

Background: Malonyl-CoA, a key molecule playing central roles in fatty acids metabolism, is associated with human diseases such as malonic aciduria.
Results: Crystal structures and functional data were determined for human malonyl-CoA decarboxylase (MCD), responsible for malonyl-CoA decarboxylation.
Conclusion: MCD catalytic activity combines half-of-the-sites reactivity with positive cooperativity modulated by intersubunit disulfide bridges.
Significance: Structural and functional information on MCD provides elements for a better understanding of the associated pathologies.
Keywords: Acetyl Coenzyme A, Fatty Acid Oxidation, Lipid Metabolism, Mitochondria, Peroxisomes, Half-of-the-sites Reactivity, Malonic Aciduria, Malonyl-CoA, Malonyl-CoA Decarboxylase
Abstract
Decarboxylation of malonyl-CoA to acetyl-CoA by malonyl-CoA decarboxylase (MCD; EC 4.1.1.9) is an essential facet in the regulation of fatty acid metabolism. The structure of human peroxisomal MCD reveals a molecular tetramer that is best described as a dimer of structural heterodimers, in which the two subunits present markedly different conformations. This molecular organization is consistent with half-of-the-sites reactivity. Each subunit has an all-helix N-terminal domain and a catalytic C-terminal domain with an acetyltransferase fold (GNAT superfamily). Intersubunit disulfide bridges, Cys-206–Cys-206 and Cys-243–Cys-243, can link the four subunits of the tetramer, imparting positive cooperativity to the catalytic process. The combination of a half-of-the-sites mechanism within each structural heterodimer and positive cooperativity in the tetramer produces a complex regulatory picture that is further complicated by the multiple intracellular locations of the enzyme. Transport into the peroxisome has been investigated by docking human MCD onto the peroxisomal import protein peroxin 5, which revealed interactions that extend beyond the C-terminal targeting motif.
Introduction
Malonyl-CoA is a key metabolite (Fig. 1A) in the synthesis, degradation, and regulation of fatty acids. It is the direct precursor of most of the carbon atoms of fatty acids (i.e. 14 of 16 carbons in palmitic acid), and its availability is the rate-determining factor of fatty acid biosynthesis. It is a degradation product of odd chain-length dicarboxylic fatty acids (1), and it inhibits the uptake of long chain fatty acids into mitochondria by carnitine acyltransferase 1, thereby inhibiting β-oxidation (Fig. 1B) (2). Mechanisms to modulate the levels of a molecule like malonyl-CoA with such a multiplicity of pivotal roles are expected, most likely involving both its synthesis and degradation. Synthesis (acetyl-CoA + ATP + CO2 → malonyl-CoA + ADP + Pi) is catalyzed by acetyl-CoA carboxylase, an enzyme that is regulated by a diversity of mechanisms, including feedback inhibition by palmitoyl-CoA and activation by citrate and by reversible phosphorylation in response to hormones. Most of malonyl-CoA is utilized by fatty acid synthase, but it can also be degraded through decarboxylation (malonyl-CoA → acetyl-CoA + CO2) catalyzed by malonyl-CoA decarboxylase (MCD).2 Surprisingly, especially in light of the potential futile cycle created by acetyl-CoA carboxylase and MCD, little is known about the mechanisms involved in the regulation of MCD. It is expected that MCD is also subject to regulation, but a clear picture has not yet been reported.
FIGURE 1.
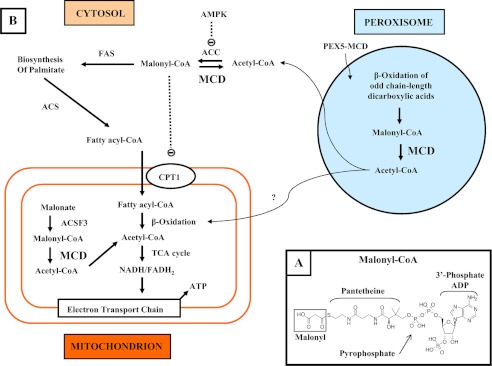
Roles of MCD in different cell compartments. A, structure of malonyl-CoA. B, levels of malonyl-CoA are regulated in the cytosol by the concerted activities of acetyl-CoA carboxylase (ACC), MCD, and AMP-activated protein kinase (AMPK). Malonyl-CoA is a substrate for fatty acid synthesis via fatty acid synthase (FAS) and acyl-CoA synthetase (ACS) and can also inhibit the carnitine acyltransferase CPT1, a key enzyme for the translocation of fatty acids to the mitochondria matrix. In mitochondria, acetyl-CoA fatty acids are oxidized in the β-oxidation and TCA pathways to produce ATP. Malonyl-CoA synthesized from malonate by ACSF3 (acyl-CoA synthetase family member 3) is decarboxylated by MCD to acetyl-CoA for entry into the TCA cycle. In peroxisomes, acetyl-CoA is also produced during the β-oxidation of odd chain-length fatty acids (light blue).
MCD is widely distributed in organisms ranging from bacteria to plants and mammals (1, 3–6), and in humans, it has been identified in heart, skeletal muscle, pancreas, liver, and kidney (1). In addition, MCD shows a broad intracellular distribution, including the cytoplasm, mitochondria, and peroxisomes of both human (1) and rat (7) liver cells. MCD deficiency in humans causes severe phenotypic consequences, including malonic aciduria, developmental delay, cardiomyopathy, hypoglycemia, and episodes of organic aciduria (2), whereas increased levels of malonyl-CoA caused by MCD inhibition in breast cancer cells induce cytotoxicity (9). Consistent with the observed mitochondrial and peroxisomal locations, the predicted sequence of human MCD contains both an N-terminal mitochondrial targeting sequence (Met-1–Ala-39) and a type 1 peroxisomal targeting signal (PTS1) in the C-terminal tripeptide (Ser-491–Lys-492–Leu-493). In rat tissues, the mitochondrial enzyme has a molecular mass of 54 kDa, whereas the peroxisomal and cytosolic isoforms have a molecular mass of 49 kDa (10). The homologous enzymes from rat, cow, and goose show 89, 83, and 71% sequence identities to human MCD, respectively.
Here, we report the crystal structure of human peroxisomal MCD. The structure reveals what at first glance appears to be a homotetrameric protein, but which in fact is a dimer of structural heterodimers. This organization and biochemical data are consistent with an enzyme subject to half-of-the-sites reactivity. Even greater regulatory complexity is suggested by the presence of disulfide bonds connecting the four subunits, which are responsible for the positive cooperativity exhibited by MCD under oxidative conditions.
EXPERIMENTAL PROCEDURES
Cloning and Mutagenesis
The open reading frame of human MCD in the cDNA clone MGC3193 (RZPD GmbH) was PCR-amplified using oligonucleotides 5′-ggggacaagtttgtacaaaaaagcaggcttcgaaaacctgtattttcagggcagcggcgcgatggacgagctgctgcgccgc-3′ (forward) and 5′-ggggaccactttgtacaagaaagctgggtgttatcagagcttgctgttcttttg-3′ (reverse) and subcloned into pET-DEST-42-N112 (EMBL Hamburg) using the Gateway recombination system (Invitrogen) to generate pMCD. Oligonucleotides 5′-cgggttacctggcattcaccgagcgaagtgcttcagaaaatcaggaggc-3′ (forward) and 5′-gcctcactgattttctgaagcacttcgctcggtgaatgccaggtaacccg-3′ (reverse) (C206S) and oligonucleotides 5′-tacagaaggtgttacttcttttctcacagctcgacccctggggagcccct-3′ (forward) and 5′-aggggctccccaggggtcgagctgtgagaaaagaagtaacaccttctgta-3′ (reverse) (C243S) were purchased from Roche Applied Science and used to mutate the MCD clone with the In-Fusion® system (Clontech) to generate plasmids pC206S, pC243S, and pC206S/pC243S, which were used to clone the amplified sequences in the pOPINM plasmid, the sequences of which were confirmed.
Expression and Purification
The maltose-binding protein (MBP)-MCD fusion protein was expressed in Escherichia coli BL21(DE3) in minimal medium (containing selenomethionine) (11). For the MBP-MCD variants, overexpression in Superior Broth (Molecular Dimensions) was induced at mid-log growth phase (A600 = 0.6) using 0.3 mm isopropyl β-d-thiogalactopyranoside at 17 °C. The cells were harvested and lysed at 4 °C by sonication in lysis buffer consisting of 1× PBS (pH 7.4), 10 mm DTT, and cOmplete EDTA-free protease inhibitor mixture (Roche Applied Science). The cell lysate was clarified by centrifugation and loaded onto a MBPTrap HP affinity column (GE Healthcare). After extensive washing with lysis buffer, the MBP-MCD fusion protein was eluted with lysis buffer containing 10 mm maltose. The MBP fusion protein was cleaved in a 1:50 mixture of fusion protein and tobacco etch virus protease at 20 °C overnight and fractionated on a Superdex 200 16/60 GL column (Amersham Biosciences) pre-equilibrated with 1× PBS (pH 7.4) and 10 mm DTT. The fractions containing MCD protein were collected; transferred to 50 mm Bistris (pH 6.5), 25 mm NaCl, and 10 mm DTT; and loaded onto a Mono Q 5/50 GL column (Amersham Biosciences). After elution with a linear gradient of NaCl, the purified protein was concentrated to 6 mg/ml in 50 mm Bistris propane (pH 6.5) and 150 mm NaCl. The purification of the variants differed only in the omission of DTT and the use of PreScission as the protease for MBP cleavage.
Crystallization, Data Collection, and Phasing
The sitting drop vapor diffusion method was used for crystal screening with commercial screening kits (Hampton Research) by mixing 100 nl of concentrated selenomethionine-labeled MCD and 100 nl of reservoir solution dispensed with a Cartesian robot at 4 and 20 °C. Triclinic crystals (space group P1) obtained in 15% PEG 2000 monomethyl ether and 0.1 m sodium acetate (pH 4.5) were optimized with 10 mm sodium citrate in 24-well plates. Data from these crystals at 3.29 Å (see Table 1), collected at European Radiation Synchrotron Facility beamline ID29 (Grenoble, France), were processed with XDS (12) and TRUNCATE (13), although attempts to find the selenium atoms were unsuccessful. Self-rotation showed three binary axes at 90° from each other, which appeared to indicate one molecular tetramer (with D2 symmetry) per asymmetric unit. However, when the structure was solved, it was found that the asymmetric unit contains in fact two molecular tetramers with a volume solvent of ∼55%. The two molecules are close to parallel, but not completely, which could explain both the self-rotation and the absence of a pseudo origin peak in the native Patterson map.
TABLE 1.
Data collection and refinement statistics obtained by single anomalous diffraction
os, outer shell; is, inner shell; r.m.s.d., root mean square deviation.
| Data collection | ||
| Space group | P6122 | P1 |
| Cell dimensions | ||
| a, b, c (Å) | 144.70, 144.70, 493.00 | 80.42, 103.31, 134.24 |
| α, β, γ | 90.00°, 90.00°, 120.00° | 95.32°, 90.22°, 94.46° |
| Unique reflections | 37,728 (2787)a | 63,973 (4164) |
| Resolution (Å) | 47.36–4.36 (4.47–4.36) | 68.41–3.29 (3.48–3.29) |
| Wavelength (Å) | 0.9791 | 0.9786 |
| Rsym (%)b | 0.17 (0.63) | 0.15 (0.37) |
| I/σI | 12.42 (2.69) | 7.87 (2.18) |
| Completeness (%) | 99.90 (99.90) | 98.20 (84.7) |
| Redundancy | 12.35 (11.30) | 1.93 (1.65) |
| 〈d″/sig〉c | 3.07os (0.81is) | 1.57os (0.87is) |
| B model refinement statistics | ||
| Resolution | 68.41–3.29 (3.48–3.29) | |
| No. of reflections | 63,973 (4164) | |
| Rcryst (%)d | 24.44 (26.47) | |
| Rfree (%)e | 26.70 (31.44) | |
| No. of residues | 3632 | |
| No. of ligands | 4 | |
| Solvent content (%) | 55 | |
| Non-H atoms | 28162 | |
| Average B-factor (Å2) | 48.02 | |
| Coordinate error (Å)f | 0.56 | |
| r.m.s.d. bonds (Å) | 0.009 | |
| r.m.s.d. angles | 1.21° | |
a Values in parentheses correspond to the highest resolution shell.
b Rsym = ΣhklΣi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl), where Ii(hkl) is the intensity of an observation and 〈I(hkl)〉 is the mean value of observations for a unique reflection.
c 〈d″/sig〉 = |Fhkl − F−h−k−l|/σ(|Fhkl − F−h−k−l|) (average of the anomalous difference d″ divided by its standard deviation).
d Rcryst = Σh|Fo(h) − Fc(h)|/Σh|Fo(h), where Fo and Fc are the observed and calculated structure factor amplitudes, respectively.
e Rfree was calculated with 5% of the data, which were excluded from the refinement.
f Based on maximum likelihood.
A ratio of 1 μl of protein to 4 μl of reservoir solution produced a second hexagonal crystal form (space group P6122) (Table 1), from which data up to 4.2 Å were collected at beamline PROXIMA 1 (SOLEIL, Paris, France). For these crystals, the data, processed also with XDS (12) and TRUNCATE (13), allowed some initial phases to be obtained with SHELXD (14). RESOLVE (15) and DM (16) were then used to produce an experimental map at 4.36 Å, applying the non-crystallographic symmetry restraints. A mask of the density corresponding to the molecular tetramer found in the asymmetric unit was used as a searching model to obtain, by molecular replacement, an initial solution for the P1 crystals. Phases were then improved and extended to 3.3 Å by density modification, mainly averaging between the 12 MCD subunits found in the asymmetric units of the two crystal forms. Model building was completed, alternating manual and automated refinement steps with Coot (17), REFMAC (18), and BUSTER (19). Docking analysis of MCD onto PEX5 (peroxin 5) was performed with HADDOCK (high ambiguity driven biomolecular docking) software (20) using the MCD and PEX5 coordinates files of Protein Data Bank codes 4F0X (this work) and 1FCH, respectively.
Kinetic Characterization
MCD activity was assayed spectrophotometrically by following the generation of NADH in the coupled reaction with malate dehydrogenase and citrate synthase (21). The reaction mixture contained 20 mm Tris (pH 8.5), 4 mm malate, 4 mm NAD+, varying amounts of malonyl-CoA (0.05–4 mm), 8.9 units of malate dehydrogenase, 3.1 units of citrate synthase, and varying concentrations of MCD in a total volume of 100 μl. The reaction was initiated by the addition of MCD, and the increase in absorbance at 340 nm was measured. The kinetic constants were determined by fitting the data to the Hill equation (Equation 1) by nonlinear least square regression using the program Origin 5.0,
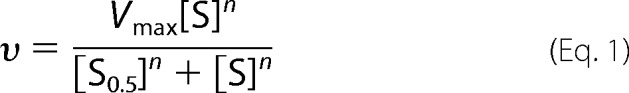 |
where the constant n is the Hill coefficient, and [S0.5] is the concentration of substrate giving 50% of the maximal velocity. Wild-type MCD and variants were oxidized prior to kinetic analysis by incubation with the specified amounts of H2O2 for 3 h at 4 °C, followed by gel filtration to remove the excess H2O2.
RESULTS
Overall Structure and Oligomeric Organization of Human MCD
The crystal structure of human peroxisomal MCD from Met-40 to Leu-493 has been solved (Fig. 2A) by single anomalous diffraction and density modification (16), combining data from triclinic and hexagonal crystal forms (Table 1). The quality of the final averaged electron density maps allowed us to identify and place the majority of the side chains despite the relatively low resolution of 3.29 Å for the triclinic crystals (Fig. 2D). Each MCD subunit is organized as an all-helical N-terminal domain and a catalytic C-terminal domain exhibiting the GNAT (GCN5-related N-acetyltransferase) fold (Protein Data Bank code 4F0X) (Fig. 2, A and B). In both crystal forms, subunits adopt two markedly different conformations (Fig. 2C), which initially was a major obstacle for model building in averaged maps and during refinement when non-crystallographic symmetry restraints were imposed. Monomers with different conformations pair with each other, across an interface with a large surface area, to produce what is essentially a structural heterodimer. The association of two such dimers (both structural heterodimers) yields the molecular tetramer (Fig. 3, A–D). The asymmetric units of the triclinic and hexagonal crystal forms contain two and one MCD tetramers, respectively. The tetrameric structure of the enzyme suggested by the crystal structure is supported by gel filtration analysis (data not shown). The two structural heterodimers in the tetramer are related by a molecular 2-fold axis (C2 molecular symmetry) with a rotation angle close to 180° (Table 2; represented as a solid arrow or an oval in Fig. 3, A–D). All other rotation axes for the superimposition of the different subunits in the tetramer (represented as dashed arrows in Fig. 3, A–C) present significant deviations from 180° (Table 2). These other rotation axes could become accurate 2-fold axes if structural differences between the two conformations adopted by subunits were not present. This would increase the symmetry of the molecular tetramer from C2 (one 2-fold axis) to D2 (three 2-fold axes perpendicular to each other).
FIGURE 2.

Overall structure of MCD subunits. Shown are ribbon (A) and secondary element (B) representations of a human MCD subunit, which is composed of an all-helix N-terminal domain (yellow) and a catalytic C-terminal domain (brown). The positions of Cys-206 and Cys-243 forming intersubunit disulfide bonds are indicated. C, MCD subunits adopt two markedly different conformations (shown in yellow/brown and green, respectively). The main differences, around the active center binding pocket, are likely related to changes in the relative orientation of the N- and C-terminal domains. A putative substrate/product fragment represented with sticks was found with partial occupancy only in subunits presenting the conformation defined as bound. D, stereo views of the 2Fo − Fc electron density maps at 1σ, corresponding to helices α7 and α8 at the end of the N-terminal domain.
FIGURE 3.

Molecular organization of MCD. A–C, three views (90° apart) of the tetrameric molecule of human MCD. In C, the view is down the only accurate molecular 2-fold axis of the tetramer (represented as an oval). This axis is represented as a solid arrow in A and B. Other axes relating the different subunits are indicated as dashed arrows. Subunits and conformations are labeled in C. D, the coordinated alternancy between subunit conformations would result in large structural rearrangements of the molecular tetramer.
TABLE 2.
Superimposition of MCD (Protein Data Bank code 4F0X) subunits and domains
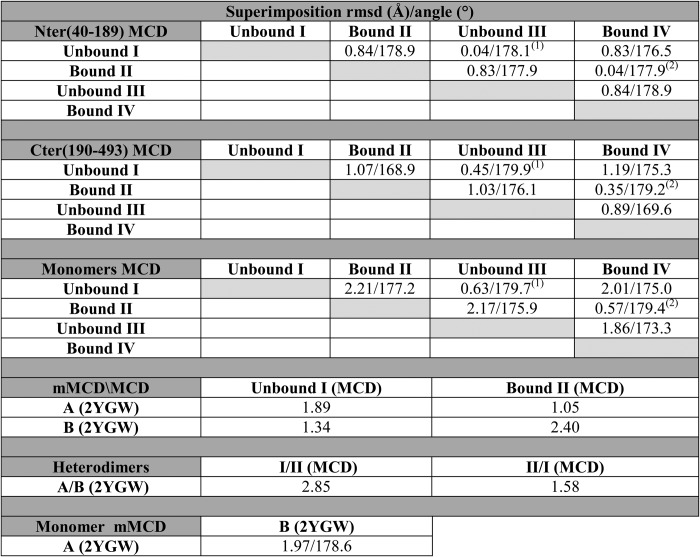
1 Unbound monomers I and III from MCD are accurately related by a 2-fold rotation.
2 Similarly for bound monomers II and IV.
The N-terminal domain of each monomer, Met-40–Trp-189, contains eight α-helices organized as a bundle of four antiparallel helices (α1–α3 and α6) with two pairs of helices inserted (α4-α5 and α7-α8). This arrangement may be the first representative of a new four-helix bundle variant because no other protein domain presenting an equivalent topology was found in a search with Dali (22). The C-terminal domain, Phe-190–Leu-493, presents a very precise GNAT domain topology (Fig. 2A), although decorated by a protruding cluster of seven helices (α13–α17, α19, and α20) (Fig. 2B). The major differences between the two conformations adopted by the subunits arise from rearrangements in the active site binding pocket (described below) and changes in the relative orientations of the N- and C-terminal domains, essentially consisting of a rotation angle difference of ∼10° for the superimposition of the N- and C-terminal domains from both types of subunits (Table 2).
While the present work was in progress, the coordinates of the crystal structure of human mitochondrial MCD (mMCD) were released (Protein Data Bank code 2YGW). The mMCD structure is based on data from an orthorhombic crystal (space group C2221) that contains a dimer in the asymmetric unit. Like the peroxisomal MCD described in this report, the dimer in mMCD is a structural heterodimer that can be superimposed onto the MCD heterodimer with root mean square deviations of 1.6 and 2.9 Å for the two possible superimpositions (Table 2). A crystal 2-fold axis completes the molecular tetramer of mMCD, making the organization of subunits nearly identical to MCD and providing independent support for the unusual structural features of the enzyme.
Active Site of Human Peroxisomal MCD
Decarboxylase activity in a GNAT domain was first described for a polyketide synthase (CurA_GNAT; Protein Data Bank codes 2REE and 2REF) having a dual decarboxylase/S-acetyltransferase activity (23). Based on this observation and notwithstanding there is no report of a GNAT protein with only decarboxylase activity, it seemed reasonable to assume that the catalytic site of MCD might resemble that of CurA_GNAT and reside in the GNAT fold (Fig. 4A). Although the overall sequence identity of MCD and CurA_GNAT is only 13% and the root mean square deviation for 181 Cα atoms aligned is 3 Å, the identity and geometry of residues essential for decarboxylation in CurA_GNAT (in particular, Thr-355 and His-389 interacting with Tyr-419) are very similar in MCD (Ser-329 and His-423 interacting with Tyr-456) (Fig. 4B).
FIGURE 4.

C-terminal domain of MCD. A, stereo views of the superimposition of MCD (represented as described for Fig. 2 (A and B) in yellow and brown for the N-terminal (N-TER) and C-terminal (C-TER) domains, respectively) onto the GNAT-like domain of the polyketide synthase CurA (shown in blue). The GNAT folds of the two proteins superimpose with great accuracy despite a very low sequence identity. B, stereo view of the MCD and CurA active centers. The organization of residues that are considered to be essential for decarboxylase activity in CurA (Thr-355 and His-389 interacting with Tyr-419) remains mostly unchanged in MCD (Ser-329 and His-423 interacting with Tyr-456). The molecule of malonyl-CoA found in the complex with CurA (Protein Data Bank code 2REF) suggests that, in MCD, the binding site for the cofactor might require some adjustments only for the base moiety.
Superimposition of MCD with GNAT complexes with CoA substrates, in particular that between CurA_GNAT and malonyl-CoA (Protein Data Bank code 2REF), indicates that CoA should bind in the opening formed between the diverging strands β4 and β5, interacting mainly with main chain atoms from β4 and α12 (Fig. 4B). One of the conformations of the heterodimer appears to facilitate binding of the malonyl-CoA substrate (the B or binding conformation), whereas binding to the other (the U or unbinding conformation) is apparently hindered. Electron density consistent with partial occupancy of the pantetheine pyrophosphate segment of CoA is found in B conformers (subunits II and IV in Figs. 3 and 5A), but not in U conformers (subunits I and III in Fig. 3). A similar situation is evident in the mMCD structure, where a region of well defined density, albeit not modeled in the Protein Data Bank file (code 2YGW), is found only in the subunit with the binding conformation, which is labeled as A in the Protein Data Bank file (Fig. 5B).
FIGURE 5.

Ligands in the active centers of subunits with the B conformation. Shown are stereo views of Fo − Fc difference electron density maps for the active centers of MCD subunits presenting the B conformation. A, for MCD (Protein Data Bank code 4F0X). B, for mMCD (code 2YGW). Density corresponds to unidentified ligands, which have been represented with the pantetheine and pyrophosphate moieties of a CoA molecule according to the superimposition shown in Fig. 4B.
The CoA fragments modeled in MCD have a sharply bent conformation with an acute angle between the two amide planes of the pantetheine moiety, similar to what is observed in CoA-GNAT complexes (Fig. 4B). One unusual feature of the CoA derivatives bound to GNAT is the interaction of the substrate phosphates with the main chain amides of the P-loop found in the CoA-binding motif ((R/Q)XXGX(G/A), where X indicates any residue) rather than with lysine or arginine side chains. In MCD, the corresponding sequence (Q299XXE302XG304) has a glutamic acid residue (Glu-302) replacing the central glycine in the standard motif. Glu-302 presents different main chain conformations in the B and U subunits. In the B conformation, Glu-302 has main chain torsional angles characteristic of a left-handed helix (Φ ∼ 60°, Ψ ∼ 20°), usually favored only by glycine residues. In addition, the side chain from Glu-302 is well defined, likely because it establishes hydrogen bonding interactions with Thr-60 in the loop between helices α1 and α2 (Tyr-54–Glu-65) from the N-terminal domain of the neighboring subunit (Fig. 6A). In the U conformation, Glu-302 adopts more relaxed main chain torsional angles (Φ ∼ −75°, Ψ ∼ 180°), and its side chain is not as well defined but appears to fill, at least partially, the site occupied by the pyrophosphate moiety of CoA in the bound conformers. In other words, the Glu-302 side chain potentially interferes with substrate binding in the U conformation (Fig. 6B). Furthermore, a shift of Glu-302 away from Thr-60 frees the Tyr-54–Glu-65 loop to become more disordered, and thus, a second part of the protein with two significantly distinct conformations is related to substrate occupancy of the catalytic site.
FIGURE 6.

Interactions between subunits in the MCD tetramer. The pyrophosphate-binding P-loop from human MCD presents major differences between subunits with B and U conformations (A and B, respectively). The standard GNAT CoA-binding motif ((R/Q)XXGX(G/A), where X indicates any residue), corresponds in MCD to Q299XXE302XG304. Replacing the central glycine of the standard motif with Glu-302 in MCD appears to have major consequences for the functioning of the P-loop. In B conformers, Glu-302 adopts a tense main chain conformation, which is stabilized by interactions with the Tyr-54–Glu-65 loop from a neighboring subunit. In the U conformers, Glu-302 adopts a relaxed main chain conformation not interacting with the Tyr-54–Gly-65 loop, which is disordered. In U conformers, the Glu-302 side chain occupies, at least in part, the site to be occupied by the pyrophosphate of a CoA, contributing to the release of the cofactor. The environment of Cys-206 and Cys-243 at the two intersubunit interfaces of the MCD tetramer (C and D) shows the feasibility of Cys-206–Cys-206 and Cys-243–Cys-243 disulfide bridges.
The binding site for the adenosine portion of CoA in MCD is most likely different from that of CurA_GNAT because a similar arrangement would involve an unfavorable steric interaction (Fig. 4B). Unfortunately, the electron density maps of both MCD and mMCD do not provide any hint about a possible alternative for the adenosine-binding location, and attempts to produce complexes of MCD by soaking crystals with several CoA derivatives were unsuccessful.
Intersubunit Interactions and Disulfide Bridges of MCD
MCD subunits in the tetramer have no interactions across the main 2-fold axis (Fig. 3C), but they do interact in two other possible ways as a result of the pseudo D2 molecular organization. Type I-II interactions (between subunits I and II and also between subunits III and IV) stabilize the formation of the structural heterodimer and consist of a large (3964 Å2), strongly hydrophobic interface involving mainly the N-terminal domains. By contrast, type I-IV interactions (between subunits I and IV and subunits II and III) consist of a much smaller interface area (1051 Å2) and involve exclusively the C-terminal domains (Figs. 3, A–C, and 6C). The proximity of cysteine residues across both interfaces suggests the possible formation of disulfide bridges (Cys-206–Cys-206 across type I-IV interfaces and Cys-243–Cys-243 across type I-II interfaces), thereby covalently linking the four monomers in the tetramer (Figs. 3C and 6, C and D). Because in our hands crystal growth required the presence of a reducing agent in the medium, probably to enhance the homogeneity of the sample, disulfide bridges are not seen in the MCD structure. However, there is evidence of partial formation of these disulfide bridges in the mMCD structure. The cysteine residues are positioned such that formation of the disulfide bonds would not require large structural changes, and main chain atoms in mMCD have unique positions despite the presence of cross-linked and uncross-linked forms.
The possible formation of disulfide bonds linking the four subunits of MCD was analyzed using nonreducing denaturing gels with samples of the enzyme maintained under reducing conditions or pretreated with hydrogen peroxide (Fig. 7). Upon H2O2 treatment of wild-type MCD, the band attributed to the monomeric species was rapidly, although not quantitatively, converted to a band with the molecular mass expected for the tetramer, without the apparent accumulation of dimers. In contrast, the C206S/C243S double variant yielded a band with an apparent molecular mass corresponding to the monomer under every condition. Surprisingly, the single cysteine-to-serine variants did not afford the expected bands attributable to the respective dimers after H2O2 oxidation: C243S MCD behaved as the wild-type enzyme and C206S MCD as the double-variant protein. It must be noted that, under nondenaturing conditions, all variants behaved as tetramers by gel filtration analysis, which indicates that the Cys-206 and Cys-243 side chains are not essential for oligomerization of the enzyme. Attempts to further characterize the molecular oligomers by mass spectrometry provided evidence only of the unlinked subunits in untreated wild-type MCD. Unfortunately and possibly due to the complexity of the samples, which yielded spectra difficult to interpret, we were not able to unambiguously detect the formation of disulfide bonds in the H2O2-treated samples (data not shown). Nevertheless, our results suggest that MCD oxidation leads to the formation of disulfide linkages between subunits as an all (tetramer)-or-nothing process, involving both Cys-206 and Cys-243.
FIGURE 7.

Formation of disulfide bridges in MCD. Shown is a SDS-polyacrylamide gel without β-mercaptoethanol of human MCD samples with and without preincubation with hydrogen peroxide. Samples with DTT added are also shown for comparison. Formation of tetramers, in both wild-type MCD and the C243S variant, appears to be a highly cooperative process because dimers were never detected, although the presence of tetramers in the C243S variant is difficult to interpret. Cys-206 is conserved in vertebrates, whereas Cys-243 is conserved only among mammals (see supplemental Fig. 1).
Steady-state Kinetic Measurements
Using a coupled enzyme assay (24), steady-state kinetic analyses were conducted on wild-type MCD and the single and double cysteine-to-serine variants (Table 3). Both wild-type MCD and the C206S/C243S double variant exhibited Michaelis-Menten saturation kinetics (Hill coefficient (n) near unity) (Table 3) with some small variations in kcat and S0.5, but almost identical catalytic efficiencies (kcat/S0.5), indicating that these cysteine residues are dispensable for function. However, although the kinetic parameters remained essentially unchanged for the double variant when it was pretreated with H2O2, the catalytic efficiency of the wild-type enzyme almost doubled at 0.2 m H2O2. Most significantly, the experimental data were now best fitted to a Hill equation in which the n value gradually increased with the concentration of the oxidizing agent used in the pretreatment, reaching a value of 1.4 at 0.2 m H2O2 (Table 3). In this respect, the single variants also exhibited divergent behaviors. C206S MCD was most similar to the C206S/C243S double variant because its catalytic efficiency did not considerably change upon preincubation with H2O2, and the Hill coefficients remained close to unity under all conditions. In contrast, the kcat/S0.5 value for the C243S variant more than doubled, and its n value increased to 1.37 at 0.2 m H2O2.
TABLE 3.
Kinetic parameters of wild-type MCD and the C206S/C243S, C206S, C243S, and E302G variants under non-oxidative and oxidative conditions
| Oxidant (H2O2) concentration | Variant | MCD kinetic parameters |
|||
|---|---|---|---|---|---|
| kcata | S0.5 (malonyl-CoA) | n (Hill coefficient) | kcat/S0.5 | ||
| s−1 | mm | m−1 s−1 | |||
| 0 m | WT | 141.2 ± 2.1 | 0.83 ± 0.03 | 1.04 ± 0.02 | 0.17 |
| 0.05 m | WT | 128.3 ± 4.6 | 0.46 ± 0.04 | 1.14 ± 0.08 | 0.28 |
| 0.1 m | WT | 135.0 ± 4.2 | 0.42 ± 0.03 | 1.28 ± 0.09 | 0.32 |
| 0.2 m | WT | 109.2 ± 3.8 | 0.35 ± 0.03 | 1.43 ± 0.12 | 0.31 |
| 0 m | C206S/C243S | 162.5 ± 7.5 | 1.16 ± 0.13 | 1.04 ± 0.06 | 0.14 |
| 0.1 m | C206S/C243S | 175.4 ± 9.2 | 0.99 ± 0.13 | 1.06 ± 0.07 | 0.18 |
| 0.2 m | C206S/C243S | 167.1 ± 7.1 | 0.81 ± 0.09 | 0.98 ± 0.05 | 0.20 |
| 0 m | C206S | 117.1 ± 1.7 | 0.73 ± 0.02 | 1.17 ± 0.02 | 0.16 |
| 0.1 m | C206S | 141.2 ± 8.8 | 1.04 ± 0.16 | 1.11 ± 0.09 | 0.14 |
| 0.2 m | C206S | 114.2 ± 4.2 | 0.79 ± 0.08 | 1.04 ± 0.06 | 0.15 |
| 0 m | C243S | 94.6 ± 2.1 | 0.58 ± 0.03 | 1.19 ± 0.05 | 0.16 |
| 0.1 m | C243S | 137.5 ± 5.4 | 0.68 ± 0.07 | 1.25 ± 0.09 | 0.20 |
| 0.2 m | C243S | 208.3 ± 2.5 | 0.56 ± 0.02 | 1.37 ± 0.04 | 0.37 |
| 0 m | E302G | 13.3 ± 8.8 | 0.22 ± 0.13 | 0.43 ± 0.07 | 0.06 |
a kcat was computed for monomers.
The side chain of Glu-302, the residue of MCD (Q299XXE302XG304) that replaces the central glycine in the consensus GNAT CoA-binding motif ((R/Q)XXGX(G/A)), undergoes a significant positional change between the B and U conformations of the monomers. The relevance of this residue in the catalytic turnover of MCD was investigated by analyzing the kinetic parameters of the E302G variant. This mutant form exhibited a moderate reduction of the S0.5 value and a 10-fold decrease in the kcat, consistent with a significantly lower overall catalytic efficiency (Table 3). However, the most notable change lies in the low Hill coefficient (n = 0.43), which suggests negative cooperativity in the mode of substrate binding by the enzyme when the Glu-302 side chain is absent.
Model of the Interaction of Human MCD with PEX5
MCD can be found in different intracellular compartments such as the cytoplasm, mitochondria, and peroxisomes. Transport of MCD into the peroxisome involves the interaction with the peroxisomal transport protein PEX5, and this interaction was investigated by superimposing the C-terminal PTS1 motif (Ser-491–Lys-492–Leu-493) found in one of the MCD subunits onto the structure of the Ser-Lys-Leu peptide in complex with PEX5 (Protein Data Bank code 1FCH) (Fig. 8, A–C). The extended conformation of the SKL tripeptide was similar in both structures, suggesting that this is a preferred conformation. However, there must be flexibility in at least the orientation of the tripeptide with respect to the MCD terminal helix α20, which ends at Asn-490 (supplemental Fig. 1), as the tripeptide is not visible in most of the MCD subunits. Thus, using SKL as an anchorage point, the optimization of the MCD orientation with respect to PEX5 to avoid steric clashes gave a very narrow window of possibilities. In all cases, the closeness of the molecular surfaces of MCD and PEX5 suggests that the specific interaction determining recognition and binding should include regions in MCD beyond the SKL motif. These putative additional interacting regions are all located in the protruding cluster of α-helices of the C-terminal domain (α13–α17, α19, and α20) (Fig. 2B). On the other hand, the corresponding interacting regions in PEX5 are not so well defined by the docking and could involve four to six different helices. The involvement of specific interactions apart from the SKL import signal is in agreement with reports indicating that proteins transported by the PTS1 peroxisomal import system present a number of structural features in addition to, but not necessarily directly linked with, the SKL signal tripeptide (25).
FIGURE 8.

Docking of human MCD onto PEX5. A, stereo view of the C-terminal PTS1 motif (Ser-491–Lys-492–Leu-493), which is seen only in one of the subunits. Electron density corresponding to the SKL tripeptide is also shown. B, docking of a MCD subunit (green) onto PEX5 (orange). C, stereo view showing the superimposition of the peroxisomal targeting tripeptide structures as determined in this work and in the complex of PEX5 with a peptide containing the SKL tripeptide (Protein Data Bank code 1FCH). The tight packing between both proteins appears to require interactions extending beyond the SKL tripeptide. In MCD, these interactions will all involve residues from the inserted cluster of helices (α14-α16 and α20) found in the C-terminal domain.
DISCUSSION
MCD subunits contain an all-helical N-terminal domain and a catalytic C-terminal domain organized in a GNAT fold. Members of the GNAT superfamily, with several thousand representatives from all kingdoms of life, act usually as N-acetyltransferases, transferring acetyl groups to primary amines. Typically, GNAT proteins have separate binding sites for the acetyl donor and acceptor substrates and catalyze the direct transfer without the participation of a covalent enzyme-substrate intermediate. The basic structure of the GNAT fold is extraordinarily conserved and serves two nearly universal functions: (i) binding the pantetheine moiety of acetyl-CoA and (ii) polarizing the carbonyl of the thioester through hydrogen bonding interactions. These general properties of GNAT proteins are applicable to CurA_GNAT (23), but only the first is applicable to MCD because polarization of the thioester carbonyl is not necessary for the conversion of malonyl-CoA to acetyl-CoA. Furthermore, because decarboxylase activity does not require an acetyl acceptor, the binding site for the acceptor molecule found in most GNAT structures is absent in MCD.
The MCD tetramer is best described as an association of two structural heterodimers, each composed of two subunits with alternate conformations, one that allows substrate/product binding and one that does not. Such a conformational arrangement is consistent with the half-of-the-sites kinetic mechanism, in which, at a given time, only one of the two sites in the heterodimer is active. This mechanism of catalysis is compatible with the Michaelis-Menten saturation kinetics observed for wild-type MCD that had not been pretreated with an oxidizing agent.
Although neither substrate nor product was added to the crystals, partial occupancy of a portion of a pantetheine chain is evident, indicating that some residual substrate-like molecule remained bound throughout the purification procedure, which suggests a stable association. However, the observation that the two subunits of the structural heterodimer present alternate conformations despite incomplete occupancy of the binding site suggests an inherent structural asymmetry that is independent of the presence of substrate. Moreover, the fact that the asymmetry is present in the structures of all available MCD molecules indicates it is not artifactually generated by crystal contacts and that the structurally distinct subunits are present in significant concentration in solution.
There is an apparent waste of catalytic power in the half-of-the-sites mechanism because only half of the potential active sites can function at a time. For enzymes catalyzing small changes in large substrates, such as in the decarboxylation of malonyl-CoA to acetyl-CoA, product and substrate have similar affinities for the enzyme active site. In this scenario, the slow release of the product would interfere with entry of the next substrate and would make product release the rate-limiting step. However, in the case of MCD, the conformational change in one subunit from binding the substrate (B conformation) to contributing to the release of the product (U conformation) is coincident with the opposite conformational changes (from U to B) in the neighboring subunit, which creates a binding site for the next substrate. In this way, the concerted conformational changes in the paired subunits actually facilitate rapid turnover despite the reduction of the number of active sites operating simultaneously. The amino acid residue in the CoA-binding motif of MCD that differs from that of the GNAT standard sequence, i.e. Glu-302, is a central element of the molecular mechanism that triggers these concerted changes. Interaction of the Glu-302 side chain carboxylate with the Tyr-54–Glu-65 loop of the adjacent subunit maintains this residue in a “tense” state that stabilizes the B conformation. In the U conformer, this interaction is broken, allowing Glu-302 to adopt a “relaxed” conformation, in which its side chain points toward the pantetheine pyrophosphate-binding site in the substrate/product pocket. This in turn changes the Tyr-54–Glu-65 loop from a more to a less ordered structure. Thus, the Glu-302 side chain acts as a kind of molecular lever, whose movement in and out of the active site, coordinated with the movement of the Tyr-54–Glu-65 loop, is used by MCD to facilitate the release of the reaction product in one subunit and the binding of a new substrate molecule in the other subunit, after every catalytic cycle. Consistent with this, the MCD variant E302G exhibits substantially altered catalytic properties, with marginally enhanced affinity for the substrate, a 10-fold reduced turnover rate, and large negative cooperativity. We interpret this last observation as an indication that removal of the Glu-302 side chain in the E302G MCD variant produces an enzyme in which the binding of substrate to one subunit largely reduces the affinity of the second subunit of the dimer, but does not completely prevent substrate binding to the second subunit as occurs in the wild-type enzyme.
The proximity and relative orientation of the two pairs of Cys-206 and Cys-243 residues across the I-IV/II-III and I-II/III-IV interfaces of the tetramer, respectively, suggest that, under oxidizing conditions, the four subunits of MCD could be connected through disulfide bonds. The appearance after H2O2 treatment of a band with the molecular mass expected for the tetramer on nonreducing denaturing gels and the mitochondrial MCD structure, in which the disulfide bonds are partially formed, are consistent with this hypothesis. However, it remains unclear why H2O2 oxidation is apparently cooperative in the sense that linked tetramers are formed preferentially to dimers, why the conversion to tetramer is apparently not quantitative even at high H2O2 concentrations, or why the dimers are not detected in the C206S or C243S single variant (Fig. 7).
The kinetic parameters of MCD are also affected by pretreatment of the enzyme with H2O2, which produces an apparent increase in positive cooperativity (larger Hill coefficients). Although our structure does not provide an obvious molecular mechanism that could explain enhanced cooperativity through disulfide bridge formation, the fact that this phenomenon was observed in the wild-type enzyme and the C243S variant, but not in the C206S or C206S/C243S variant, suggests that the Cys-206–Cys-206 disulfide bridge across the I-IV and II-III interfaces is the more critical of the two cross-links for imparting the cooperative effect.
MCD can be found in several intracellular locations, the including cytoplasm, mitochondria, and peroxisomes (1, 7). The first two are reducing compartments, and protein disulfide bonds are rarely found in such environments (26). In turn, peroxisomes have been shown to be net producers of H2O2 (8).
We have not investigated the in vivo significance of the cooperative effect and the increased catalytic efficiency caused by MCD oxidation and possibly mediated by the cross-linking of the four subunits in the tetramer through disulfide bridges. However, our results indicate that the distinct redox environments in which the enzyme can be found modulate its catalytic behavior and may contribute to the complex regulatory picture presented by an enzyme that controls the levels of malonyl-CoA, a key metabolite of fatty acid metabolism.
Acknowledgments
We thank Dr. Andrew W. Thompson (SOLEIL Synchrotron) for the excellent support provided during data collection and data processing and Dr. Nicholas S. Berrow (IRB Barcelona Protein Production Facility) for the work related to the MCD variants.
This work was supported by Discovery Grant 9600 from the Natural Sciences and Engineering Research Council of Canada and the Canada Research Chair Program (to P. C. L.) and by Grant BFU2012-36827 from the Ministerio de Ciencia e Innovación (MICINN) and Grant SGR2009-00327 from the Generalitat de Catalunya (to I. F.).

This article contains supplemental Fig. 1.
The atomic coordinates and structure factors (code 4F0X) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- MCD
- malonyl-CoA decarboxylase
- mMCD
- mitochondrial MCD
- MBP
- maltose-binding protein
- Bistris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane.
REFERENCES
- 1. Sacksteder K. A., Morrell J. C., Wanders R. J., Matalon R., Gould S. J. (1999) MCD encodes peroxisomal and cytoplasmic forms of malonyl-CoA decarboxylase and is mutated in malonyl-CoA decarboxylase deficiency. J. Biol. Chem. 274, 24461–24468 [DOI] [PubMed] [Google Scholar]
- 2. Haan E. A., Scholem R. D., Croll H. B., Brown G. K. (1986) Malonyl-coenzyme A decarboxylase deficiency. Clinical and biochemical findings in a second child with a more severe enzyme defect. Eur. J. Pediatr. 144, 567–570 [DOI] [PubMed] [Google Scholar]
- 3. Hayaishi O. (1955) Enzymatic decarboxylation of malonic acid. J. Biol. Chem. 215, 125–136 [PubMed] [Google Scholar]
- 4. Wolfe J. B., Rittenberg S. C. (1954) Malonate decarboxylation by Pseudomonas fluorescens. III. The role of acetyl coenzyme A. J. Biol. Chem. 209, 885–892 [PubMed] [Google Scholar]
- 5. Hatch M. D., Stumpf P. K. (1962) Fat metabolism in higher plants. XVII. Metabolism of malonic acid and its α-substituted derivatives in plants. Plant Physiol. 37, 121–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lynen F. (1962) Fatty acid synthesis from malonyl CoA. Methods Enzymol. 5, 443–451 [Google Scholar]
- 7. Nakada H. I., Wolfe J. B., Wick A. N. (1957) Degradation of malonic acid by rat tissues. J. Biol. Chem. 226, 145–152 [PubMed] [Google Scholar]
- 8. Nyathi Y., Baker A. (2006) Plant peroxisomes as a source of signalling molecules. Biochim. Biophys. Acta 1763, 1478–1495 [DOI] [PubMed] [Google Scholar]
- 9. Zhou W., Tu Y., Simpson P. J., Kuhajda F. P. (2009) Malonyl-CoA decarboxylase inhibition is selectively cytotoxic to human breast cancer cells. Oncogene 28, 2979–2987 [DOI] [PubMed] [Google Scholar]
- 10. Joly E., Bendayan M., Roduit R., Saha A. K., Ruderman N. B., Prentki M. (2005) Malonyl-CoA decarboxylase is present in the cytosolic, mitochondrial and peroxisomal compartments of rat hepatocytes. FEBS Lett. 579, 6581–6586 [DOI] [PubMed] [Google Scholar]
- 11. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 12. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. French S., Wilson K. (1978) On the treatment of negative intensity observations. Acta Crystallogr. Sect. A 34, 517–525 [Google Scholar]
- 14. Sheldrick G. M. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Terwilliger T. C., Berendzen J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cowtan K. (1994) Joint CCP4 and ESF-EACBM newsletter on protein crystallography. Protein Crystallogr. 31, 34–38 [Google Scholar]
- 17. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 18. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 19. Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S. M., Bricogne G. (2004) Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D Biol. Crystallogr. 60, 2210–2221 [DOI] [PubMed] [Google Scholar]
- 20. de Vries S. J., van Dijk M., Bonvin A. M. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 [DOI] [PubMed] [Google Scholar]
- 21. Kolattukudy P. E., Poulose A. J., Kim Y. S. (1981) Malonyl-CoA decarboxylase from avian, mammalian, and microbial sources. Methods Enzymol. 71, 150–163 [DOI] [PubMed] [Google Scholar]
- 22. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gu L., Geders T. W., Wang B., Gerwick W. H., Håkansson K., Smith J. L., Sherman D. H. (2007) GNAT-like strategy for polyketide chain initiation. Science 318, 970–974 [DOI] [PubMed] [Google Scholar]
- 24. Kim Y. S., Kolattukudy P. E. (1978) Purification and properties of malonyl-CoA decarboxylase from rat liver mitochondria and its immunological comparison with the enzymes from rat brain, heart, and mammary gland. Arch. Biochem. Biophys. 190, 234–246 [DOI] [PubMed] [Google Scholar]
- 25. Brocard C., Hartig A. (2006) Peroxisome targeting signal 1: is it really a simple tripeptide? Biochim. Biophys. Acta 1763, 1565–1573 [DOI] [PubMed] [Google Scholar]
- 26. Hanson G. T., Aggeler R., Oglesbee D., Cannon M., Capaldi R. A., Tsien R. Y., Remington S. J. (2004) Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 279, 13044–13053 [DOI] [PubMed] [Google Scholar]