

Abstract
Background
For many diseases such as cancer where phosphorylation-dependent signaling is the foundation of disease onset and progression, single-gene testing and genomic profiling alone are not sufficient in providing most critical information. The reason for this is that in these activated pathways the signaling changes and drug resistance are often not directly correlated with changes in protein expression levels. In order to obtain the essential information needed to evaluate pathway activation or the effects of certain drugs and therapies on the molecular level, the analysis of changes in protein phosphorylation is critical.
Methods
Existing approaches do not differentiate clinical disease subtypes on the protein and signaling pathway level, and therefore hamper the predictive management of the disease and the selection of therapeutic targets.
Conclusions
The mini-review examines the impact of emerging systems biology tools and the possibility of applying phosphoproteomics to clinical research.
Keywords: Phosphoproteomics, Mass spectrometry, Signaling
Personalized medicine redefines clinical research
We are currently living through a revolution in healthcare and medicine that will significantly influence our day-to-day lives in the future. The primary shift is occurring from a single-target treatment and one-fits-all strategy of therapeutic intervention, to a much more personalized medicine that precisely defines molecular misregulations on an individual level and can pinpoint those patients that would benefit most from a particular treatment regime [1]. One clear example is that, it could significantly increase survival rates, reduce toxic effects, and improve cancer patients’ quality of life, if a physician can match individual patients with their therapies designed specifically to treat the individualized molecular profile of their cancer.
One of the prime storylines that perfectly exemplifies the beginning of such a transformation has been the introduction of Gleevac (imatinib) for treatment of chronic myelogenous leukemia (CML) [2-5]. CML is characterized by overproliferation of myeloid cells and usually onset by the abnormal increase in phosphorylation-dependent signaling of a tyrosine kinase ABL through its genetic fusion with BCR. It was the first drug developed for direct and specific kinase inhibition, demonstrating potent activity with low toxicity levels due to its targeted treatment toward individuals with the presence of the oncogenic BCR-ABL fusion. The striking success of imatinib has validated the great utility of using kinase inhibitors for cancer treatment and opened the doors for multiple new compounds being developed, currently accounting for over 30% of drug development resources [6].
However, the real game-changer in attitudes of drug developers and healthcare providers came through a realization that despite imatinib being singularly effective in regressing CML in many of patients, some of those affected still display resistance to the drug, while majority of others (~90%) form resistance within 5-y period [7]. Outside of BCR-ABL point mutations, further molecular analyses into these phenomena have uncovered dozens of signaling pathways that are affected by CML, such as the activation of Src family tyrosine kinases [8, 9]. In many cases, Src is often responsible for progression of the disease into more aggressive stages, as well as for overcoming the BCR-ABL inhibition. As a confirmation of this, a second-generation CML kinase inhibitor, dasatinib, which has been developed as an answer to imatinib resistance, has a potent inhibitory effect on both BCR-ABL and Src kinases [10, 11]. In addition, a number of other mechanisms of drug resistance have been observed that involved evolutionary changes in signaling pathways to compensate for BCR-ABL inactivity [12]. As an example, findings that dual inhibitions of other downstream kinases in addition to BCR-ABL, such as mTOR [13] or MEK1/2 [14], can significantly decrease CML progenitor growth. This example highlights a much generalized concept, synthetic lethality, where one needs to also account for oncogene-addicted cells that often employ other pathways to activate downstream targets and promote their own survival. Thus, inhibition of these secondary pathways should be a necessary treatment option to further impede the disease progression [15, 16].
Here, CML serves as just one example of how a complicated signaling pathway can cause the progression of a disease through multiple networks, feedback loops, and compensation mechanisms. In fact, leukemia actually represents relatively a much simple case to understand and treat in comparison to solid tumors. In carcinomas, such as breast, colon or lung, it is incredibly difficult to find specific targets that would cause a significant tumor remission in vivo. Any current therapies only produce a short-lived response and eventual resistance to the treatment [17]. What has become more evident over the past years is that the to-date identified therapeutic targets alone do not provide anticipated results, particularly when it comes to receptors on cells as drug targets. It would be much more beneficial to turn the focus to the downstream signaling cascades that could offer more predictive outcomes.
What makes therapy development even more difficult are the observations that signaling pathways activated in one individual tumor or sub-category do not automatically translate into another person’s seemingly identical tumor due to the highly heterogeneous nature of the disease [18]. Currently, for many carcinomas, the pathological parameters for stratification include size, morphological pattern, metastasis presence, degree of differentiation, and detection of oncogenic marker genetic mutations. However, these strategies are not nearly sufficient for true classification of a disease and singular tumors, and do little to predict proper therapeutic treatment or individual response to it. It is imperative, therefore, to have the necessary tools to study multiple molecular pathways simultaneously in each individual case in order obtain a more comprehensive picture about the system and the true effects of drug treatment.
Systems biology for clinical research
Systems biology strategies seem to be the logical choice for personalized medicine, which embraces a comprehensive high-throughput analysis of many important individual features on a molecular level, such as genomic, transcriptomic, proteomic, phosphoproteomic, and metabolomic examinations. Such an all-inclusive multidimensional “omics’ analysis would provide an insight into not only target mutations and morphological differences, but, more importantly, into the effected signaling networks and molecular differentiation, enabling pre-symptomatic diagnosis, molecular classification, tailored therapy formulation, and evaluation of patient response (Fig. 1). The new integrative set of tools that would allow this should be fast, highly accurate, and enable simultaneous detection of all of the necessary molecular components from a small clinical sample.
Figure 1.

A systems biology approach for comprehensive and multidimensional clinical sample analysis.
To date, only one component of the “omics” approach – genomic profiling - has been significantly utilized for predictive measurements and disease classification, likely due to the ease of amplification, detection, and quantitation. The genome-based strategy has been used for numerous clinical analyses, such as lymphoma classification [19] and breast cancer transcript analysis [20]. While single gene testing and genomic profiling have become important tools to characterize predictive diagnostic features, it is far from adequate to reflect the complex, dynamic nature of many diseases. First of all, mRNA abundance profile does not always correspond to the protein expression levels. It has been previously demonstrated that only up to 40% of mRNA levels can accurately predict the changes in protein expression [21, 22]. Second, the genomic and transcriptomic profiling offer no insights into protein interactome patterns, subcellular localization, or their functional status. Finally, and more importantly, utilizing only genomic profiling, all information about protein post-translation modifications (the driver of signal transduction) is missing. This is especially critical in carcinomas, where abnormal regulation of phosphorylation is the main cause and marker of onset and progression of the disease. Because proteins are the workhorse of the molecular networks responsible for most cellular processes, proteomic analysis offers a more in-depth understanding of molecular abnormalities. As phosphorylation-based signaling molecules are becoming the most significant target for cancer and many other disease treatments, phosphorylation and enzyme (kinase/phosphatase) activity profiling should emerge as a critical element in therapy selection.
Pathway profiling and phosphoproteomics will become an integral part of clinical research for personalized medicine and preventative healthcare with multiple attractive features. It will enable identification and recognition of disease biomarkers, will allow sub-classification of individual cancers through comprehensive analysis of effected signaling pathways, will empower discovery of new targets and treatment options, will elucidate the poorly-understood mechanisms of drug resistance, and will facilitate the monitoring of the tumor response to treatments [23]. While currently phosphoproteomic analyses are primarily utilized in an R&D setting, the development of new clinically-friendly tools will finally bring this powerful oncoproteomic profiling strategy into the medical setting.
Current status in phosphoproteomics
Phosphoproteomic platforms that allow simultaneous detection and quantification of multiple molecular markers within cellular signaling microenvironment would be the most valuable and attractive systems. While a number of technologies have become available for partial phosphoproteomics profiling studies [24], the most successfully utilized to date have been protein array platforms and LC-MS based quantitative phosphoproteomics studies.
Over the past decade, protein microarrays have been demonstrated as a very powerful tool in comprehensive analysis of signaling networks. They provide a researcher with the ability to quantitatively monitor known signaling targets throughout various treatments and environments in a flexible, multiplexed, and time-efficient manner. Reverse phase protein microarrays (RPPMA) have become particularly popular for this screening purpose due to robustness and reproducibility of the platform, and the ability to use very small amounts of clinical samples – a typical bottleneck in these analyses [25, 26]. Already, certain clinical studies have begun appearing in the literature that utilize RPPMA for therapeutic studies and detailed disease classification. As an example, Wulfkuhle et.al. has recently published a multiplexed phosphoproteomic RPPMA analysis of 25 distinct human breast cancer surgical specimen, which allowed certain level of sub-classification of these tumors based on phosphorylation profile of the targeted markers [27]. It is easy to envision that with much larger sample sizes, such multiplexed phosphoproteomic analyses could provide an abundance of clinically-relevant information.
Nonetheless, despite the evident utility of the protein arrays, they are greatly limited by the requirement of good-quality highly selective antibodies, which are currently available only for a limited number of proteins. The problem is even more significant for detection of phosphorylation. Though there have been attempts to create antibody arrays that detect phosphorylation changes in certain target proteins [28-30], they are generally limited by the availability of phosphosite-specific antibodies. These assays typically detect only well-characterized phosphorylation events, while providing no information about the overall phosphorylation profile of the targets. While some assays have used general phospho-tyrosine antibodies to analyze the changes in global pTyr levels of specific proteins, this approach is still not sufficient enough, as it targets only tyrosine phosphorylation. In many cases, some of the most important information can be obtained from the phosphorylation analysis of the downstream signaling networks, such as Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways, which are saturated with serine/threonine phospho-sites important for cancer progression [31, 32]. In addition, it has been also demonstrated on multiple occasions that the detection of a single or few well-characterized phospho-sites in a protein of interest using phosphospecific antibodies is not sufficient. Often, relevant protein targets have multiple, sometimes dozens, of important phosphorylation sites, as is the case with Raf, Erk, MEK, Bim, mTOR and others. In some cases, the location of phosphorylation can have completely different or opposite effects [31, 32]. While strategies like single reaction monitoring (SRM) and multiple reaction monitoring (MRM) can overcome the troublesome issues of poor antibody quality and availability, they still require a priori knowledge of the phosphorylation sites on target proteins. Therefore, though this strategy has been successfully demonstrated to allow effective phosphopeptide quantitation in many instances [33-35], it cannot be routinely used for discovery of new phospho-targets or profiling of multiple lesser-known sites. To overcome this, high-throughput large-scale phosphoproteomic analysis using LC-MS can be utilized, allowing for un-biased simultaneous detection and quantification of phosphorylation events in discovery mode.
Since the great advancements in mass spectrometry instrumentation, large-scale phosphoproteomic experiments have become the standard in unbiased high-throughput analysis. There have been hundreds of studies published on successful use of quantitative MS-based phosphoproteomics in analysis of clinically-relevant samples [34, 36-39]. The demonstrated LC-MS utility ranges from examination of cancer-induced signaling pathways, to biomarker discovery, to identification of new therapeutic targets, to elucidation of causes for drug resistance, to uncovering of novel potential synthetic lethality targets for poly-therapy regimes. The significant advantage of this unbiased approach lies in its ability to not only scrutinize the targeted hypothesis-based signaling pathways of interest, but also uncover new targets and networks with previously unknown involvement in particular cellular processes [40]. Quantitative phosphoproteomics has become the unprecedented source of new high-throughput information and the workhorse in signal transduction studies in both cell lines and clinical samples.
Though these large-scale phosphoproteomics experiments, in our opinion, are currently the most optimal way of profiling phosphorylation changes, it is not a feasible approach for most clinical research due to high cost and lengthy time associated with large scale experiments. Because an extremely large number of phosphorylated proteins are present in a single sample (as many as one-third of proteins can be phosphorylated simultaneously;the phosphorylation events that are of most interest to a researcher or a clinician can become overwhelmed by non-changing “house-keeping” phosphorylated proteins. This presents enormous challenges in bioinformatics and data validation. Additionally, a typical mass spectrometry-based phosphoproteomic experiment also requires relatively large amount of sample, which is difficult to obtain in clinical setting.
A viable phosphoproteomic platform for clinical research?
We envision that a new platform is needed for clinical research. It integrates the best qualities of protein arrays and MS-based phosphoproteomics, which allows multiplexed detection of changes in target protein phosphorylation, followed by rapid mass-spectrometry profiling of the effected phosphoproteins with the goal of phosphosite localization. Protein arrays could be generated with covalently linked antibodies against known molecular targets, biomarkers, and other proteins observed to be controlled by phosphorylation in a system of interest. After various treatments, the target proteins can be immunoprecipitated using the generated protein array plates. The changes in the phosphorylation levels of the bound proteins across different sample types and treatment options can be detected using a universal phosphorylation method such as infrared fluorophore-based pIMAGO reagent developed in our group [41, 42]. The pIMAGO (phosphoimaging) reagent is a water-soluble nanopolymer-based molecule multi-functionalized with titanium metal ions, enabling strong, selective and unbiased binding to any phosphoprotein, and with multiple fluorescent molecules that provide enhanced fluorescence detection. Simultaneously, pIMAGO detection can be multiplexed with another general protein antibody, which will allow a more direct assessment of changes in the level of phosphorylation against the total protein amount. Following the detection and selection of the proteins with observable changes in phosphorylation, the effected wells can be treated with a protease such as trypsin, and the resulting peptides are subjected to a phosphopeptide enrichment technology to isolate phosphopeptides. An efficient enrichment is highly desirable. The previously demonstrated high selectivity (>95%) and recovery (>90%) of PolyMAC (Polymer-based Metal Affinity Capture) enrichment makes it well-suited for optimal retrieval of these modified peptides [43]. Finally, the isolated phosphopeptides can be rapidly identified by LC-MS and the phosphorylated residues localized (overall strategy is presented in Fig. 2).
Figure 2.
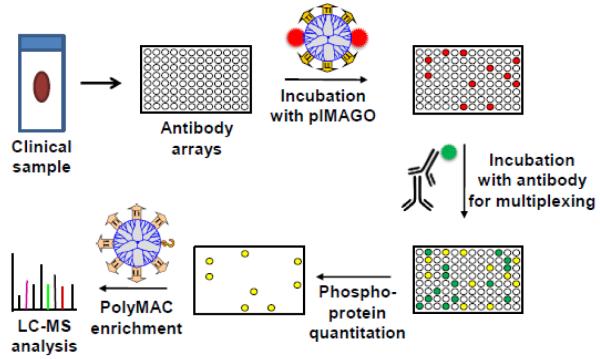
Integrated phosphoproteomics approach that combines molecular target enrichment using antibody microarrays, pIMAGO-based detection and quantitation of phosphorylation, and LC-MS analysis of effected phosphoproteins.
This strategy would have the inherent benefit of an unbiased detection of possible phosphorylation changes in protein markers of interest through antibody arrays without the need to analyze every phosphoprotein by mass spectrometry in the first step, thus allowing for targeted analyses only of those proteins involved in the signaling events. While still far-fetched, such an approach provides an example of a valuable platform for impartial and balanced high-throughput phosphoproteomic analysis of clinically-relevant samples.
Is clinical research ready for phosphoproteomics?
One of the great challenges in the application of phosphoproteomics to clinical sample analysis is the transition from controlled laboratory settings to highly heterogeneous environment of clinical sample collection and preservation. Non-standardized handling of samples can often result in significant variability of results, and thus hinder interpretation and subsequent treatment. This is particularly true for phosphoproteomics, where a simple and commonly-used formalin fixation is not a viable approach due to the slow preservation process, which allows ample time for unwanted ex vivo protease and phosphatase activities. This concern was to some extent addressed by the recent publication from Mueller et.al., who used a combination of enzyme inhibitors, permeation enhancers, reversible cross-linkers, and a balanced buffer to improve phosphorylation profile stabilization while still allowing for efficient histology analysis [44]. Nonetheless, their one-step biomarker and histology preservative approach still uses chemical interference, thus potentially introducing undesirable artifacts into the analysis.
As a result, snap-freezing remains the most preferred approach for proteomic sample preservation. While effective, this strategy cannot be universal, as it cannot constantly be practiced in the surgical room where the primary attention has been on the patient and not the sample preservation. Requirement for liquid nitrogen or dry ice for storage and transportation is often another limiting factor. In response, new techniques are being continuously introduced that allow effective sample preservation instantly without a negative effect on the total protein levels or amount of phosphorylation. Among them, the most notable protocol is based on the inactivation of enzymes through heat and pressure, originally introduced by Svensson et.al. [45], and already made into a successful commercial product [46]. While this method has great potential for phosphoproteome stabilization, it was shown to negatively affect mRNA levels. Thus, the search for the most optimal universal sample collection and preservation approach for comprehensive systems biology analysis is still undergoing. Without radical improvements, this might well be the major hurdle to bring phosphoproteomics to clinical research.
Overall, significant advancements in the proteomics and phosphoproteomics technologies in recent years have provided us with a wealth of information regarding biochemical signaling networks and disease progression pathways. Integration of these strategies into the comprehensive and integrative systems biology analysis of all relevant biomolecules would generate an incredibly valuable tool for disease sub-classification, formulation of targeted therapy, and in-depth examination of treatment outcomes and possible drug resistance [47]. A publication by Michael Snyder’s groups this year has examined the possibility of using a systems biology approach to molecular profiling of a single individual using their integrative personal “omics” profile (iPOP) strategy [48]. The preliminary observations have already demonstrated the power of detailed dynamic “omics” analysis on the molecular and physiological scale, producing interesting results with significant medical implications. It can be easily envisioned that the introduction of phosphoproteomics will add another dimension that could provide the ultimate tool to characterize cellular states for personalized medicine. The task is enormous but, with much at stake, analytical chemists and clinical researchers need to work together to face the challenge.
Highlights.
-
○
Personalized medicine redefines current and future clinical research
-
○
Phosphoproteomics measures signaling pathways for better description of cellular states
-
○
Current phosphoproteomics platforms are not appropriate for clinical research
-
○
A new platform embracing protein arrays and targeted proteomics is proposed for clinical research
Acknowledgment
This project was funded in part by an NSF CAREER award CHE-0645020, National Institutes of Health grant 1R01GM088317, and National Institute of Food and Agriculture (NIFA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- [1].Hood L, Flores M. A personal view on systems medicine and the emergence of proactive P4 medicine: predictive, preventive, personalized and participatory. N Biotechnol. 2012;29:613. doi: 10.1016/j.nbt.2012.03.004. [DOI] [PubMed] [Google Scholar]
- [2].Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Regenass U, Lydon NB. Selective inhibition of the platelet-derived growth factor signal transduction pathway by a protein-tyrosine kinase inhibitor of the 2-phenylaminopyrimidine class. Proc Natl Acad Sci U S A. 1995;92:2558. doi: 10.1073/pnas.92.7.2558. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [3].Druker BJ, Lydon NB. Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J Clin Invest. 2000;105:3. doi: 10.1172/JCI9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mauro MJ, Druker BJ. STI571: targeting BCR-ABL as therapy for CML. Oncologist. 2001;6:233. doi: 10.1634/theoncologist.6-3-233. [DOI] [PubMed] [Google Scholar]
- [5].Sherbenou DW, Druker BJ. Applying the discovery of the Philadelphia chromosome. J Clin Invest. 2007;117:2067. doi: 10.1172/JCI31988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Knight KA, Shokat KM. Features of selective kinase inhibitors. Chem Biol. 2005;12:621. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- [7].Juan WC, Ong ST. The role of protein phosphorylation in therapy resistance and disease progression in chronic myelogenous leukemia. Prog Mol Biol Transl Sci. 2012;106:107. doi: 10.1016/B978-0-12-396456-4.00007-9. [DOI] [PubMed] [Google Scholar]
- [8].Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, Fabbro D, Hallek M, Van Etten RA, Li S. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004;36:453. doi: 10.1038/ng1343. [DOI] [PubMed] [Google Scholar]
- [9].Hu Y, Swerdlow S, Duffy TM, Weinmann R, Lee FY, Li S. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci U S A. 2006;103:16870. doi: 10.1073/pnas.0606509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].O’Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65:4500. doi: 10.1158/0008-5472.CAN-05-0259. [DOI] [PubMed] [Google Scholar]
- [11].Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- [12].Rubin BP, Duensing A. Mechanisms of resistance to small molecule kinase inhibition in the treatment of solid tumors. Lab Invest. 2006;86:981. doi: 10.1038/labinvest.3700466. [DOI] [PubMed] [Google Scholar]
- [13].Carayol N, Vakana E, Sassano A, Kaur S, Goussetis DJ, Glaser H, et al. Critical roles for mTORC2- and rapamycin-insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proc Natl Acad Sci U S A. 2010;107:12469. doi: 10.1073/pnas.1005114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chu S, Holtz S, Gupta M, Bhatia R. BCR/ABL kinase inhibition by imatinib mesylate enhances MAP kinase activity in chronic myelogenous leukemia CD34+ cells. Blood. 2004;103:3167. doi: 10.1182/blood-2003-04-1271. [DOI] [PubMed] [Google Scholar]
- [15].Chan DA, Giaccia AJ. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov. 2011;10:351. doi: 10.1038/nrd3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- [17].Haura EB. From modules to medicine: How modular domains and their associated networks can enable personalized medicine. FEBS Lett. 2012;586:2580. doi: 10.1016/j.febslet.2012.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Riedel RF. Targeted agents for sarcoma: is individualized therapy possible in such a diverse tumor type? Semin Oncol. 2011;38(Suppl 3):S30. doi: 10.1053/j.seminoncol.2011.09.003. [DOI] [PubMed] [Google Scholar]
- [19].Mischel PS, Shai R, Shi T, Horvath S, Lu KV, Choe G, et al. Identification of molecular subtypes of glioblastoma by gene expression profiling. Oncogene. 2003;22:2361. doi: 10.1038/sj.onc.1206344. [DOI] [PubMed] [Google Scholar]
- [20].Li M, Wang IX, Li Y, Bruzel A, Richards AL, Toung JM, Cheung VG. Widespread RNA and DNA sequence differences in the human transcriptome. Science. 2011;333:53. doi: 10.1126/science.1207018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ideker T, Thorsson V, Ranish JA, Christmas R, Buhler J, Eng JK, et al. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 2001;292:929. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- [22].Tian Q, Stepaniants SB, Mao M, Weng L, Feetham MC, Doyle MJ, et al. Integrated genomic and proteomic analyses of gene expression in Mammalian cells. Mol Cell Proteomics. 2004;3:960. doi: 10.1074/mcp.M400055-MCP200. [DOI] [PubMed] [Google Scholar]
- [23].Macek B, Mann M, Olsen JV. Global and site-specific quantitative phosphoproteomics: principles and applications. Annu Rev Pharmacol Toxicol. 2009;49:199. doi: 10.1146/annurev.pharmtox.011008.145606. [DOI] [PubMed] [Google Scholar]
- [24].Tan HT, Lee YH, Chung MC. Cancer proteomics. Mass Spectrom Rev. 31:583. doi: 10.1002/mas.20356. [DOI] [PubMed] [Google Scholar]
- [25].Mueller C, Liotta LA, Espina V. Reverse phase protein microarrays advance to use in clinical trials. Mol Oncol. 2010;4:461. doi: 10.1016/j.molonc.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Malinowsky K, Wolff C, Ergin B, Berg D, Becker KF. Deciphering signaling pathways in clinical tissues for personalized medicine using protein microarrays. J Cell Physiol. 2010;225:364. doi: 10.1002/jcp.22307. [DOI] [PubMed] [Google Scholar]
- [27].Wulfkuhle JD, Speer R, Pierobon M, Laird J, Espina V, Deng J, et al. Multiplexed cell signaling analysis of human breast cancer applications for personalized therapy. J Proteome Res. 2008;7:1508. doi: 10.1021/pr7008127. [DOI] [PubMed] [Google Scholar]
- [28].Gembitsky DS, Lawlor K, Jacovina A, Yaneva M, Tempst P. A prototype antibody microarray platform to monitor changes in protein tyrosine phosphorylation. Mol Cell Proteomics. 2004;3:1102. doi: 10.1074/mcp.M400075-MCP200. [DOI] [PubMed] [Google Scholar]
- [29].Nielsen UB, Cardone MH, Sinskey AJ, MacBeath G, Sorger PK. Profiling receptor tyrosine kinase activation by using Ab microarrays. Proc Natl Acad Sci U S A. 2003;100:9330. doi: 10.1073/pnas.1633513100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, Kornblau SM. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5:2512. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- [31].Steelman LS, Abrams SL, Whelan J, Bertrand FE, Ludwig DE, Basecke J, et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia. 2008;22:686. doi: 10.1038/leu.2008.26. [DOI] [PubMed] [Google Scholar]
- [32].Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P, et al. Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY) 2011;3:192. doi: 10.18632/aging.100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cox DM, Zhong F, Du M, Duchoslav E, Sakuma T, McDermott JC. Multiple reaction monitoring as a method for identifying protein posttranslational modifications. J Biomol Tech. 2005;16:83. [PMC free article] [PubMed] [Google Scholar]
- [34].Narumi R, Murakami T, Kuga T, Adachi J, Shiromizu T, Muraoka S, et al. A Strategy for Large-Scale Phosphoproteomics and SRM-Based Validation of Human Breast Cancer Tissue Samples. J Proteome Res. 2012 doi: 10.1021/pr3005474. [DOI] [PubMed] [Google Scholar]
- [35].Domanski D, Murphy LC, Borchers CH. Assay development for the determination of phosphorylation stoichiometry using multiple reaction monitoring methods with and without phosphatase treatment: application to breast cancer signaling pathways. Anal Chem. 2010;82:5610. doi: 10.1021/ac1005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Pan C, Olsen JV, Daub H, Mann M. Global effects of kinase inhibitors on signaling networks revealed by quantitative phosphoproteomics. Mol Cell Proteomics. 2009;8:2796. doi: 10.1074/mcp.M900285-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rubbi L, Titz B, Brown L, Galvan E, Komisopoulou E, Chen SS. Global phosphoproteomics reveals crosstalk between Bcr-Abl and negative feedback mechanisms controlling Src signaling. Sci Signal. 2011;4:18. doi: 10.1126/scisignal.2001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Xue L, Wang WH, Iliuk A, Hu L, Galan JA, Yu S, et al. Sensitive kinase assay linked with phosphoproteomics for identifying direct kinase substrates. Proc Natl Acad Sci U S A. 2012;109:5615. doi: 10.1073/pnas.1119418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Andersen JN, Sathyanarayanan S, Di Bacco A, Chi A, Zhang T, Chen AH, et al. Pathway-based identification of biomarkers for targeted therapeutics: personalized oncology with PI3K pathway inhibitors. Sci Transl Med. 2010;2:43ra55. doi: 10.1126/scitranslmed.3001065. [DOI] [PubMed] [Google Scholar]
- [40].Alcolea MP, Casado P, Rodriguez-Prados JC, Vanhaesebroeck B, Cutillas PR. Phosphoproteomic analysis of leukemia cells under basal and drug-treated conditions identifies markers of kinase pathway activation and mechanisms of resistance. Mol Cell Proteomics. 2012;11:453. doi: 10.1074/mcp.M112.017483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Iliuk A, Martinez JS, Hall MC, Tao WA. Phosphorylation assay based on multifunctionalized soluble nanopolymer. Anal Chem. 2011;83:2767. doi: 10.1021/ac2000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Iliuk A, Liu XS, Xue L, Liu X, Tao WA. Chemical visualization of phosphoproteomes on membrane. Mol Cell Proteomics. 2012;11:629. doi: 10.1074/mcp.O112.018010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Iliuk AB, Martin VA, Alicie BM, Geahlen RL, Tao WA. In-depth analyses of kinase-dependent tyrosine phosphoproteomes based on metal ion functionalized soluble nanopolymers. Mol Cell Proteomics. 2010;9:2162. doi: 10.1074/mcp.M110.000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mueller C, Edmiston KH, Carpenter C, Gaffney E, Ryan C, Ward R, et al. One-step preservation of phosphoproteins and tissue morphology at room temperature for diagnostic and research specimens. PLoS One. 2011;6:e23780. doi: 10.1371/journal.pone.0023780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Svensson M, Boren M, Skold K, Falth M, Sjogren B, Andersson N, et al. Heat stabilization of the tissue proteome: a new technology for improved proteomics. J Proteome Res. 2009;8:974. doi: 10.1021/pr8006446. [DOI] [PubMed] [Google Scholar]
- [46].Rountree CB, Van Kirk CA, You H, Ding W, Dang H, VanGuilder HD, Freeman WM. Clinical application for the preservation of phospho-proteins through in-situ tissue stabilization. Proteome Sci. 2010;8:61. doi: 10.1186/1477-5956-8-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tian Q, Price ND, Hood L. Systems cancer medicine: towards realization of predictive, preventive, personalized and participatory (P4) medicine. J Intern Med. 2011;271:111. doi: 10.1111/j.1365-2796.2011.02498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen R, Mias GI, Li-Pook-Than J, Jiang L, Lam HY, Miriami E, et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell. 2012;148:1293. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]