

Abstract
Nanoparticles (NPs) are being extensively studied as carriers for drug delivery, but they often have limited penetration inside tumor. We envision that by targeting an endocytic receptor on cell surface, the uptake of NPs can be significantly enhanced through receptor mediated endocytosis. In addition, if the receptor is recycled to cell surface, the NP cargo can be transported out of the cells, which are then taken up by neighboring cells thus enhancing solid tumor penetration. To validate our hypothesis, in the first of two articles, we report the synthesis of doxorubicin (DOX)-loaded, hyaluronan (HA) coated silica nanoparticles (SNP) containing a highly fluorescent core to target CD44, a receptor expressed on cancer cell surface. HA was conjugated onto amine-functionalized SNPs prepared through an oil/water microemulsion method. The immobilization of the cytotoxic drug DOX was achieved through an acid sensitive hydrazone linkage. The NPs were fully characterized by transmission electron microscopy (TEM), dynamic light scattering (DLS), zeta potential measurements, thermal gravimetric analysis (TGA), UV-vis absorbance, and nuclear magnetic resonance (NMR). Initial biological evaluation experiments demonstrated that compared to ligand-free SNPs, the uptake of HA-SNP by the CD44-expressing SKOV-3 ovarian cancer cells was significantly enhanced when evaluated in the 2D monolayer cell culture. Mechanistic studies suggested that cellular uptake of HA-SNP was mainly through CD44 mediated endocytosis. HA-SNPs with DOX immobilized were endocytosed efficiently by the SKOV-3 cells as well. The enhanced tumor penetration and drug delivery properties of HA-SNP will be evaluated in 3D tumor models in the subsequent paper.
Keywords: CD44, fluorescent nanoparticles, hyaluronan, transcytosis, tumor penetration
1. Introduction
Chemotherapy is one of the major methods for cancer treatment. Traditionally, drugs are administered orally or intravenously resulting in their systemic distribution inside the body. To enhance the percentage of drugs reaching the diseased site and reduce offsite side effects and toxicities, drug delivery using NPs is undergoing intensive research.1–4 There are two general approaches to target tumor. The first strategy is termed passive targeting, which takes advantage of the enhanced permeability and retention (EPR) effect.5, 6 Due to the uncontrolled growth, tumor tissues are often characterized by a leaky vasculature that permits the escape of nanometer sized particles from the blood vessels into the tumor. However, the rapid growth also leads to incomplete development of the lymphatic drainage system resulting in high interstitial pressure. This restricts the free diffusion of NPs and drugs in the tumor mass with limited infiltration to areas not in the immediate vicinity of the vasculature.7–12 The limited drug penetration of solid tumor is one of the potential mechanisms of resistance to chemotherapeutic agents.13, 14 In addition, due to the lack of inherent affinity between the passively targeted NPs and tumor cells, the uptake of NPs is often low and non-specific, which results in suboptimal drug load inside the cells. To improve tumor uptake, active targeting approaches have been developed by modifying NPs with affinity ligands, which are selectively recognized by receptors over-expressed on the surface of tumor cells.3 While such an approach can significantly enhance the binding and uptake of NPs, large amounts of NPs will be absorbed by tumor cells closer to the vasculature of solid tumors due to the high affinity between targeting ligands and tumor cells. This will lead to a rapid reduction in the concentrations of NP/drug conjugates as the distance from the vasculature increases, resulting in decreased killing of distal tumor cells. Furthermore, a small population of cells within some tumors, which are referred to as cancer stem cells or tumor initiating cells, possess the ability to self-renew and develop into new cancer cells.15, 16 Some purported cancer stem cells are believed to be residing in the core of the tumor and the inability to eradicate cancer stem cells is hypothesized to be associated with tumor recurrence and chemotherapy failure. Therefore, it is important to be able to deliver NP/drug deeply inside the tumor for a more effective tumor therapy.
CD44 is a cell surface receptor expressed on the surface of a variety of cancer cells, and is known to mediate cancer cell interaction with the extracellular matrix. CD44 plays important roles in cancer development, drug resistance and metastasis.17–20 In addition, it is found highly expressed on cancer stem cells21 for many types of tumors including breast,22 colorectal23 and ovarian cancer.24, 25 The major endogenous ligand for CD44 is HA,26, 27 a naturally existing polysaccharide.28 The binding of CD44 and HA is under tight physiological control. On the vast majority of normal cells, CD44 exists at a low affinity state with little interactions with HA, which requires inflammatory signals to be activated.29 In contrast, many tumor-derived cells constitutively express CD44 in the high affinity state capable of HA binding,30 rendering it an attractive target for anti-tumor drug delivery.
To enhance NP penetration of tumor, we propose an approach utilizing the transcytosis process mediated by a cell surface receptor, i.e., CD44. In this manuscript, we describe the synthesis and characterization of DOX-loaded HA-coated fluorescent SNPs, and provide evidence that illustrates the CD44 mediated endocytosis of HA-SNPs by cancer cells. Detailed results on transcytosis leading to enhanced penetration of the targeted NPs in 3D cancer models will be presented in the subsequent manuscript.31
2. Materials and Methods
2.1. Materials and instrumentation
All chemical were reagent grade and were used as received from the manufacturers. Bovine serum albumin (BSA), 3-(trihydroxysilyl)propylmethylphosphonate (THPMP), cyclohexane, 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT), N-methylmorpholine (NMM), N,N-diethylaminopropyl carbodiimide hydrochloride (EDCI), fetal bovine serum (FBS) and sodium chloride were purchased from Sigma-Aldrich. Triton® X-100 was purchased from Supleco. 28–30% Ammonium hydroxide (NH4OH) and 30% hydrogen peroxide were purchased from CCI. Amberlite® IR 120 hydrogen form (Amberlite H+) was purchased from Fluka. ADH, 3,3′,5,5′-tetramethylbenzidine (TMB), n-hexanol, 3-(aminopropyl)-triethoxysilane (APTES), and tetraethylorthosilicate (TEOS) were purchased from Acros Organics. HA (31 kDa) was purchased from Lifecore Biomedicals. Doxorubicin hydrochloride was purchased from Shanghai FChemicals Technology Co. SKOV-3 cell line was purchased from American Type Culture Collection (ATCC). Phosphate buffered saline (PBS), Dulbecco’s Modified Eagle Medium (DMEM), fluorescein isothiocyanate (FITC), sodium pyruvate (100 mM), glutamine, and Penicillin-Streptomycin (Pen Strep) mixture were purchased from Invitrogen. Ultrafiltration membranes were purchased from Millipore. Dialysis tubing was obtained from BioDesign Inc. The Hermes-1 antibody was purchased from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biology, Iowa City, IA 52242. SKOV-3 cell line was cultured in DMEM supplemented with 10% inactivated FBS, 1% Pen-Strep mixture, glutamine (2 mM) and sodium pyruvate (1mM). DLS and zeta potential measurements were performed on a Zetasizer Nano zs apparatus (Malvern, UK). TEM images for nanoparticles were collected on a JEM-2200FS operating at 200 kV using Gatan multiscan CCD camera with Digital Micrograph imaging software. TEM images for NP-treated cells were obtained on JEOL100 CXII microscope. TGA was carried on a Thermal Advantage (TA-Instruments-Waters LLC) TGA-Q500 series and the samples were burned under nitrogen. Fluorescence activated cell sorting (FACS) experiments were conducted on a BD Vantage SE flow cytometer. NMR spectra of DOX-HA-SNP were collected on a Varian Avance 900 spectrometer (900 MHz) while those of HA polymer and free DOX were collected on a Varian Inova 500 (500 MHz). All confocal laser microscopy images were collected on an Olympus FluoView 1000 LSM confocal microscope. UV-vis measurements were carried out on a UV-4001 spectrometer (Hitachi High-Technologies Co., Japan).
2.2. Competitive enzyme linked immunosorbent assay (ELISA) assay
IgG-Fc (100 μL) was added to the wells of a 96-well microtiter plate, and the plate was stored at 4 °C overnight after which the excess IgG-Fc was removed and the wells were washed with 0.5% PBS-Tween 20 (PBST). The wells were blocked by adding 5% BSA in PBS (200 μL), and the plate was incubated at 37 °C for 2 h. The wells were washed as above. CD44-Fc γ chimera (0.2 μg/well, 100 μL) in PBS was added to the wells, and the plate was incubated at 37 °C for 45 min. The wells were then washed using 0.05% PBST. Biotinated HA (b-HA)32 was added to each well to bind with the immobilized CD44. To generate the IC50 curve, various concentrations of HA-SNPs (4.88 × 10−3, 1.95 × 10−2, 7.82 × 10−2, 0.3125, 1.25, 5, and 20μg-HA/mL) were added to each well to compete with b-HA for CD44 binding. The plate was stored at room temperature for 2 h. The wells were then washed using 0.05% PBST. Avidin-HRP (1:2000 dilution, 100 μL) in 0.2% BSA-PBS was added to each well and the plate was stored at room temperature for 1 h. The wells were washed using 0.05% PBST and twice with PBS buffer. A fresh TMB solution was prepared by dissolving TMB (5 mg) in DMSO (2 ml), followed by addition of citrate phosphate buffer (18 mL). 30% H2O2 (20 μL) was added just before use. TMB solution (100 μL) was added to the wells, and the plate was stored at room temperature in the dark for 15 min, or until the blue color appears. The reaction was then quenched by adding 0.5 M H2SO4 (50 μL) to each well with positive wells turning yellow. Optical absorbance was directly measured through the bottom of the microtiter plate using an automated plate reader (Bio-Rad) at 450 nm.
2.3. Qualitative determination of NP uptake by laser confocal microscopy
SKOV-3 cells (2 × 105 cells/well) were cultured in a 4-well chambered plate at 37 °C and 5% CO2 for 24 h. The culture media was removed and the cells were washed with PBS (2 times). FITC-doped SNPs with equivalent fluorescence in serum-free DMEM were added. The cells were incubated with the NPs for 5 h. Lysotracker red (1 μM, 50 μl/well) was added 1 h before completion of incubation. The supernatant was removed. The cells were washed twice with PBS, and fixed with 10% formalin (0.5 ml/well) for 15 min. Formalin was removed and the cells were washed twice with PBS. 4′,6-Diamidino-2-phenylindole (DAPI, 300 nM, 300 μl/well) was added, and the cells were incubated for 4–5 min. The supernatant was removed, and the cells were washed with PBS and water. The plate was covered by an aluminum foil and stored at 4 °C till imaging time.
2.4. Evidence for HA-SNP uptake by TEM
SKOV-3 cells were cultured in 100 mm cell culture plate at 37 °C and 5% CO2 till it reached 70–80% confluency (5 × 106 cells/plate). The growth media was removed and the cells were washed with PBS. HA-SNP (85 μg-NP/ml; 10 ml) in serum free DMEM was added and the cells were incubated for 18 h. The NPs were removed, and the cells were washed with PBS three times. The cells were collected by trypsinization followed by centrifugation (2500 rpm, 4 °C) for 5 min. The supernatant was removed and the fixing reagent (0.5 ml) was added (paraformaldehyde/glutaraldehyde 2.5 % each in 0.1 M sodium cacodylate buffer pH 7.4). The cells were incubated in the fixing reagent for 18 h at 4 °C. The cells were centrifuged, embedded in 2% agarose, treated with 1% osmium tetraoxide in 0.1 M sodium cacodylate buffer and dehydrated in a series of acetone dilutions. The cells were embedded in Poly/Bed 812 resin and sections were made on copper grids. Images were collected on TEM.
2.5. Evidence for CD44-dependence for efficient HA-SNP binding
100,000 cells were transferred to Eppendorf tubes, and washed with PBS. The cells were fixed with 10% formalin (200 μl/tube) for 15–20 min. Formalin was removed and the cells were washed with PBS (3 times). Hermes-1 (rat antihuman CD44 IgG2a mAb) (2 μg or 4 μg in 100 μl PBS) or KM81 (rat antimouse CD44 IgG2a mAb) (4 μg in 100 μl PBS) was added to some tubes, while the others received PBS (100 μl). The cells were incubated with the antibody for 2 h at room temperature. HA-SNP (1.7 mg/ml, 10 μl) and SNP (0.7 mg/ml, 10 μl) (equivalent fluorescence) were added to the antibody-free and antibody-containing tubes. The cells were incubated for 90 min. A tube that did not receive antibody or NPs was used as a control. Following washing, PBS (400 μl) was added to all tubes, and the cells were transferred to FACS tubes. Fluorescence was assessed on a flow cytometer.
3. Results and discussion
3.1. Synthesis and characterization of HA-SNPs and DOX-HA-SNPs
Several types of HA-NPs including gold NPs,33, 34 magnetic NPs,32, 35, 36 liposomes37–42 and polymersomes43–56 have been utilized for innovative applications in tumor imaging and drug delivery studies.57–59 In this work, we synthesized HA coated fluorescent SNPs to aid cellular tracking. There are several potential advantages associated with this new type of HA-NP. The SNPs can be rendered highly fluorescent by encapsulating high concentrations of a fluorophore such as FITC in the core of the NP. Having the fluorophore buried in the core can avoid the concern that if the hydrophobic fluorophore is attached on the external surface of NPs, it may alter NP properties and affect the NP interactions with cells.
The preparation of a SNP with a FITC core (Fig. 1a) started from conjugation of FITC with APTES. The resulting FITC-APTES was co-polymerized with TEOS to form SNPs in an oil-water microemulsion60 created by mixing cyclohexane (oil), Triton X-100 (surfactant), n-hexanol (co-surfactant) with water. However, the fluorescence of these NPs was low, which was attributed to the inefficient FITC incorporation presumably due to the slower polymerization rate of FITC-APTES compared to TEOS.61 To overcome this problem, FITC-APTES was allowed to self-polymerize first in the oil-water microemulsion. After the reaction had proceeded for six hours, TEOS was added to the reaction mixture to coat the fluorescent core. This step-wise polymerization protocol led to highly fluorescent SNPs, with the core shell structures of the NPs clearly visible from the TEM images (Fig. 1b). The NPs were functionalized by the Stöber reaction to immobilize APTES on SNP surface, which was followed by attaching THPMP to enhance water solubility leading to SNPs with diameters around 88 nm (Fig. S1a) and a zeta potential of −40.4 mV (Table S1). The negative value of zeta potential indicates that negative charges dominate the SNP surface.
Figure 1.

a) Synthesis of the fluorescent SNP, HA-SNP, and DOX-HA-SNP. b) TEM image of HA-SNP showing the core-shell structure of the NP core (the scale bar is 100 nm). c) Pictures of HA-SNP (left) and DOX-HA-SNP (right) solutions. The orange color of DOX-HA-SNP solution suggested the successful incorporation of DOX on the NPs.
The HA-SNPs were produced by immobilizing HA (31 kDa) onto SNPs through amide bond formation promoted by CDMT.62, 63 Naturally existing HA has molecular weight ranging from 10 kDa to 10 MDa depending on its source and purification procedures.28 CD44 contains a single HA binding domain, which can accommodate a HA oligosaccharide up to hexasaccharide in length.64 On cell surface, there are multiple copies of CD44, which can be simultaneously engaged in binding a HA polymer. Although the avidity of low molecular weight HA towards cell surface CD44 receptors is lower than that of the corresponding high molecular weight counterpart, it can still compete with endogenous HA for receptor binding.65, 66 Low molecular weight HA has been shown to be endocytosed by cells much faster.67 Furthermore, it was easier to separate the unreacted low molecular weight HA from the SNPs during purification of HA-SNPs. These considerations led us to select the HA with 31 kDa molecular weight to modify SNPs. The HA-SNPs had hydrodynamic diameters of 112 nm and zeta potential of −52.0 mV similar to SNPs (Fig. S1a, Table S1). TGA revealed that 31% of the weight of HA-SNPs was due to HA (Fig. S1b). HA on HA-SNPs retained its biological recognition with CD44 as HA-SNPs competitively inhibited the binding between a biotinated HA polymer with CD44 immobilized on an ELISA plate (Fig. 2).
Figure 2.
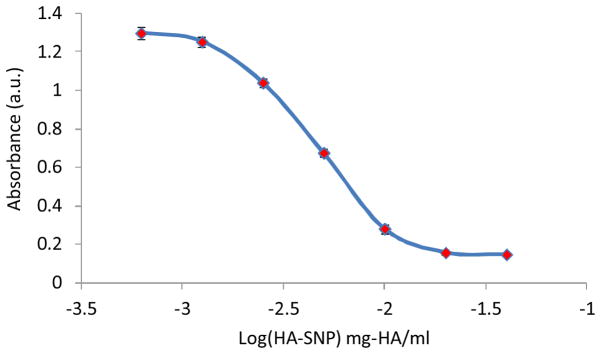
HA-SNP competitively inhibited the binding of a biotinated-HA to CD44 immobilized on a microtiter plate. Each data point is an average reading of at least three wells.
To test the application in drug delivery, a chemotherapeutic drug DOX was utilized as a model since it has been shown to have limited penetration in solid tumors.13, 68 To immobilize DOX, HA-SNP was first functionalized with adipic dihydrazide (ADH-HA-SNP) (Fig. 1a). Conjugating ADH to the carboxyl groups of HA was performed in the presence of fivefold excess of ADH at pH 4.5–5 and 0.2 equivalent of EDCI to reduce interparticle crosslinking by ADH and keep the majority of carboxylic acids of HA free. DOX was then conjugated to the ADH-HA-SNP through a hydrazone linkage between the C-13 ketone group of DOX and the hydrazide.69 A large excess of DOX was used to ensure all hydrazides were consumed. The success of the reaction was evident from the NP color change from yellow to orange upon DOX incubation with ADH-HA-SNP followed by thorough washing (Fig. 1c). UV-vis (Fig. S1c) and NMR measurements (Fig. S2) confirmed the presence of DOX on HA-SNP with a loading of 0.6% DOX by weight. The comparable zeta potential values of HA-SNP and DOX-HA-SNP suggested that functionalizing HA-SNP with DOX did not affect the surface characteristics of NPs much (Table S1).
For drug delivery applications, it is important that the drug/NP conjugate is stable under physiological conditions with little premature release of the therapeutic cargo. Once the conjugates reach the tumor, the drug needs to be released to exert its cytotoxic effects. Previously, we have shown that DOX linked with HA functionalized superparamagnetic magnetite NPs through the hydrazone linker was stable at pH 7.4, and was released when pH dropped to 4.5, the acidic environment typically encountered in late endosomes and lysosomes.32 The DOX release was not due to HA backbone cleavage at this mild acidic pH. We performed similar experiments with DOX-HA-SNP. DOX attached on DOX-HA-SNP was stable at pH 7.4 with only a very small amount of DOX found in the incubation media (Fig. 3). In contrast, at pH 4.5, DOX was rapidly cleaved from DOX-HA-SNP and released in the media (Fig. 3). This suggested that DOX can be freed from the SNPs once they are internalized by the cells and reach late endosomes and lysosomes.
Figure 3.
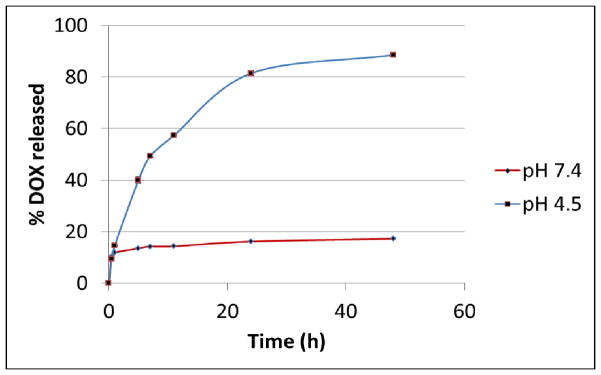
In vitro DOX release from DOX-HA-SNP at pH 7.4 and 4.5. Significant DOX release was observed at pH 4.5.
3.2. Binding and internalization of HA-SNPs by CD44 expressing SKOV-3 ovarian cancer cells
With HA-SNPs in hand, their interactions with the CD44 expressing ovarian cancer cell line SKOV-370 were examined first using confocal microscopy. The cells were incubated with NPs for 5 hours followed by washing to remove all the unbound particles. Confocal microscopy showed that the SNPs without HA were mainly located on the cell surface with little internalization presumably due to non-specific binding (Fig. 4a). The introduction of HA onto SNPs greatly enhanced NP uptake as extensive intracellular uptake was observed when HA-SNPs were incubated with the cells. The NPs appeared as bright green spots in the cytoplasm of the cells and excellent co-localization with a lysosome tracker Lysotracker red (Fig. 4b). To confirm the uptake of HA-SNPs into cancer cells, the HA-SNP loaded cells were imaged by TEM (Fig. 4c–e and Fig. S3). Many HA-SNPs were observed clustered in vesicles presumably endosomes and/or lysosomes (Fig. 4d). Interestingly, some HA-SNPs appeared to be dispersed in cytoplasm, probably due to their escape from the endosomes (Fig. 4e). Weakly basic drugs such as DOX can be sequestered in acidic endosomes thus reducing their efficacies.71, 72 The ability of the NPs to escape from the endosomes/lysosomes can enhance the delivery of drugs to the cytoplasm.
Figure 4.

Laser confocal microscopy images collected for cell incubated with (a) SNP and (b) HA-SNP, and stained with DAPI and Lysotracker red. (a1, b1) overlay DIC images and FITC channel, (a2, b2) DAPI channel showing location of the nucleus, (a3, b3) FITC channel showing location of SNPs, (a4, b4) Texas red channel showing Lysotracker red, and (a5, b5) an overlay of DIC images, FITC, DAPI, and Texas red channels. These images clearly showed the SNPs were mainly located on the outer membrane of the cells, while HA-SNPs were mainly inside the cells. (c–e) TEM images showing the intracellular distribution of HA-SNPs in SKOV-3 cells. (c) Uptake of NPs by cancer cells (Scale bar 2 μm). (d) NPs inside vesicles (Scale bar 100 nm). (e) NPs in the cytoplasm (Scale bar 500 nm).
The amounts of NPs taken up by SKOV-3 cells were quantified by flow cytometry (Fig. 5). The fluorescence intensities of SKOV-3 cells upon incubation with HA-SNPs were 8 times higher than those incubated with SNP (Fig. 5a). This suggested that HA significantly enhanced cellular binding and uptake of the NPs. The uptake of HA-SNPs by SKOV-3 was concentration and time dependent with the maximum intracellular fluorescence reached at 12 hours (Fig. 5b,c). The kinetics of uptake was also confirmed by confocal microscopy with significant increase of intracellular fluorescence over 12 hours (Fig. 5d).
Figure 5.

(a) Flow cytometry analysis showed that the binding and uptake of HA-SNP by SKOV-3 cells was much higher than that by SNP (HA-SNP and SNP with equal fluorescent intensities were incubated with the cells). (b) Time dependent uptake of HA-SNP by SKOV-3 cancer cells as determined by flow cytometry. The uptake of HA-SNP reached a maximum after 12 h. c) Concentration dependent uptake of HA-SNP by SKOV-3 cancer cells as determined by flow cytometry (1x = 80 μg NP/mL). d) Confocal microscopy images supporting the time dependent uptake of HA-SNP by SKOV-3 cells. (d1–d7) shows the overlay of the DAPI and FITC channels, while (d8) shows the overlay of DAPI and FITC channels on DIC image.
In order to demonstrate the role of CD44 in HA-SNP and SKOV-3 cell interactions, blocking experiments were performed using anti-CD44 antibodies. Hermes-1, a rat anti-human CD44 IgG2a antibody, is known to recognize the HA binding domain of CD44 and to competitively block HA binding with CD44.42, 73 While Hermes-1 (2 μg) did not affect the binding of SNP to SKOV-3 cells much, it reduced HA-SNP binding to the cells by about 80% (Fig. 6a). Further reduction of HA-SNP binding was observed with 4 μg of Hermes-1. As a control, a subtype matched rat IgG2a antibody KM81 was tested in the same setting, which displayed no influence on HA-SNP binding with SKOV-3 cells at all (Fig. 6b). Therefore, the interactions between HA-SNPs and SKOV-3 were largely through CD44.
Figure 6.

Flow cytometry histograms showing the binding of SNP and HA-SNP to CD44 expressing SKOV-3 cells in the absence and presence of (a) the antiCD44 mAb Hermes-1 or (b) the isotype matched KM81. Hermes-1 led to 80% reduction in HA-SNP but not SNP binding to SKOV-3 cells. On the other hand, KM81 had no effect on the binding of either SNP or HA-SNP. (c) Flow cytometry showing the effect of β-CD on the uptake of HA-SNP. Pre-treating the cells with β-CD reduced the uptake of HA-SNP by 85% demonstrating that uptake of HA-SNP by SKOV-3 cells was mediated through cholesterol rich lipid rafts. (d) Flow cytometry showing the relative binding/uptake of HA-SNP and SNP by SKOV-3 cells at 37 °C compared to those at 4 °C. The binding/uptake of HA-SNP, but not SNP, was energy dependent. For this experiment, higher concentration of SNP than HA-SNP was used for cellular incubation due to the low uptake of SNP.
It has been shown that the endocytosis of CD44-HA complexes goes through cholesterol rich lipid rafts rather than caveolae or clathrin-coated pits.37, 65, 74, 75 Addition of a cholesterol binder, i.e., β-cyclodextrin, during NP incubation should disrupt the lipid rafts and CD44 mediated HA uptake.37 Indeed, the cellular uptake of HA-SNP dropped by 85% in the presence of β-cyclodextrin (Fig. 6c). As receptor mediated endocytosis is energy dependent, we examined the effect of temperature on cellular uptake. Incubation of SKOV-3 with HA-SNP at 4°C drastically reduced the amount of NPs bound and internalized compared to that at 37°C (Fig. 6d). In contrast, cellular binding of SNP was not affected much by incubation temperature (Fig. 6d). These results were consistent with the notion that HA-SNP uptake by SKOV-3 cells was through CD44 mediated endocytosis.37, 75
3.3. Internalization of DOX-HA-SNP by SKOV-3 cells in 2D cell culture
Having demonstrated CD44 mediated uptake of HA-SNP, we investigated interactions of DOX-HA-SNP with SKOV-3 cancer cells in the traditional monolayer 2D culture. Upon incubation with DOX-HA-SNPs for 24 hours, the cells were imaged with confocal microscopy. Similar to those acquired with HA-SNPs (Fig. 5d), much green color from the SNPs was found in the cytoplasm confirming DOX-HA-SNPs were internalized (Fig. 7b). Extensive red color due to DOX was observed in both cytoplasm and the nucleus suggesting some DOX was released, which diffused to the nucleus (Fig. 7c). Importantly, DOX was also present on HA-SNP as evident from the bright yellow spots due to the overlay of green fluorescence from SNP and red color from DOX (Fig. 7d–f). This indicates that some DOX was still retained on the SNPs after cellular uptake, which could be exocytosed to enhance drug penetration as to be demonstrated in the subsequent paper.31
Figure 7.
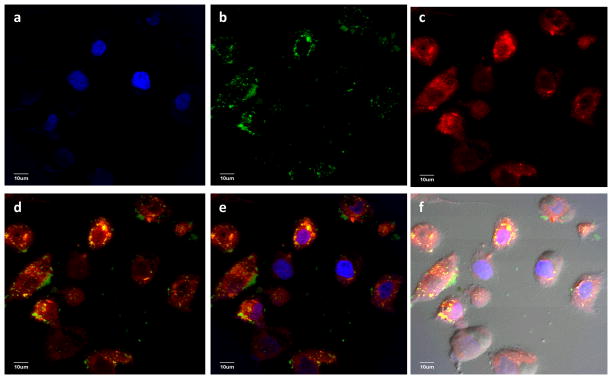
Laser confocal microscopy images collected for SKOV-3 cell incubated with DOX-HA-SNPs for 24 hours after extensive wash to remove the unbound particles. (a) DAPI channel showing location of nucleus; (b) FITC channel showing location of SNPs; (c) Texas red channel showing location of DOX; (d) an overlay of (b) and (c); (e) an overlay of (a), (b), and (c); (f) An overlay of DIC image and (e). These images clearly demonstrated that DOX-HA-SNPs were internalized inside the cells and not all DOX was released from the NPs as evident from the yellow spots in d–f.
4. Conclusions
HA-SNPs were synthesized as potential drug carriers with enhanced tumor penetration abilities. HA significantly enhanced NP uptake by the CD44 expressing SKOV-3 ovarian cancer cells as compared to unfunctionalized SNPs. The collective analysis of the uptake experiments conducted in the presence of an anti-CD44 mAb (Hermes-1), lipid raft disrupter (β-cyclodextrin), or at low temperatures (4 °C) suggested that HA-SNPs were mainly taken up through CD44 mediated endocytosis. DOX immobilized on HA-SNPs were also efficiently delivered to SKOV-3 cells. The ability of HA coating to enhance the penetration potential of SNPs in tumors by transcytosis will be described in the following manuscript.
Supplementary Material
Acknowledgments
This work was supported by NIH (R01CA149451) and an NSF CAREER award.
Footnotes
Electronic supplementary information (ESI) available: detailed experimental procedures on synthesis and characterization nanoparticles, confocal studies, flow cytometry, and supporting figures and tables. See DOI: #####.
References
- 1.Wang AZ, Langer R, Farokhzad OC. Annu Rev Med. 2012;63:185–198. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]
- 2.Adair JH, Parette MP, Altınoğlu EI, Kester M. ACS Nano. 2010;4:4967–4970. doi: 10.1021/nn102324e. [DOI] [PubMed] [Google Scholar]
- 3.Byrne JD, Betancourt T, Brannon-Peppas L. Adv Drug Del Rev. 2008;60:1615–1626. doi: 10.1016/j.addr.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Cho K, Wang X, Nie S, Chen Z, Shin DM. Clin Cancer Res. 2008;14:1310–1316. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- 5.Fang J, Nakamura H, Maeda H. Adv Drug Del Rev. 2011;63:136–151. doi: 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 6.Maeda H. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 7.Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK. Cancer Res. 1994;54:3352–3356. [PubMed] [Google Scholar]
- 8.Minchinton AI, Tannock IF. Nature Rev Cancer. 2006;6:583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 9.Heldin CH, Rubin K, Pietras K, Oestman A. Nat Rev Cancer. 2004;4:806–813. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- 10.Kim JJ, Tannock IF. Nat Rev Cancer. 2005;5:516–525. doi: 10.1038/nrc1650. [DOI] [PubMed] [Google Scholar]
- 11.Jain RK, Tong RT, Munn LL. Cancer Res. 2007;67:2729–2735. doi: 10.1158/0008-5472.CAN-06-4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jain RK. Cancer Res. 1990;50:814s–819s. [PubMed] [Google Scholar]
- 13.Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. Clin Cancer Res. 2005;11:8782–8788. doi: 10.1158/1078-0432.CCR-05-1664. [DOI] [PubMed] [Google Scholar]
- 14.Tannock IF, Lee CM, Tunggal JK, Cowan DSM, Egorin MJ. Clin Cancer Res. 2002;8:878–884. [PubMed] [Google Scholar]
- 15.Sarkar B, Dosch J, Simeone DM. Chem Rev. 2009;109:3200–3208. doi: 10.1021/cr9000397. [DOI] [PubMed] [Google Scholar]
- 16.Visvader JE, Lindeman GJ. Nature Rev Cancer. 2008;8:755–769. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 17.Ween MP, Oehler MK, Ricciardelli C. Int J Mol Sci. 2011;12:1009–1029. doi: 10.3390/ijms12021009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marhaba R, Zöller M. J Mol Histol. 2004;35:211–231. doi: 10.1023/b:hijo.0000032354.94213.69. [DOI] [PubMed] [Google Scholar]
- 19.Draffin JE, McFarlane S, Hill A, Johnston PG, Waugh DJJ. Cancer Res. 2004;64:5702–5711. doi: 10.1158/0008-5472.CAN-04-0389. [DOI] [PubMed] [Google Scholar]
- 20.Naor D, Nedvetzki S, Golan I, Melnik L, Faitelson Y. Cric Rev Clin Lab Sci. 2002;39:527–579. doi: 10.1080/10408360290795574. [DOI] [PubMed] [Google Scholar]
- 21.Zöller M. Nature Rev Cancer. 2011;11:254–267. doi: 10.1038/nrc3023. [DOI] [PubMed] [Google Scholar]
- 22.Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, Halushka MK, Sukumar S, Parker LM, Anderson KS, Harris LN, Garber JE, Richardson AL, Schnitt SJ, Nikolsky Y, Gelman RS, Polya K. Cancer Cell. 2007;11:259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 23.Yeung TM, Gandhi SC, Wilding JL, Muschel R, Bodmer WF. Proc Natl Acad Sci USA. 2010;107:3722–3727. doi: 10.1073/pnas.0915135107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng W, Liu T, Wan X, Gao Y, Wang H. FEBS J. 2012;279:2047–2059. doi: 10.1111/j.1742-4658.2012.08589.x. [DOI] [PubMed] [Google Scholar]
- 25.Alvero AB, Fu HH, Holmberg J, Visintin R, Mor L, Marquina CC, Oidtman J, Silasi DA, Mor G. Stem Cells. 2009;27:2405–2413. doi: 10.1002/stem.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lesley J, Hascall VC, Tammi M, Hyman R. J Biol Chem. 2000;275:26967–26975. doi: 10.1074/jbc.M002527200. [DOI] [PubMed] [Google Scholar]
- 27.Lesley J, He Q, Miyake K, Hamman A, Hyman R, Kincade PW. J Exp Med. 1992;175:257–266. doi: 10.1084/jem.175.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lapcik L, Jr, Lapcik L, De Smedt S, Demeester J, Chabrecek P. Chem Rev. 1998;98:2663–2684. doi: 10.1021/cr941199z. [DOI] [PubMed] [Google Scholar]
- 29.Maiti A, Maki G, Johnson P. Science. 1998;282:941–943. doi: 10.1126/science.282.5390.941. [DOI] [PubMed] [Google Scholar]
- 30.Cichy J, Puré E. J Cell Biol. 2003;161:839–843. doi: 10.1083/jcb.200302098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El-Dakdouki MH, Puré E, Huang X. Nanoscale. 2013 doi: 10.1039/c3nr90022c. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.El-Dakdouki MH, Zhu DC, El-boubbou K, Kamat M, Chen J, Li W, Huang X. Biomacromolecules. 2012;13:1144–1151. doi: 10.1021/bm300046h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu W, Shen J, Banerjee P, Zhou S. Biomaterials. 2010;31:7555–7566. doi: 10.1016/j.biomaterials.2010.06.030. [DOI] [PubMed] [Google Scholar]
- 34.Lee H, Lee K, Kim IK, Park TG. Biomaterials. 2008;29:4709–4718. doi: 10.1016/j.biomaterials.2008.08.038. [DOI] [PubMed] [Google Scholar]
- 35.Lee Y, Lee H, Kim YB, Kim J, Hyeon T, Park H, Messersmith PB, Park TG. Adv Mater. 2008;20:4154–4157. doi: 10.1002/adma.200800756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar A, Sahoo B, Montpetit A, Behera S, Lockey RF, Mohapatra SS. Nanomedicine. 2007;3:132–137. doi: 10.1016/j.nano.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 37.Qhattal HS, Liu X. Mol Pharmaceutics. 2011;8:1233–1246. doi: 10.1021/mp2000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rivkin I, Cohen K, Koffler J, Melikhov D, Peer D, Margalit R. Biomaterials. 2010;31:7106–7114. doi: 10.1016/j.biomaterials.2010.05.067. [DOI] [PubMed] [Google Scholar]
- 39.Peer D, Margalit R. Neoplasia. 2004;6:343–353. doi: 10.1593/neo.03460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bachar G, Cohen K, Hod R, Feinmesser R, Mizrachi A, Shpitzer T, Katz O, Peer D. Biomaterials. 2011;32:4840–4848. doi: 10.1016/j.biomaterials.2011.03.040. [DOI] [PubMed] [Google Scholar]
- 41.Eliaz RE, Nir S, Marty C, Szoka FC., Jr Cancer Res. 2004;64:711–718. doi: 10.1158/0008-5472.can-03-0654. [DOI] [PubMed] [Google Scholar]
- 42.Surace C, Arpicco S, Dufay-Wojcicki A, Marsaud V, Bouclier C, Clay D, Cattel L, Renoir JM, Fattal E. Mol Pharmaceutics. 2009;6:1062–1073. doi: 10.1021/mp800215d. [DOI] [PubMed] [Google Scholar]
- 43.Cho HJ, Yoon IS, Yoon HY, Koo H, Jin YJ, Ko SH, Shim JS, Kim KS, Kwon IC, Kim DD. Biomaterials. 2012;33:1190–1200. doi: 10.1016/j.biomaterials.2011.10.064. [DOI] [PubMed] [Google Scholar]
- 44.Upadhyay KK, Bhatt AN, Mishra AK, Dwarakanath BS, Jain S, Schatz C, Le Meins JF, Farooque A, Chandraiah G, Jain AK, Misra A, Lecommandoux S. Biomaterials. 2010;31:2882–2892. doi: 10.1016/j.biomaterials.2009.12.043. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Zhu X, Zhang X, Liu B, Huang L. Mol Therapeutics. 2010;18:1650–1656. doi: 10.1038/mt.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen MS, Cai S, Xie Y, Forrest ML. Am J Surg. 2009;198:781–786. doi: 10.1016/j.amjsurg.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jeong YI, Kim ST, Jin SG, Ryu HH, Jin YH, Jung TY, Kim IY, Jung S. J Pharm Sci. 2008;97:1268–1276. doi: 10.1002/jps.21103. [DOI] [PubMed] [Google Scholar]
- 48.Xin D, Wang Y, Xiang J. Pharm Res. 2009;27:380–389. doi: 10.1007/s11095-009-9997-9. [DOI] [PubMed] [Google Scholar]
- 49.Choi KY, Min KH, Yoon HY, Kim K, Park JH, Kwon IC, Choi K, Jeong SY. Biomaterials. 2011;32:1880–1889. doi: 10.1016/j.biomaterials.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 50.Laroui H, Grossin L, Leonard M, Stoltz J-F, Gillet P, Netter P, Dellacherie E. Biomacromolecules. 2007;8:3879–3885. doi: 10.1021/bm700836y. [DOI] [PubMed] [Google Scholar]
- 51.Yim H, Na K. Biomacromolecules. 2010;11:2387–2393. doi: 10.1021/bm100562z. [DOI] [PubMed] [Google Scholar]
- 52.He M, Zhao Z, Yin L, Tang C, Yin C. Int J Pharm. 2009;373:165–173. doi: 10.1016/j.ijpharm.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 53.Yadav AK, Mishra P, Mishra AK, Mishra P, Jain S, Agrawal GP. Nanomedicine. 2007;3:246–257. doi: 10.1016/j.nano.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 54.Hyung W, Ko H, Park J, Lim E, Park SB, Park YJ, Yoon HG, Suh JS, Haam S, Huh YM. Biotechnol Bioeng. 2007;99:442–454. doi: 10.1002/bit.21578. [DOI] [PubMed] [Google Scholar]
- 55.Lee H, Lee K, Park TG. Bioconjugate Chem. 2008;19:1319–1325. doi: 10.1021/bc8000485. [DOI] [PubMed] [Google Scholar]
- 56.Pitarresi G, Craparo EF, Palumbo FS, Carlisi B, Giammona G. Biomacromolecules. 2007;8:1890–1898. doi: 10.1021/bm070224a. [DOI] [PubMed] [Google Scholar]
- 57.El-Dakdouki MH, Huang X. Petite and sweet: glyco-nanotechnology as a bridge to new medicines. In: Huang X, Barchi J, editors. ACS Symposium Series. 2011. pp. 181–213. and references cited therein. [Google Scholar]
- 58.Prestwich GD. J Control Release. 2011;155:193–199. doi: 10.1016/j.jconrel.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Platt VM, Szoka FC., Jr Mol Pharmaceutics. 2008;5:474–486. doi: 10.1021/mp800024g. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Santra S, Yang H, Dutta D, Stanley JT, Holloway PH, Tan W, Moudgil BM, Mericle RA. Chem Commun. 2004:2810–2811. doi: 10.1039/b411916a. [DOI] [PubMed] [Google Scholar]
- 61.Wang L, Tan W. Nano Lett. 2006;6:84–88. doi: 10.1021/nl052105b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bergman K, Elvingson C, Hilborn J, Svensk G, Bowden T. Biomacromolecules. 2007;8:2190–2195. doi: 10.1021/bm0701604. [DOI] [PubMed] [Google Scholar]
- 63.Kamat M, El-Boubbou K, Zhu DC, Lansdell T, Lu X, Li W, Huang X. Bioconjugate Chem. 2010;21:2128–2135. doi: 10.1021/bc100354m. [DOI] [PubMed] [Google Scholar]
- 64.Banerji S, Wright AJ, Noble M, Mahoney DJ, Campbell ID, Day AJ, Jackson DG. Nature Struc Mol Biol. 2007;14:234–239. doi: 10.1038/nsmb1201. [DOI] [PubMed] [Google Scholar]
- 65.Tammi R, Rilla K, Pienimaki JP, MacCallum DK, Hogg M, Luukkonen M, Hascall VC, Tammi M. J Biol Chem. 2001;276:35111–35122. doi: 10.1074/jbc.M103481200. [DOI] [PubMed] [Google Scholar]
- 66.Tammi R, MacCallum D, Hascall VC, Pienimäki JP, Hyttinen M, Tammi M. J Biol Chem. 1998;273:28878–28888. doi: 10.1074/jbc.273.44.28878. [DOI] [PubMed] [Google Scholar]
- 67.Ouasti S, Kingham PJ, Terenghi G, Tirelli N. Biomaterials. 2012;33:1120–1134. doi: 10.1016/j.biomaterials.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 68.Gerweck LE, Vijayappa S, Kozin S. Mol Cancer Ther. 2006;5:1275–1279. doi: 10.1158/1535-7163.MCT-06-0024. [DOI] [PubMed] [Google Scholar]
- 69.Wang F, Wang YC, Dou S, Xiong MH, Sun TM, Wang J. ACS Nano. 2011;5:3679–3692. doi: 10.1021/nn200007z. [DOI] [PubMed] [Google Scholar]
- 70.Bourguignon LYW, Zhu H, Chu A, Iida N, Zhang L, Hung MC. J Biol Chem. 1997;272:27913–27918. doi: 10.1074/jbc.272.44.27913. [DOI] [PubMed] [Google Scholar]
- 71.Lee CM, Tannock IF. Br J Cancer. 2006;94:863–869. doi: 10.1038/sj.bjc.6603010. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hurwitz SJ, Terashima M, Mizunuma N, Slapak CA. Blood. 1997;89:3745–3754. [PubMed] [Google Scholar]
- 73.Li L, Heldin CH, Heldin P. J Biol Chem. 2006;281:26512–26519. doi: 10.1074/jbc.M605607200. [DOI] [PubMed] [Google Scholar]
- 74.Lee JL, Wang MJ, Sudhir PR, Chen JY. Mol Cell Biol. 2008;28:5710–5723. doi: 10.1128/MCB.00186-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thankamony SP, Knudson W. J Biol Chem. 2006;281:34601–34609. doi: 10.1074/jbc.M601530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.