complexes capable of supporting the maturation of caspase-1 in response to unique, but overlapping, danger and pathogen signals. To date, these include NLRP1, NLRP3, NLRC4, NLRP6, NLRP7 and absent in melanoma 2 (AIM2)(5, 9–16), although studies of NLRC5 and NLRP7 were performed in human cells with gene knockdown strategies and NLRP1 by assembly in a cell free system. Similar to many of the NLRs capable of forming an inflammasome, NLRP1 contains a pyrin domain that can bind ASC, a nucleotide binding domain (NBD or NATCH) responsible for intramolecular folding and nucleotide triphosphate-dependent oligomerization, a function to find domain (FIIND), a CARD domain that binds pro-caspase-1, and a leucine-rich repeat domain (LRR). The LRR domain is believed to confer the sensitivity of a given inflammasome to a particular pathogen or alteration in cellular homeostasis, although direct evidence for this is not available. While the list of agents that stimulate NLR/AIM2-dependent caspase-1 activation continues to expand, the intracellular trigger for protein oligomerization remains unclear. To date, an in vitro protein reconstitution assay showing formation of the NLRP1 inflammasome complex in response to MDP treatment is the best display of stimulus-induced complex formation (6). Much of the information regarding the activation of these human inflammasomes has been garnered from experiments carried out in mice lacking the orthologous NLR gene, however, this has not been achieved for NLRP1.
In mouse, three orthologs of the human NLRP1 gene have been identified, termed Nlrp1a, Nlrp1b, and Nlrp1c (17). Using genetic studies, Boyden and Dietrich showed that the locus encoding the Nlrp1 genes confers the sensitivity of some mouse lines to the lethal toxin of Bacillus anthracis (17). Anthrax causes rapid tissue necrosis, circulatory collapse, and acute respiratory failure due to fluid retention in the lung (18). Many of these pathologies can be recapitulated in animal models by an exposure to Lethal Toxin (LT), which is a combination of lethal factor (LF) and protective antigen (PA)(19), indicating that intoxication is mediated, at least in part, by these factors. The mouse haplotype that confers sensitivity to LT also carries mutations in the Nlrp1a and Nlrp1c genes that render them unlikely to direct the expression of a functional protein (17). This suggests that it is the Nlrp1b allele carried by some mouse strains that initiates the response to LT.
Here we report the generation of a mouse line lacking Nlrp1b expression. Using cells from this line, we show that LT, but not MDP, can trigger the assembly of the murine NLRP1 inflammasome, leading not only to caspase-1 activation and IL-1β production, but also to pyroptosis. These events are sufficient to result in acute lung injury that escalates and leads to morbidity within three days of activation of the NLRP1 inflammasome.
Materials and Methods
Mice
A vector was designed to inactivate the mouse Nlrp1b locus by removing a 16 kb segment DNA extending from the middle of exon 3, which encodes the ATP binding NACHT domain, to the promoter region of the gene and replacing it with a neomycin resistance marker. An Xba I restriction digest site was added at the 3′ end of the Neor cassette allowing detection of the mutant locus by Southern blot analysis. 129 ES cells carrying the correctly modified Nlrp1b allele were mated with 129S6 mice (Taconic Farms Inc., Hudson, NY) to maintain the mutation on this inbred genetic background.
The Nlrp3 −/− mouse line was generated by targeted mutagenesis in 129 ES cells using a replacement type vector, which removed all Nlrp3 exons and replaced this segment of DNA with the neomycin resistance gene.
The Casp1−/− mouse line was generated in 129 ES cells using a replacement type targeting construct. Recombination with the endogenous Casp1/4 locus removed the entire coding regions of both genes as well as 8 kb of DNA 5′ of Casp1 and 6 kb 3′ of Casp4. Chimeras generated from the targeted ES cells were bred directly to 129S6 mice to maintain the mutation on this genetic background.
The P2X7−/− mouse line was moved to the BALB/cByJ background by six consecutive crosses to BALB mice (20).
All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals as well as the Institutional Animal Care and Use Committee guidelines of the University of North Carolina at Chapel Hill.
Reverse Transcription PCR
RNA was isolated from intestine of wild-type 129 and Nlrp1b−/− mice with RNABee (Tel-Test Inc., Friendswood, TX). RNA was reverse transcribed with Superscript III (Invitrogen, Life Technologies, Grand Island, NY, USA) follow by PCR using primer sets specific for Nlrp1a, Nlrp1b and Nlrp1c. Amplified products were visualized by gel electrophoresis.
Cell Culture
Bone marrow-derived macrophages (BMDM) were prepared by harvesting marrow from femurs and tibias of 129+/+ and Nlrp1b−/− mice. Marrow was cultured for 7 days in IMDM containing 15% L-cell-conditioned medium, 10% FBS, 2 mM L-glutamine, 150 U/mL of penicillin, 150 μg/mL of streptomycin, and 57.2 μM 2-mercaptoethanol (all from GIBCO Life Technologies, Grand Island, NY, USA). BMDM were incubated in OPTIMEM plus Glutamax (GIBCO Life Technologies, Grand Island, NY, USA) containing varying concentrations of lethal toxin (LT; List Biological Laboratories, Inc., Campbell, CA, USA) for 4 hours and cell viability was assessed with the WST-1 reagent (F. Hoffmann-La Roche AG, Basel, Switzerland). Peritoneal macrophages were elicited from wild-type, Nlrp1b−/−, Nlrp3−/− and Casp−1−/− mice with an i.p. injection of 4 ml of 4% thioglycollate in PBS. 72 hours after injection, the peritoneal cavity was lavaged with DMEM and cells were plated and incubated for one hour. Adherent cells were then washed twice with cold PBS and cultured in DMEM containing 10% FBS, 2 mM L-glutamine, 150 U/mL of penicillin, 150 μg/mL of streptomycin, and 57.2 μM 2-mercaptoethanol (all from GIBCO Life Technologies, Grand Island, NY, USA). For LT treatments, macrophages were primed overnight with 100 ng/ml ultra-pure LPS (Invitrogen, Life Technologies, Grand Island, NY, USA), washed with PBS, and incubated with OPTIMEM plus Glutamax containing either PBS or 1 μg/ml LT for 6 hours. Cytotoxicity was determined by measuring LDH release in the cell supernatant by colorimetric assay following the manufacturer’s protocol (Clontech laboratories, Mountain View, CA). For MDP, PGN, TiO2, and L18-MDP treatments, cells were primed with 100 ng/ml ultrapure LPS for 6 hours, washed with PBS, and incubated in OPTIMEM, 5 mg/mL TiO2, 5 mg/mL TiO2 + 10 mg/mL MDP, 5 mg/mL TiO2 + 20 mg/mL PGN, 10 mg/mL MDP, 20 mg/mL PGN, or 1 mg/ml L18-MDP for 16 hours.
MDP Treatment of Mice
Mice received an i.p. injection of 5 μg LPS in 200 μl of PBS. 3 hours after LPS administration, mice were i.p. injected with 50 μg of L18-MDP in 200 μl PBS. Serum and peritoneal lavage fluid was collected 1.5 hours post-injection with L18-MDP.
LPS/LT and LPS/ATP treatments of Mice
129+/+ and Nlrp1b −/− mice were treated with 0.3 mg/kg body weight LPS intratracheally. Two hours later, mice were treated with vehicle, 20 μg LT, or 50 μl 100 mM ATP intratracheally. Two hours after the second treatment, mice were sacrificed and their lungs lavaged with HBSS.
Lethal Toxin Treatment of Mice
Nlrp1b−/−, Il1β−/−, Casp−1−/−, Nlrp3−/− and appropriate 129+/+ control mice were dosed intratracheally with vehicle or 20 μg LT and BAL fluid was collected at various time points. The total number of cells in the BAL fluid was determined using a hemocytometer.
Protein analyses
IL-1β, IL-1α, KC, MIP-2 and CXCL15/lungkine levels were detected by ELISA as recommended by the manufacturer (eBioscience, San Diego, CA and R&D Systems, Minneapolis, MN). Albumin levels were evaluated by colorimetric assay according to the manufacturer (Bethyl Laboratories, Montgomery, TX). BAL cell samples were processed to isolate myeloperoxidase (MPO) enzyme, and its catalytic activity was evaluated by colorimetric assay as a measure of neutrophil recruitment into the lungs. LDH release was measured by colorimetric assay following the manufacturer’s protocol (Clontech Laboratories, Mountain View, CA). Results were compared to the total LDH released from cells treated with 1% Triton-X (100% lysis control). For western blot analysis, protein from BAL fluid was concentrated using a Microcon YM-30 column (Millipore, Billerica, MA) and subsequently quantified by BCA assay (Thermo Scientific Pierce, Rockford, IL). 20 μg of total protein was separated by SDS/PAGE, transferred onto a PVDF membrane, and incubated with IL-1β (AF-401-NA, R&D Systems, Minneapolis, MN), caspase-1 (sc-514, Santa Cruz Biotechnology Inc., Santa Cruz, CA), HMGB1 (ab12029, Abcam Inc., Cambridge, MA), or GAPDH (#3683, Cell Signaling Technology, Danvers, MA, USA) antibodies.
Histology
Lungs were inflated with 10% formalin via a tracheal cannula, removed from the thoracic cavity, and fixed overnight in formalin. Sections of the left lobe of the lung were stained with hematoxylin-eosin. Digital images of the sections were captured under bright field mode using Olympus BX61 Upright Fluorescence Microscope (Olympus, Center Valley, PA, USA) and Velocity software (PerkinElmer Life and Analytical Sciences, Waltham, MA, USA). Whole lung sections and at least four images of representative areas of each section were captured. For 2x images, a background image was digitally subtracted using ImageJ software (National Institutes of Health, Bethesda, MD).
FACS analysis
BAL was collected two hours after intratracheal instillation of 20 μg LT or vehicle. Cells were stained with fluorescently labeled caspase inhibitors FAM-YVAD-FMK (caspase-1) or FAM-DVED-FMK (caspase-3) and with 7-Amino-Actinomycin (7-AAD, BD Bioscience, Franklin Lakes, NJ USA) according to manufacturer’s instructions (Cell Technology, Inc., Mountain View, CA, USA). For whole lung cell suspension, isolated lungs were cut into pieces and incubated for 1 h in digestion medium (RPMI, collagenase IV 12 mg/ml, DNase I 10mg/ml) at 37°C. A single cell suspension was prepared by passing through 70 μm cell strainer (BD Bioscience, Franklin Lakes, NJ USA). Red blood cells were removed by incubation in lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA). Cell were incubated with Fc block, and stained for CD45, Mac-I, and GR-1 or CCR-2 (all from BD Bioscience, Franklin Lakes, NJ USA). FACS analysis was done using a Beckman Coulter CyAn ADP Analyzer (Beckman Coulter, Inc., Brea, CA). Data were analyzed using FloJo (TreeStar, Ashland, OR) software.
Statistical analyses
Data is represented as mean ± standard error of the mean. A student’s t-test, one-way ANOVA with Tukey’s posttest, or two-way ANOVA with Bonferroni posttest were used to determine the statistical probability of differences between experimental cohorts. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ***, p < 0.0001.
Results
Generation of mice lacking Nlrp1b
The mouse Nlrp1 locus contains three closely related genes designated Nlrp1b, Nlrp1c, and Nlrp1a. To generate a mouse lacking NLRP1 function, we took advantage of the fact that, in the 129 mouse, Nlrp1a and Nlrp1c do not encode functional proteins. Therefore, inactivation of the functional Nlrp1b gene is expected to yield an Nlrp1−/− animal. A vector was designed that, upon homologous recombination at the Nlrp1b locus, results in the replacement of the 5′ portion of exon 3 as well as exons 1 and 2 and the promoter region of the gene with a neomycin resistance marker. Using Southern blot analysis, we confirmed the generation and transmission of the mutant Nlrp1b allele (Fig. 1B). The mutant allele was maintained on the 129 genetic background by breeding chimeras generated from the modified ES cells directly to 129S6 mice. The impact of the Nlrp1b mutation was, therefore, studied here in co-isogenic cohorts of animals. No differences were noted in the growth or the behavior or fertility of the Nlrp1b−/− mouse line, and expression analysis indicated that the mutation generated a null allele. Both gross and histological analysis revealed no lesions and no changes in the development. RNA analysis confirmed the absence of Nlrp1a-c transcripts (Fig. 1C).
Figure 1. Generation of a Nlrp1 deficient mouse line.

(A) A schematic of the endogenous Nlrp1b locus, the targeting vector designed to disrupt the gene and the expected structure of the gene after homologous recombination. (B) Southern blot analysis of DNA from ES cells (upper panel) and from tail biopsies of pups generated from the intercross of mice carrying the modified Nlrp1b locus (lower panel) verified correct recombination of the plasmid with the endogenous locus and transmission of this allele through the germline. The expected 9.5kb XbaI fragment is observed in wild type ES cells and in mice carrying a wild type Nlrplb allele. Homologous recombination introduces a novel XbaI site reducing the size of the DNA fragment that binds to the probe to 7.7 kb. (C) RNA was prepared from the jejunum of 129 and Nlrp1b−/− mice and expression of all three Nlrp1 genes was examined by PCR using primers specific for each gene. As expected, 129S6 mice express only Nlrp1b, and this expression was not observed on analysis of RNA prepared from mice homozygous for the mutant allele.
Nlrp1b−/− macrophages are protected from LT induced cell death
Genetic studies indicate that expression of a functional Nlrp1b allele confers sensitivity of 129S6 cells to anthrax lethal toxin (LT)(17). If this is indeed the case, and if the targeting event created a null allele as expected based on RNA analysis of tissue from the Nlrp1b−/− line, then macrophages from mice homozygous for the mutant allele should be resistant to LT. To test this, we first cultured macrophages from the bone marrow (BMDM) of 129S6 (129+/+) and Nlrp1b−/− animals and examined their viability in the presence of increasing amounts of LT. A decrease in the viability of 129+/+ macrophages was noted even upon exposure to very low levels (1 ng/mL) of LT. At doses greater than 10 ng/mL, virtually all cells succumbed to the toxin. In contrast, no decrease in viability of the Nlrp1b−/− cells was observed following exposure to increasing doses of toxin (Fig. 2A).
Figure 2. Lethal toxin mediated cell death and IL-1β and IL-1α cytokine release from macrophages.

(A) BMDMs from 129+/+ and Nlrp1b −/− mice were incubated with LT for 4 hours and cell viability was determined with the WST-1 reagent. (B–D) Thioglycolate-elicited peritoneal macrophages were primed with 100 ng/ml ultra-pure LPS overnight and subsequently incubated for 6 hours in presence of 1 μg/ml LT. Cell death was determined by LDH activity in supernatant and expressed as the percentage of total cellular LDH activity (B). IL-1β (C) and IL-1α (D) release to supernatant was detected by ELISA. n=4 for 129+/+, Nlrp1b −/−, Nlrp3 −/−; n=3 for Casp-1 −/−. * p<0.05, ** p<0.01, *** p<0.001
LT mediated cell death and IL-1β and IL-1α release are caspase-1 dependent but NLRP3 independent
We next determined whether the LT mediated cell death coincided with the release of IL-1β and IL-1α. Thioglycolate-elicited peritoneal macrophages were collected and treated for 12 hour with LPS to induce expression of cytokines prior to exposure to LT. Six hours after the addition of LT, the cell supernatant was collected. LT mediated cell death was assessed by measuring the levels of the cytoplasmic enzyme, lactate dehydrogenase (LDH), released into the medium. IL-1β and IL-1α release were determined by ELISA. LDH levels in the supernatant collected from 129+/+ cultures corresponded to 75% of total cellular LDH content. Only low levels of LDH (0.8%) were detected in the supernatant from Nlrp1b−/− macrophages. Casp−1−/− macrophages also released small amounts of LDH compared to 129+/+ cells (9.7%; Fig. 2B). Although the levels of LDH released by the Casp−1−/− cultures were higher than those observed in the Nlrp1b−/− cultures, these results indicated that cell death is largely NLRP1b and caspase-1 dependent. NLRP1b has been reported to collaborate with other NLR proteins (21). We therefore examined the possibility that, while NLRP1b was required for initiating the response to LT, subsequent recruitment of NLRP3 was required for caspase-1 activation and cell death. To test this, an Nlrp3 deficient, 129S6 co-isogenic mouse line was generated by homologous recombination. Nlrp3−/− thioglycolate-elicited macrophages were collected and treated with LPS followed by LT. No significant difference in the survival of LT-treated 129+/+ and Nlrp3−/− cells was observed (Fig. 2B). Production and release of IL-1β and IL-1α by LT-treated cells were detected by ELISA in supernatants collected from 129+/+, Nlrp1b−/−, Nlrp3−/−, and Casp1−/− macrophages. While levels of IL-1β and IL-1α in the supernatant collected from the LT-treated Nlrp3−/− cells did not significantly differ from LT-treated 129+/+ cells, in Casp1−/− and Nlrp1b−/− cells, the release of IL-1β and IL-1α was attenuated to level similar to PBS-treated controls. (Fig. 2C, D).
NLRP3, not NLRP1b, is essential for MDP and PGN mediated IL-1β release
In vitro assembly of the human NLRP1 inflammasome and activation of caspase-1 can be initiated by exposure of inflammasome components to bacterial peptidoglycans (6). We therefore asked whether NLRP1b was necessary for inflammasome assembly and processing of IL-1β by mouse macrophages in response to peptidoglycan (PGN) or its degradation product muramyl dipeptide (MDP). Peritoneal macrophages were collected from 129+/+, Nlrp1b−/−, Nlrp3−/− and Casp1−/− mice. Cells were treated with MDP and PGN alone or in combination with titanium dioxide (TiO2). TiO2 has been reported to bind to peptidoglycans, increasing their uptake by macrophages (21). Consistent with this report, the release of IL-1β in response to PGN and MDP in the absence of TiO2 was below the level of detection. The addition of TiO2 together with either PGN or MDP resulted in a robust release of IL-1β that was approximately twice that observed with TiO2 alone. However, this response was unaltered in the Nlrp1b−/− cells. Instead, the response to TiO2 alone or TiO2 in combination with MDP or PGN was dependent on NLRP3 expression. As expected, the maturation and release of IL-1β was not observed in Casp1−/− cells (Fig. 3A). To further verify the normal response of Nlrp1b−/− macrophages to MDP, we examined the response of 129+/+, Nlpr1b−/−, Nlrp3−/− and Casp1−/− macrophages to L18-MDP. L-18-MDP is a lipidated form of MDP that improves uptake of the molecule. Again, the response of the peritoneal macrophages to MDP was dependent on NLRP3 but not NLRP1b (Fig. 3B). In vivo studies on 129+/+ and Nlrp1b −/− mice exposed to L18-MDP also failed to support a role for NLRP1b in the response to MDP (Fig. 3C and D).
Figure 3. NLRP1b is not required for IL-1β release induced by MDP or PGN.

(A) Thioglycolate-elicited peritoneal macrophages were primed with 100 ng/ml ultrapure LPS for 6 hours and incubated in OPTIMEM with indicated concentration of TiO2, MDP, PGN or combinations of those stimuli for 16 hours. IL-1β from supernatants was detected by ELISA, n=4. (B) Thioglycolate-elicited peritoneal macrophages were primed as in (A) follow by treatment with 1 mg/ml L18-MDP, 5 mg/ml TiO2 or 5 mg/ml TiO2 and 10 mg/ml MDP for 16 h. IL-1β was detected by ELISA in supernatants, n=4 (C, D) 129+/+ (black) and Nlrp1b −/− (white) mice were injected i.p. with 5 μg LPS. 3 hours later mice were i.p. injected with 50 μg of L18-MDP. Serum and peritoneal lavage fluid (PLF) was collected 1.5 hours postinjection with L18-MDP. The total number of inflammatory cells in peritoneal lavage fluid (60% neutrophils) (C) and IL-1β in the serum and PLF measured by ELISA (D) were determined, n = 13 for 129+/+, 14 for Nlrp1b −/−, # p<0.0001.
In vivo LT mediated processing and release of IL-1β is Nlrp1b-dependent
We next verified that NLRP1b could mediate IL-1β processing in vivo. To test this, pro-IL-1β expression in the lung was induced by intratracheal (i.t.) instillation of LPS at 0 h and followed by LT (LPS/LT) or ATP (LPS/ATP) treatment at 2 h to induce IL-1β processing. Bronchoalveolar lavage (BAL) fluid was collected at 4 h, and levels of mature IL-1β were determined by ELISA. While no IL-1β was observed in the BAL fluid of the vehicle-treated animals, treatment with LPS/ATP resulted in high levels of IL-1β in the lavage fluid of 129+/+ and Nlrp1b−/− animals. LPS/LT treatment resulted in release of IL-1β in 129+/+ mice but not in mice lacking NLRP1b (Fig. 4A). Similarly to IL-1β, the release of IL-1α following LPS/LT treatment was dependent on NLRP1b (Fig. 4B). To verify that the IL-1β present in the BAL fluid represented processed cytokine, proteins were analyzed by western blot. Processed IL-1β as a p17 fragment was detected in the BAL fluid from 129+/+ mice treated with LPS/LT, but not in BAL collected from Nlrp1b−/− animals (Fig. 4C). Consistent with this, amount of processed caspase-1 in the BAL fluid from the Nlrp1b−/− animals treated with LT was substantially reduced compared to that observed in the 129+/+ LT-treated animals (Fig. 4C).
Figure 4. Nlrp1b is required for lethal toxin mediated release of IL-1β and IL-1α in the lung.
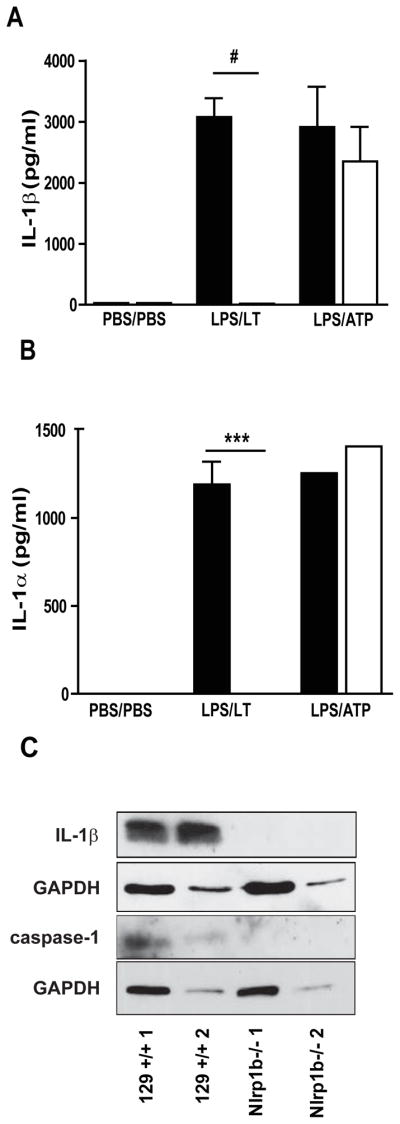
129 +/+ (black) and Nlrp1b −/− (white) mice were treated with 0.3 mg/kg LPS or PBS i.t. at 0 h, and with PBS, 1 μg LT or 50 μl of 100 mM ATP i.t. at 2 h. BAL fluid was collected at 4 h after the initial treatment. IL-1β (A) and IL-1α (B) in the BAL determined by ELISA. (C) Processed IL-1β and caspase-1 proteins from concentrated BAL fluid analyzed by western blot, 20μg of protein per line, 2 representative results from 5 independent experiments shown.*** p<0.001, # p<0.0001, n=1 for PBS, n=10 for LPS/LT, n=4 for LPS/ATP.
Mice lacking NLRP1 are protected from LT mediated acute lung injury
We next asked whether, in addition to altering the processing and release of IL-1β, the absence of NLRP1b would protect mice from the development of LT-induced acute lung injury. 129+/+ and Nlrp1b−/− mice were instilled with LT, and the progression of disease was monitored over 24 hours. LT induced a rapid and progressive disease in the 129+/+ mice that was characterized by a dramatic increase in KC, MIP-2, and HMGB1 levels. A small increase in IL-1β and CXCL15 were also measured in the LT-treated animals (Fig. 5A–E). A parallel increase in the cellularity of the BAL fluid was observed in mice that received the toxin. However, cell numbers in the BAL fluid peaked at 6 hours, and then remained largely unchanged throughout the course of the disease (Fig. 5F). The failure to observe a progressive increase in cell numbers as the health of the mice declined likely reflects a balance between cell death and cell recruitment. Consistent with this interpretation, the BAL fluid collected from animals with more advanced disease contained increasing levels of fragmented cells that were difficult to categorize based on morphological criteria (data not shown). Furthermore, LDH levels in the BAL fluid continued to increase throughout the experiment, while the number of intact cells present in the BAL fluid remained largely unaltered (Fig. 5G). After 24 hours, alterations in lung permeability, determined by measurement of the levels of albumin in the BAL fluid, became apparent (Fig. 5H). Recruitment of cells into the airways of the Nlrp1b−/− mice was significantly lower than that observed in 129+/+ mice. This was consistent with the failure of LT to lead to increase in levels of KC, MIP-2, IL-1β, CXCL15, or HMGB1. The latter is an indication of cell death and has been previously observed in Klebsiella pneumonia infected Nlrp3−/− mice (22). Levels of LDH in the BAL fluid were significantly lower than those observed in the 129+/+ animals and did not significantly increase during the course of the experiments. The permeability of the lungs was not altered, as albumin levels remained low throughout the experiment (Fig. 5A–H). The majority of the cells recruited to the lung after exposure to LT are neutrophils and their recruitment is attenuated in the mice lacking NLRP1 as shown by quantitative analysis of myeloperoxidase levels in cell pellet obtained from the BAL at 48 and 72 h post LT-treatment (5I). Similarly, FACS analysis of single cell suspensions of whole lung revealed that the NLRP1 deficient mice had significantly lower numbers of total lung neutrophils even at this earlier time point (16 h) (5J, K). The recruitment of CCR2+ monocytes has been noted in models of lung injury and inflammation (23). We therefore examined the recruitment of these cells to the lung in single cell preparations of whole lung using antibodies specific for this population. As can be seen in figure 5L–M, inflammatory monocyte numbers were significantly reduce in the lungs of the NLRP1 deficient animals compare to 129+/+.
Figure 5. Nlrp1b−/− mice are protected from lethal toxin-induced lung injury.

129 +/+ (black) and Nlrp1b −/− (white) mice were treated with 20 μg LT i.t. BAL fluid was collected at 0 h, 2 h, 6 h, 16 h, and 24 h after treatment. MIP-2 (A), KC (B), IL-1β (C), and CXCL15/lungkine (D) in the BAL fluid were detected by ELISA. (E) Western blot of HMGB1 protein in concentrated BAL fluid; GAPDH was used as a loading control, 20μg of protein per line. Results shown are representative of 3 independent experiments. (F) The total number of inflammatory cells in BAL fluid. (G) Cell death in BAL fluid assessed by LDH activity. (H) Vascular permeability evaluated as a concentration of albumin in the BAL fluid, measured by colorimetric assay. (I) Myeloperoxidase (MPO) level in BAL cell pellet collected from 129+/+ and Nlrp1b−/− mice 48 and 72 h after LT treatment. (J–N) FACS analysis of whole lung cell suspension from 129+/+ (J, L) and Nlrp1b−/− mice 16 h post-LT treatment (K,M). CD 45+ cells were gated and analyzed for Mac-1 and GR-1 epitope (J, K) or Mac-1 and CCR-2 epitope (L, M). (J–M) representative from 3 independent experiment, quantitative analysis of CD45+Mac-1+CCR2+ cells (N). n=2 per genotype at 0h, n=5 per genotype at 2h, n=6 per genotype at 6h, n=3 per genotype at 16h, n=7 per genotype at 24–72 h. * p < 0.05, ** p < 0.01, *** p < 0.001.
Progression of Acute Lung injury in Nlrp1+/+ animals
To further compare the progression of lung disease in 129+/+ and Nlrp1b−/− animals, we evaluated changes in the structure of the lungs collected from mice at various time points after LT exposure. Histological changes were assessed by analysis of hematoxylin-eosin stained tissue sections prepared from the left lobe. As expected, no difference was observed in lungs collected from vehicle-treated Nlrp1−/− and 129+/+ animals (Fig. 6A and E). Exposure to LT induced diffuse lung injury, and the extent of the damage became more apparent in samples collected at later time points, especially 72 h after exposure to LT. Visualization of whole lung sections showed diffuse alveolar damage and evidence of hemorrhaging in multiple regions of the lobe (Fig. 6B–D). Neutrophils and macrophages could be detected in the alveolar walls and interstitial spaces surrounding the alveolar airways (Fig. 6I). However, consistent with the cellularity of the BAL fluid, the number of cells in the alveoli did not increase notably as the injury progressed. Deposition of proteinous fibers in the airways was observed, consistent with significant trapping of red blood cells within the alveolar spaces (Fig. 6J, K). While inflammatory infiltrates were observed in the lymphatics, the airway epithelial cells remained largely intact. Remarkably, lungs of Nlrp1b−/− mice were largely spared from injury (Figs. 6F–H). Some increase in inflammatory cell numbers was observed at 48 and 72 h but was less extensive compared to 129+/+ mice. The airways and alveolar architecture were maintained, and no hemorrhaging was observed even at 72 h (Fig. 6L–N). Consistent with these histological changes, 129+/+ mice exhibited significant inflammation, compromised of barrier function, lethargy, and decline in body temperature, and displayed morbidity requiring euthanasia (data not shown). In contrast, mice lacking NLRP1b were largely protected from the sequel of events that followed exposure of the lung to LT. Even at 72 h post-treatment, Nlrp1−/− mice appeared normal and could not be distinguished from untreated littermates.
Figure 6. Histological analysis of lethal toxin-induced lung injury.

Micrographs of H&E-stained sections of lungs harvested from 129+/+ and Nlrp1b−/− mice after treatment with 20 μg LT i.t. Lungs were harvested at the indicated times (A) – (H), at 48 h (I, L) or at 72 h in (J, K, M and N). Scale bars represent 250 μm for (A – H) and 10 μm for (I – N). Original magnification was 2 x for (A – H) and 40x for (I – N).
LT mediated lung injury is dependent upon caspase-1 activation
Since exposure of the mouse airways to LT was accompanied by a modest, NLRP1b-dependent increase in IL-1β measured in the BAL fluid (Fig. 5C), we further assessed the contribution of this NLRP1b pathway to the development of acute lung disease. We generated cohorts of mice that expressed the 129+/+ 129S6 Nlrplb gene, which confers the ability to respond to LT, but lacked Casp-1 or Il1β genes. The 129S6 mouse also carries a mutant Casp-4 (alias Casp-11) (24). Thus all the animals included in the study also lack expression of Casp-4. These mice and their Casp-1 +/+ or Il1β +/+ littermates were exposed i.t. to LT, and the development of lung disease in the two groups of animals was compared. If the lung disease observed in the LT-treated animals is the consequence of pyroptosis, a caspase-1 mediated cell death, we would expect that the Casp−1−/− mice would be protected from the toxin. Indeed, the lack of caspase-1 protected the mice from LT induced lung inflammation. Inflammatory cell recruitment was reduced in Casp-1−/− mice exposed to LT compared to Casp-1+/+ mice (Fig. 7A). Cell death was also significantly reduced in Casp-1−/− mice (Fig. 7B), as expected based on our in vitro analyses of Casp-1−/− macrophages (Fig. 2B). Correspondingly, vascular permeability was less severe in Casp-1−/− mice compared to Casp-1+/+ mice, although the difference did not reach significance by 72 h (Fig. 7C). Levels of the pro-inflammatory mediators KC and CXCL15 were also significantly lower in Casp-1 deficient mice (Fig. 7D and E). In contrast, the lack of IL-1β failed to protect mice from the development of disease. No significant differences in inflammation or disease pathology were observed between Il-1b+/+ and Il-1b−/− mice (Fig. 7F and G and data not shown).
Figure 7. Lethal toxin-induced lung injury is dependent on caspase-1 but not IL-1β.

(A–E) Casp-1+/+ (black) and Casp-1 −/− (shade) mice were treated with PBS as a vehicle or 20 μg LT i.t. BAL fluid was collected at 72 h after treatment. (A) The total number of inflammatory cells collected in the BAL fluid. (B) Cell death in BAL fluid determined by LDH activity in supernatants and expressed as the percentage of total cellular LDH activity. (C) Vascular permeability evaluated as a concentration of albumin in the BAL fluid, measured by colorimetric assay. KC (D) and CXCL15/lungkine (E) in the BAL fluid were detected by ELISA. n = 2 mice per genotype for vehicle treatment and n = 10 mice per genotype for LT treatment. (F, G) Il1β+/+ (black) and Il1β−/−(striped) mice were treated as above. (F) The total number of inflammatory cells collected in BAL fluid. (G) The percentage of weight lost 72 h after treatment. n= 2 per genotype for vehicle treatment and n = 9 mice per genotype for LT treatment. * p < 0.05; ** p < 0.01; # p < 0.0001.
LT induces pyroptosis of alveolar macrophages
We next examined whether caspase-1 activity was critical in the initiation and progression of lung disease in the 129+/+ LT-treated animals. Mice were treated with LT, and cells present in the airways were collected by BAL at 2 hours after treatment. The viability of the cells was determined by staining with 7-AAD. Over 50% of the cells collected from 129+/+ animals were non-viable based on this criterion, while greater than 95% of the cells collected from the Nlr1b−/− or Casp-1−/− mice failed to take up the dye (Fig. 8A). Thus, again, Casp-1−/− and Nlrp1b−/− cells were protected from cell death induced by LT. To determine if catalytic activity of caspase-1 mediated cell death in 129+/+ animals, we stained the airway cells collected by BAL with the fluorescently-labeled caspase-1 preferring substrate and inhibitor, FAM-YVAD-FMK. This inhibitor binds irreversibly to activated caspase-1. Nearly all the cells collected from the 129+/+ animals were stained with the caspase-1 inhibitor, while no significant change in the mean fluorescent intensity was measured in the cells collected from similarly treated Nlrp1b−/− animals. As expected, no caspase-1 activity was detected in cells collected in the BAL fluid of Casp-1−/− mice. We next determined whether LT either directly or indirectly resulted in activation of caspase-3. Only a small shift in the mean fluorescent intensity was observed in BAL cells from 129+/+ animals, but no cells from Casp-1−/− or Nlrp1b−/− animals were stained with the caspase-3 preferring fluorescent inhibitor FAM-DVED-FMK (Fig. 8B). Caspase-1 activation in macrophages can occur downstream of cell death in response to the leakage of some of the intracellular contents from dying cells. For example, the accumulation of ATP and/or uric acid in the extracellular milieu stimulates caspase-1 activity in nearby cells, and this activation is dependent upon NLRP3, but not NLRP1 (25–27). However, we did not observe any attenuation of lung disease due to the absence of the NLRP3 inflammasome in Nalp3−/− mice (data not shown), suggesting that, similar to LT induced cell death in macrophages in vitro (Fig. 2), LT induced pyroptosis in alveolar macrophages is NLRP1b and caspase-1 dependent but NLRP3 independent. Similarly, loss of P2X7 did not alter the development of severe lung disease after Nlrp1b activation.
Figure 8. Lethal toxin activates caspase-1 in the lung independently on P2X7 receptor.

(A–B) 129+/+, Nlrp1b−/−, and Casp-1−/− mice were treated with 20 μg of LT by i.t. BAL fluid was collected 2 h after treatment, and cells were stained with fluorescently labeled caspase inhibitors and with 7-Amino-Actinomycin (7-AAD), n=4 per genotype. Cells were analyzed on FACS and compared to untreated controls. (A) Data is expressed as the difference in median fluorescent intensity (MFI) in the LT-treated mice and untreated mice. (B) A representative overlay of the untreated (gray filled) and LT-treated (black line) cell populations. (C–F) BALB/c or P2X7−/− mice were treated with 20 μg LT i.t. BAL fluid was collected at 72 h after treatment. (C) The total number of inflammatory cells collected in the BAL fluid. (D) Vascular permeability evaluated as a concentration of protein in the BAL fluid, measured by BCA assay. (E) Cell death in BAL fluid determined by LDH activity in supernatants. (F) KC in the BAL fluid was detected by ELISA, n=5 mice per genotype. * p < 0.05; ** p < 0.01 ***p<0.001 compared to WT.
Discussion
Early studies using human cell free reconstitution showed that NLRP1, in the presence of muramyl dipeptide (MDP), could form a complex with caspases-1 capable of augmenting autoactivation of the caspase and subsequent cleavage of pro IL-1β into its active form (6). ASC enhanced this activity but was not essential for caspase-1 cleavage. The requirement for MDP to initiate the assembly of the complex suggested that at least one of the functions of the NLRP1 inflammasome was intracellular surveillance for this bacterial cell wall component. These initial observations linking MDP and the NLRP1 inflammasome were supported by indirect evidence collected from a number of studies that followed. Mouse macrophages lacking NOD2 failed to produce IL-1β in response to MDP, clearly defining a role for this protein in the response to MDP (21). This observation was consistent with earlier studies that showed that introduction of NOD2 into cell lines allowed these cells to respond to MDP (28, 29). To reconcile this with the cell free studies showing MDP dependent assembly of the NLRP1 inflammasome, IL-1β production in response to MDP was examined in the human monocyte line, THP-1, as well as HEK293T cells transfected with various components of the inflammasome. In this study, complicity between NOD2 and NLRP1 in IL-1β release was supported by demonstrating association of NLRP1 with NOD2 using co-immunoprecipitation strategies after MDP exposure as well as demonstration of a decrease in release of IL-1β after RNAi knockdown of NLRP1 expression. MDP has also been used to stimulate NLRP1 inflammasome assembly in studies focused on identifying regulators of NLRP1 activity, again using THP-1 cells. While NLRP1 dependent maturation and release of IL-1β is also reported in this study, interpretation of these studies is complicated by the treatment of cells with ATP, a potent stimulator of NLRP3 inflammasome assembly (4).
Species conservation in the ability of a particular PAMP to trigger a specific inflammasome has generally been observed, albeit in the case of humans these studies are heavily dependent on use of the THP-1 cell line. We were, therefore, surprised to observe no difference in IL-1β release between wild type and NLRP1 deficient cells in response to MDP. This was true even when, as in previous studies, we utilized TiO2 to increase uptake of MDP by peritoneal macrophages. Lack of NLRP1 did not alter the response to L18-MDP, a derivative with improved cellular uptake. In vivo studies also failed to define a role for NLRP1 in the response to this bacterial wall component. In all of these models, increase in IL-1β release was entirely dependent on Nlrp3 expression. No significant increase in release of IL-1β was observed from macrophages isolated from the 129 Nlrp1−/− co-isogenic mice after stimulation with MDP, and Nlrp3−/− mice were protected from elevations in serum IL-1β after MDP exposure. Thus our results support a model in which NLRP3 rather than NLRP1, alone or in cooperation with NOD2, mediates IL-1β processing and release in response to MDP. Our findings are consistent with a previous study examining the response of NOD2 and NLRP3 deficient bone marrow derived macrophages to MDP (30). No increase in IL-1β release was observed after stimulation of either Nod2−/− or Nlrp3−/− cells, suggesting a possible collaboration between these two NLRs in caspase-1 activation and maturation in response to MDP exposure. These findings together with our results raise the possibility that there are species differences in assignment of functions to inflammasomes: the importance of NLRP3 in the response to MDP being limited to mouse, with NLRP1 carrying out the orthologous function in human cells. However this interpretation is not consistent with previous studies using a human ectopic expression system in which MDP was shown to stimulate the NLRP3 (31). Furthermore, Martionon et al show that cells from Muckle-Wells patients, who carry gain of function mutations in NLRP3, display increased IL-1β secretion in the presence of MDP. Taken together, our study and the differences between previous reports, depending on the system used in the study, suggest that the role of NLRP1 in the response to MDP needs to be re-evaluated.
Given the failure of MDP to stimulate the inflammasome, we used anthrax lethal toxin (LT) to define events following NLRP1 inflammasome assembly in peritoneal macrophages and in vivo in the lung. Consistent with previous observations (32), the most dramatic consequence of exposure of macrophages to LT was cell death as monitored by increase in cellular dehydrogenase levels in vitro in culture supernatant. In vivo this was manifested as an increase in LDH in the BALF collected from LT treated animals. When cells were treated with LPS prior to exposure to LT, IL-1β release was also observed. Caspase-1 deficient cells and Casp1−/− mice are largely protected. As 129 mice lack a functional Casp-4 (formerly Casp-11) gene, it is clear that activation of caspase-1 by the NLRP1 inflammasome is sufficient for both IL-1β processing and cell death (24). The dependence of cell death on caspase-1 and the accompanying release of cytokines indicate that NLRP1b activation can lead to macrophage pyroptosis. Activation of this pathway is also observed in vivo. Exposure of the mouse airways to LT resulted in activation of caspase-1 in alveolar macrophages, resulting in cell death. Again, the accompanying increase in IL-1β release was only observed if the animals had been exposed to LPS, which stimulates transcription of this cytokine.
The cellular events critical for engagement of the NLRP1 inflammasome that follow LT exposure have not been defined, nor is it clear why the Nlrp1b allele carried by a number of inbred mouse lines, including BALB/c and 129, is uniquely activated by this toxin. LT is a zinc metalloproteinase, and cleavage of MEK by this proteinase and subsequent alteration in the activity of ERK, JnK and p38 pathways has been documented by a number of groups (33–36). These events do not distinguish the LT resistant strains such as C57BL/6 from LT sensitive strains such as 129. LT inactivation of MEK occurs equally in both 129 and C57BL/6 cells, even though the latter cells do not undergo rapid pyroptosis, but rather after an extended period of time begin to undergo apoptosis (35, 37). Interestingly, it was recently discovered that the N-terminal domain of NLRP1 from LT- sensitive strain is cleaved by LT and that the cleavage is essential for NLRP1 inflammasome activation, caspase-1 activation, IL-1β production and pyroptotic macrophage death. This observation suggests that allelic differences lead to alterations in the sensitivity of NLRP1 to cleavage by proteases. In this case the 129 and BALB alleles show dramatic increase in sensitivity to lethal toxin, but it is equally likely that the many common coding variants in the mouse and human gene alter the sensitivity of NLRP1 to other proteases, thus modifying the activation of this inflammasome in response to various innate stimuli (38),(39). It is apparent that, once assembly of the Nlrp1b inflammasome is triggered, it leads to cellular events that mimic those of other inflammasomes, namely caspase-1 recruitment and autoactivation and IL-1β maturation and release. Cell death requires caspase-1. However we cannot rule out the possibility that the other activities of LT leave the cells vulnerable to pyroptosis.
Similarly to LT exposure, infection with virulent strains of Bacillus anthracis results in caspase-1 activation and IL-1β production and release. However, contrary to LT treatment, caspase-1 as well as IL-1β protects infected animals against B. anthraxis infection (40). Although it was shown that LT is a major virulence factor during B. anthracis infection, other virulence factor are probably involved during infection with B. anthracis compare to sterile induction of inflammation by LT. This is supported by recent data, showing that mice lacking major components of other inflammasomes (Nlrc4) and receptor for MDP (Nod2) are critical for protection from virulent B. anthracis infection (40).
In the lung, the activation of NLRP1 has catastrophic consequences, resulting in changes that mimic some aspects of acute respiratory distress (ARDS)(41). A notable feature of this inflammation is that, unlike many other sterile models of ARDS disease, NLRP1 activation results in progressive damage to the lung. In contrast, exposure of mouse lungs to endotoxin results in a rapid induction of chemokines followed by an influx of neutrophils/monocytes and leukocytes. However the inflammation rapidly resolves, and the majority of animals show no evidence of disease at 72 hours after treatment. NLRP1 activation also results in a rapid induction of chemokines, including those most relevant for neutrophil recruitment KC (CXCL1) and MIP-2 (42). Lungkine, (CXCL15) which is expressed at high levels by lung airway epithelial cells (43) and is protective in bacterial pneumonia is also found at higher levels in the inflamed lung (44). However, unlike LPS induced models of lung injury, damage continues to accumulate, with visible lesions apparent upon necropsy 72 hours after NLRP1 activation. It is likely, therefore, that the initial pyroptosis of the resident macrophages initiates events that overwhelm the homeostatic mechanisms of the lung. Consistent with this, the dramatic loss in barrier function becomes apparent long after the initial activation of the inflammasome by LT. An important clue to the mechanism(s) leading to organ failure is the steady increase in LDH levels in the BALF. This indicates that after the initial NLRP1 dependent pyroptosis, cell death in the lungs continues and in fact increases. Susceptible cells may include structural cells of the lung and cells recruited by the initial pyroptosis of resident macrophages, neutrophils and monocytes. Consistent with pyroptosis of resident cells high levels of HMGB1 are observed in the BAL immediately after exposure to LT. Elevated levels observed at later time points with continued detection of IL-1β suggest that pyroptosis continues. In these cases it is possible that cell death is mediated by DAMPs (damage associated molecular patterns) released when the cells undergo the NLRP1 dependent pyroptosis in response to LT.
Although it is well established that IL-1β can induce inflammation by stimulating resident immune cells to produce chemokines (45), the IL-1β observed in this model does not contribute significantly to the development of ARDS like disease. In contrast, the lungs of the Casp1−/− mice were protected, suggesting that caspase-1 activation and the resulting cell death, rather than IL-1β processing, is the critical outcome of NLRP1 activation in this model. We have not yet identified the critical factors/cytokines released by the initial NLRP1 dependent pyroptosis that lead to escalation in lung injury. ATP is released upon cell death and could lead to activation of the NLRP3 inflammasome in neighboring cells, inducing further cytokine release and pyroptosis. A role for this pathway is not supported by the observation that neither the Nlrp3−/− or the P2X7−/− mice were protected from lung damage. It also makes it unlikely that uric acid released from dying cells contributes to further tissue damage, as this DAMP has also been shown to rely on the NLRP3 inflammasome. HMGB1 released from pyroptotic cells can induced acute inflammation characterized by increased cytokine production and neutrophil recruitment (46). In animal models of systemic injury, inhibition of HMGB-1 is reported to attenuate pulmonary endothelial barrier dysfunction. Thus it is possible that release of this intracellular protein contributes to loss of vascular integrity, marked by hemorrhaging and increase in serum proteins in the airways, observed in this NLRP1 dependent model of ARDS. Further studies using specific inhibitors of factors released upon initial activation of the NLRP1 inflammasome as well as mice in which NLRP1 expression is limited to specific cell populations should allow further definition of the events that lead from initial activation of NLRP1 to acute and progressive lung disease.
Acknowledgments
This work was supported by the National Institutes of Health grant U19 A1077437 (to B.H.K.)
We thank Anne Latour for embryonic stem cell work, Darren Hursey for molecular biology, and Rebecca Dye, Peter Repenning, and Will Barker for assistance in breeding lines and genotyping mice.
References
- 1.Hlaing T, Guo RF, Dilley KA, Loussia JM, Morrish TA, Shi MM, Vincenz C, Ward PA. Molecular cloning and characterization of DEFCAP-L and -S, two isoforms of a novel member of the mammalian Ced-4 family of apoptosis proteins. J Biol Chem. 2001;276:9230–9238. doi: 10.1074/jbc.M009853200. [DOI] [PubMed] [Google Scholar]
- 2.Chu ZL, Pio F, Xie Z, Welsh K, Krajewska M, Krajewski S, Godzik A, Reed JC. A novel enhancer of the Apaf1 apoptosome involved in cytochrome c-dependent caspase activation and apoptosis. J Biol Chem. 2001;276:9239–9245. doi: 10.1074/jbc.M006309200. [DOI] [PubMed] [Google Scholar]
- 3.Yuan JY, Horvitz HR. The Caenorhabditis elegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev Biol. 1990;138:33–41. doi: 10.1016/0012-1606(90)90174-h. [DOI] [PubMed] [Google Scholar]
- 4.Bruey JM, Bruey-Sedano N, Luciano F, Zhai D, Balpai R, Xu C, Kress CL, Bailly-Maitre B, Li X, Osterman A, Matsuzawa S, Terskikh AV, Faustin B, Reed JC. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell. 2007;129:45–56. doi: 10.1016/j.cell.2007.01.045. [DOI] [PubMed] [Google Scholar]
- 5.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 6.Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I, Reed JC. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25:713–724. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 7.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nunez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miao EA, I, Leaf A, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sutterwala FS, Ogura Y, Zamboni DS, Roy CR, Flavell RA. NALP3: a key player in caspase-1 activation. J Endotoxin Res. 2006;12:251–256. doi: 10.1177/09680519060120040701. [DOI] [PubMed] [Google Scholar]
- 10.Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, Grant EP, Nunez G. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 11.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol. 2006;7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 12.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L, Rojanasakul Y, Stehlik C. An NLRP7-Containing Inflammasome Mediates Recognition of Microbial Lipopeptides in Human Macrophages. Immunity. 2012 doi: 10.1016/j.immuni.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis BK, Roberts RA, Huang MT, Willingham SB, Conti BJ, Brickey WJ, Barker BR, Kwan M, Taxman DJ, Accavitti-Loper MA, Duncan JA, Ting JP. Cutting edge: NLRC5-dependent activation of the inflammasome. J Immunol. 2011;186:1333–1337. doi: 10.4049/jimmunol.1003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 18.Quintiliani R, Jr, Quintiliani R. Inhalational anthrax and bioterrorism. Curr Opin Pulm Med. 2003;9:221–226. doi: 10.1097/00063198-200305000-00011. [DOI] [PubMed] [Google Scholar]
- 19.Moayeri M, Haines D, Young HA, Leppla SH. Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J Clin Invest. 2003;112:670–682. doi: 10.1172/JCI17991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Solle M, Labasi J, Perregaux DG, Stam E, Petrushova N, Koller BH, Griffiths RJ, Gabel CA. Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem. 2001;276:125–132. doi: 10.1074/jbc.M006781200. [DOI] [PubMed] [Google Scholar]
- 21.Hsu LC, Ali SR, McGillivray S, Tseng PH, Mariathasan S, Humke EW, Eckmann L, Powell JJ, Nizet V, Dixit VM, Karin M. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci U S A. 2008;105:7803–7808. doi: 10.1073/pnas.0802726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Willingham SB, I, Allen C, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ, Duncan JA, Ting JP. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol. 2009;183:2008–2015. doi: 10.4049/jimmunol.0900138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nature reviews Immunology. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 24.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 25.Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327:296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- 26.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 27.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 28.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, Foster SJ, Moran AP, Fernandez-Luna JL, Nunez G. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 29.Girardin SE, I, Boneca G, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 30.Pan Q, Mathison J, Fearns C, Kravchenko VV, Da Silva Correia J, Hoffman HM, Kobayashi KS, Bertin J, Grant EP, Coyle AJ, Sutterwala FS, Ogura Y, Flavell RA, Ulevitch RJ. MDP-induced interleukin-1beta processing requires Nod2 and CIAS1/NALP3. J Leukoc Biol. 2007;82:177–183. doi: 10.1189/jlb.1006627. [DOI] [PubMed] [Google Scholar]
- 31.Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14:1929–1934. doi: 10.1016/j.cub.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 32.Friedlander AM. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J Biol Chem. 1986;261:7123–7126. [PubMed] [Google Scholar]
- 33.Klimpel KR, Arora N, Leppla SH. Anthrax toxin lethal factor contains a zinc metalloprotease consensus sequence which is required for lethal toxin activity. Mol Microbiol. 1994;13:1093–1100. doi: 10.1111/j.1365-2958.1994.tb00500.x. [DOI] [PubMed] [Google Scholar]
- 34.Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, Vande Woude GF. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 35.Park JM, Greten FR, Li ZW, Karin M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 2002;297:2048–2051. doi: 10.1126/science.1073163. [DOI] [PubMed] [Google Scholar]
- 36.Vitale G, Pellizzari R, Recchi C, Napolitani G, Mock M, Montecucco C. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem Biophys Res Commun. 1998;248:706–711. doi: 10.1006/bbrc.1998.9040. [DOI] [PubMed] [Google Scholar]
- 37.Muehlbauer SM, Evering TH, Bonuccelli G, Squires RC, Ashton AW, Porcelli SA, Lisanti MP, Brojatsch J. Anthrax lethal toxin kills macrophages in a strain-specific manner by apoptosis or caspase-1-mediated necrosis. Cell Cycle. 2007;6:758–766. doi: 10.4161/cc.6.6.3991. [DOI] [PubMed] [Google Scholar]
- 38.Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH, Moayeri M. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS pathogens. 2012;8:e1002638. doi: 10.1371/journal.ppat.1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frew BC, V, Joag R, Mogridge J. Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS pathogens. 2012;8:e1002659. doi: 10.1371/journal.ppat.1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ali SR, Timmer AM, Bilgrami S, Park EJ, Eckmann L, Nizet V, Karin M. Anthrax toxin induces macrophage death by p38 MAPK inhibition but leads to inflammasome activation via ATP leakage. Immunity. 2011;35:34–44. doi: 10.1016/j.immuni.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44:725–738. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17:293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rossi DL, Hurst SD, Xu Y, Wang W, Menon S, Coffman RL, Zlotnik A. Lungkine, a novel CXC chemokine, specifically expressed by lung bronchoepithelial cells. J Immunol. 1999;162:5490–5497. [PubMed] [Google Scholar]
- 44.Chen SC, Mehrad B, Deng JC, Vassileva G, Manfra DJ, Cook DN, Wiekowski MT, Zlotnik A, Standiford TJ, Lira SA. Impaired pulmonary host defense in mice lacking expression of the CXC chemokine lungkine. J Immunol. 2001;166:3362–3368. doi: 10.4049/jimmunol.166.5.3362. [DOI] [PubMed] [Google Scholar]
- 45.Ulich TR, Yin SM, Guo KZ, del Castillo J, Eisenberg SP, Thompson RC. The intratracheal administration of endotoxin and cytokines. III. The interleukin-1 (IL-1) receptor antagonist inhibits endotoxin- and IL-1-induced acute inflammation. Am J Pathol. 1991;138:521–524. [PMC free article] [PubMed] [Google Scholar]
- 46.Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMG-1 as a mediator of acute lung inflammation. J Immunol. 2000;165:2950–2954. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]