

Abstract
We assessed the geographical distribution of C9orf72 G4C2 expansions in a pan-European frontotemporal lobar degeneration (FTLD) cohort (n = 1,205), ascertained by the European Early-Onset Dementia (EOD) consortium. Next, we performed a meta-analysis of our data and that of other European studies, together 2,668 patients from 15 Western European countries. The frequency of the C9orf72 expansions in Western Europe was 9.98% in overall FTLD, with 18.52% in familial, and 6.26% in sporadic FTLD patients. Outliers were Finland and Sweden with overall frequencies of respectively 29.33% and 20.73%, but also Spain with 25.49%. In contrast, prevalence in Germany was limited to 4.82%. In addition, we studied the role of intermediate repeats (7–24 repeat units), which are strongly correlated with the risk haplotype, on disease and C9orf72 expression. In vitro reporter gene expression studies demonstrated significantly decreased transcriptional activity of C9orf72 with increasing number of normal repeat units, indicating that intermediate repeats might act as predisposing alleles and in favor of the loss-of-function disease mechanism. Further, we observed a significantly increased frequency of short indels in the GC-rich low complexity sequence adjacent to the G4C2 repeat in C9orf72 expansion carriers (P < 0.001) with the most common indel creating one long contiguous imperfect G4C2 repeat, which is likely more prone to replication slippage and pathological expansion.
Keywords: FTLD, C9orf72, repeat expansion, intermediate alleles, European Early-Onset Dementia consortium
Introduction
A pathological expansion of a hexanucleotide G4C2 repeat in the promoter region of the gene C9orf72 (MIM #614260) was recently identified as the long sought-after underlying gene defect [Dejesus-Hernandez et al., 2011; Gijselinck et al., 2012; Renton et al., 2011] of linkage [Boxer et al., 2011; Gijselinck et al., 2010; Luty et al., 2008; Le Ber et al., 2009; Morita et al., 2006; Pearson et al., 2011; Vance et al., 2006; Valdmanis et al., 2008] and association [Laaksovirta et al., 2010; Shatunov et al., 2010; van Es et al., 2009; Van Deerlin et al., 2010] of frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) to the chromosome 9p21 region [Van Langenhove et al., 2012a]. In the Flanders-Belgian population, we calculated that the pathological G4C2 expansion is the second most common genetic cause of FTLD [Gijselinck et al., 2012; Van Langenhove et al., 2012b]. Particularly in the subgroup of familial patients presenting with ALS also (FTLD–ALS), C9orf72 was the first causal gene explaining up to 85.71% of patients [Gijselinck et al., 2012; Van Langenhove et al., 2012b]. Since then, several patient cohorts of different geographical regions have been screened for this repeat expansion mutation establishing C9orf72 as a major gene for FTLD with frequencies of 7%–11% in total FTLD and 12%–25% in familial FTLD patients [Boeve et al., 2012; DeJesus-Hernandez et al., 2011; Ferrari et al., 2012; Gijselinck et al., 2012; Hsiung et al., 2012; Mahoney et al., 2012; Majounie 2012; Renton et al., 2011; Simon-Sanchez et al., 2012; Snowden et al., 2012].
Different possible disease mechanisms have been proposed including haploinsufficiency and RNA toxicity [Dejesus-Hernandez et al., 2011; Gijselinck et al., 2012] and extensive genotype–phenotype correlation studies are being reported [Al-Sarraj et al., 2011; Arighi et al., 2012; Bigio, 2012; Boeve et al., 2012; Chio et al., 2012; Dejesus-Hernandez et al., 2011; Ferrari et al., 2012; Gijselinck et al., 2012; Hsiung et al., 2012; Majounie et al., 2012; Mahoney et al., 2012; Murray et al., 2011; Renton et al., 2011; Simon-Sanchez et al., 2012; Snowden et al., 2012; Troakes et al., 2011; Whitwell et al., 2012]. However, very little or nothing is known about the mutation spectrum, the genomic mechanism by which the G4C2 repeat is expanding, and the impact of repeat length on disease susceptibility and gene expression.
In the present study, we aimed at expanding our C9orf72 observations in the Flanders-Belgian FTLD and FTLD–ALS cohort (n = 360) with a larger European cohort of 845 FTLD and FTLD–ALS patients, in which we determined the geographical distribution and prevalence of the pathological G4C2 expansion. Furthermore, we provide the first evidence for a role of G4C2 intermediate repeat length on C9orf72 expression and hypothesize on the genomic mechanisms favoring pathological expansion of the G4C2 repeat.
Materials and Methods
Study Populations
The European FTLD cohort was collected through the European Early-Onset Dementia (EOD) consortium (Supp. Table S1). The European EOD consortium was launched in August 2011 to centralize and harmonize epidemiological, clinical, and biological data together with biomaterial of EOD patients throughout the Europe to stimulate high-profile translational dementia research. Supp. Table S1 describes the number of patients per country and per clinical subgroup contributed by the European EOD consortium members. We received DNA and clinical and demographic information on 917 unrelated FTLD and FTLD–ALS patients as well as histopathology data of 46 patients obtained at autopsy. The 917 patients also included 10 patients from Wallonia, the French speaking part of Belgium, and six more patients from Italy, Spain, and Sweden, which were referred for clinical genetic testing to the Diagnostic Service Facility in our Department of Molecular Genetics (DMG DSF). Patients had been diagnosed according to established clinical diagnostic Neary criteria [Neary et al., 1998] and to the Mackenzie consensus criteria for neuropathology diagnosis [Mackenzie et al., 2010].
The Flanders-Belgian cohort consisted of 337 unrelated patients with FTLD and 23 with FTLD–ALS. These patients were recruited through the Belgian Neurology (BELNEU) consortium, a collaboration with neurologists affiliated to nine different specialized memory clinics and neurology departments in Belgium [Gijselinck et al., 2012; Van Langenhove et al., 2012b] (Supp. Table S2). In addition to the patient cohort, a Flanders-Belgian control cohort was assembled (n = 1,083). For more detailed description see Supp. Materials and Methods.
Histopathology of C9orf72 Expansion Carriers
From 11 C9orf72 G4C2 expansion carriers, formalin-fixed brain was available for immunohistochemistry. Five micrometer slices were obtained from frontal cortex, temporal neocortex, hippocampus, area striata, neostriatum, mesencephalon, pons, and cerebellum. Of seven cases, additional samples were provided from thalamus and spinal cord. Slides were stained against Ubiquitin, p62, hyperphosphorylated tau, β-amyloid, TDP43, and FUS. For technical details see Supp. Materials and Methods.
C9orf72 G4C2 Genotyping assays
We developed an alternative repeat-primed PCR assay (reverse RP-PCR; Fig. 1) and a short tandem repeat (STR) fragment length assay with flanking primers optimized for alleles with high GC content (STR-PCR; Fig. 1) allowing reliable identification of G4C2 expansion carriers and exact sizing of normal lengths. These assays were performed in both cohorts and in relatives of the younger generation of index patients carrying an intermediate repeat allele or a variation in the flanking LCS without expansion.
Figure 1.
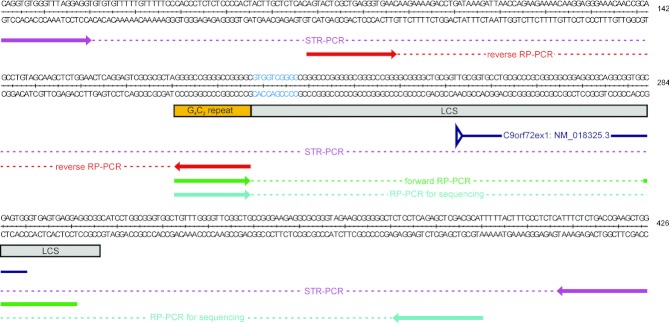
Genotyping assays to characterize the C9orf72 region and G4C2 repeat. The C9orf72 G4C2 repeat (yellow box) is located upstream of the first exon of isoform NM_018325.3 (dark blue arrow) and adjacent to a GC-rich low-complexity sequence (LCS; light grey box) with their nucleotide sequences shown above. The sequence of the recurrent 10-bp deletion g.26747_26756delGTGGTCGGGG (Table 4, Supp. Figure S1), we observed in the LCS, is indicated in blue. Below the sequence, the primers with their corresponding PCR amplicons are shown for each of the PCR genotyping assays: STR-PCR in pink, forward RP-PCR in green, reverse RP-PCR in red and RP-PCR for sequencing in blue.
For technical details on primers and amplification protocols see Supp. Materials and Methods.
Sequencing of the C9orf72 GC-Rich Low Complexity Sequence
We used the product of an alternative forward RP-PCR (RP-PCR for sequencing; Fig. 1) and sequenced the low complexity sequence (LCS) with the locus-specific reverse primer.
The Flanders-Belgian patient (n = 317) and control (n = 752) cohorts and 57 expansion carriers and 114 nonexpansion carriers of the European cohort were successfully screened. Cosegregation of LCS variations with the presence of a G4C2 expansion was analyzed in two available families. For technical details see Supp. Materials and Methods.
C9orf72 Exon Sequencing and Dosage Analysis
Both cohorts were screened for coding and splice-site mutations in C9orf72. The frequency of rare mutations was determined in 400 controls. Further, we screened the Flanders-Belgian cohort for exonic deletions or duplications using the Multiplex Amplicon Quantification technique [Kumps et al., 2010]. For technical details see Supp. Materials and Methods.
Genetic Association Studies
We calculated association with disease of C9orf72 intermediate G4C2 alleles and of SNP rs2814707 tagging the chromosome 9p21 risk haplotype stratifying for the presence or absence of C9orf72 intermediate repeats. Odds ratios (OR) with 95% confidence interval (CI) were calculated in a logistic regression model, adjusted for age and gender. In addition, we studied the correlation between the minor risk T-allele and intermediate repeat length. Further, in patients of both cohorts without a G4C2 expansion, we calculated correlation between age at onset and normal repeat length using a Kruskal–Wallis test. Further, we compared age at onset between short (2–6) and intermediate (7–24) repeat length in a Kaplan–Meier survival analysis. All analyses were done in IBM SPSS Statistics 20 (IBM Corporation, Armonk, NY, USA). For details see the Supp. Materials and Methods.
Luciferase Reporter Assays
We selected a 2 kb C9orf72 promoter fragment (chr9:27,572,414–27,574,451; NCBIBuild37 – hg19) containing the G4C2 repeat and enriched for histone marks, DNaseI hypersensitivity clusters, and transcription factor binding sites based on ENCODE transcription data [Gijselinck et al., 2012]. The fragment was obtained by PCR of individuals carrying different numbers of normal repeat units (2, 9, 17, and 24 units) using primers with flanked attB-sites. PCR products were cloned into the pDONR221 vector (Invitrogen, Life Technologies, Grand Island, NY, USA) by a BP recombination reaction (Invitrogen, Life Technologies, Grand Island, NY, USA), and the integrity of all inserts was confirmed by sequence analysis. Correct entry clones were selected and cloned into an in-house developed promoterless destination vector containing the Gaussia luciferase reporter gene downstream of a Gateway cassette, by use of a LR recombination reaction (Invitrogen, Life Technologies, Grand Island, NY, USA). Human HEK293T cells were propagated and seeded for transient transfection in 24-well tissue-culture dishes, at 2 × 105 cells per well, and were allowed to recover for 24 hr. Cells were cotransfected with 40 ng of pSV40-CLuc plasmid that encodes the Cypridina luciferase gene with a SV40 promoter (New England Biolabs, Ipswich, MA, USA) and 1,000 ng of three independent C9orf72 promoter constructs per unit, with use of 2.4 μl Lipofectamine 2000 (Invitrogen, Life Technologies, Grand Island, NY, USA), in duplo. After 24 hr, Gaussia luciferase activities (LAG) and Cypridina luciferase activities (LAC) were measured in duplo in the growth medium using a BioLux Gaussia and Cypridina Luciferase Assay Kit (New England Biolabs, Ipswich, MA, USA) and a Veritas Microplate Luminometer with Dual Reagent Injectors Luminometer (Promega, Madison, WI, USA). To correct for transfection efficiency and DNA uptake, the relative luciferase activity (RLA) was calculated as RLA = LAG/LAC. This experiment was repeated three times resulting in 36 measurements for each construct. RLAs between different repeat lengths were calculated by a Mann–Whitney U test. For details see Supp. Materials and Methods.
Results
The European EOD Consortium
The European cohort included 917 unrelated patients, of which 845 had a possible or probable diagnosis of FTLD (n = 781) or FTLD–ALS (n = 64; Table 1). In an additional 72 other patients, clinical presentation showed indications of FTLD together with symptomatology of other neurodegenerative brain diseases such as Alzheimer or Parkinson disease. A pathological diagnosis on autopsied brain was obtained for 45 patients, comprising FTLD-TDP (n = 28), FTLD-MND-TDP (n = 15), FTLD-UPS (n = 1), and FTLD-TAU (n = 1) diagnoses. Information on family history of disease was available for 609 (72.07%) of the 845 patients, of which 251 had a positive family history of disease and 358 were considered sporadic patients (Table 2). Average onset age and range were comparable between FTLD (62.7 ± 9.0, range 28–88 years) and FTLD–ALS patient groups (60.9 ± 9.8, range 31–83 years).
Table 1.
Descriptive Characteristics of the European and Flanders-Belgian Cohorts
| Clinical diagnosis cohorts | Total n | Familial n (%) | Disease onset ± SD (range) | Pathology n (%) |
|---|---|---|---|---|
| European cohort | ||||
| Total | 845 | 274 (32.43) | 62.5 ± 9.0 (28–88) | 45 (5.33) |
| FTLD | 781 | 251 (32.14) | 62.7 ± 9.0 (28–88) | 28 (3.59) |
| FTLD–ALS | 64 | 23 (35.94) | 60.9 ± 9.8 (31–83) | 17 (26.56) |
| Othera | 72 | 40 (55.56) | 60.0 ± 12.1 (35–95) | N.A. |
| Flanders-Belgian cohort | ||||
| Total | 360 | 108 (30.00) | 62.9 ± 9.7 (29–85) | 24 (6.67) |
| FTLD | 337 | 101 (29.97) | 63.0 ± 9.7 (29–85) | 21 (6.23) |
| FTLD–ALS | 23 | 7 (30.43) | 62.7 ± 10.0 (39–75) | 3 (13.04) |
| Combined European and Flanders-Belgian cohorts | ||||
| Total | 1205 | 382 (31.70) | 62.7 ± 9.2 (28–88) | 70 (5.81) |
| FTLD | 1118 | 352 (31.48) | 62.8 ± 9.2 (28–88) | 49 (4.38) |
| FTLD–ALS | 87 | 30 (34.48) | 61.4 ± 9.8 (31–83) | 21 (24.14) |
This group of other 72 patients was not included in the total because they did not fulfill the criteria for possible or probable diagnosis.
Table 2.
Frequencies of the C9orf72 Pathological G4C2 Expansion in the European and Flanders-Belgian Cohorts
| FTLD | FTLD–ALS | Total | ||||
|---|---|---|---|---|---|---|
| European cohort | ||||||
| Total | 50/781 | 6.40% | 23/64 | 35.94% | 73/845 | 8.64% |
| Familial | 21/251 | 8.37% | 11/23 | 47.83% | 32/274 | 11.68% |
| Sporadic | 13/358 | 3.63% | 5/27 | 18.52% | 18/385 | 4.68% |
| Othera | 16/172 | 9.30% | 7/14 | 50.00% | 23/186 | 12.37% |
| Flanders-Belgian cohort | ||||||
| Total | 21/337 | 6.23% | 7/23 | 30.43% | 28/360 | 7.78% |
| Familial | 12/101 | 11.88% | 6/7 | 85.71% | 18/108 | 16.67% |
| Sporadic | 9/236 | 3.81% | 1/16 | 6.25% | 10/252 | 3.97% |
| Combined European and Flanders-Belgian cohorts | ||||||
| Total | 71/1118 | 6.35% | 30/87 | 34.48% | 101/1205 | 8.38% |
| Familial | 33/352 | 9.38% | 17/30 | 56.67% | 50/382 | 13.09% |
| Sporadic | 22/594 | 3.70% | 6/43 | 13.95% | 28/637 | 4.40% |
| Othera | 16/172 | 9.30% | 7/14 | 50.00% | 23/186 | 12.37% |
Other includes patients of which there was no family history data available in the European cohort.
C9orf72 Pathological G4C2 Expansions
To determine the impact and distribution of the G4C2 expansion across Europe, G4C2 repeat lengths were determined by forward RP-PCR (Fig. 1). A G4C2 expansion was observed in 8.64% (73/845) of the European cohort (Table 2). Per clinical subgroup, 6.40% of FTLD and 35.94% of FTLD–ALS patients carried a pathological G4C2 expansion. In familial patients, the overall frequency increased to 11.68%, with frequencies of 8.37% (21/251) in FTLD, and 47.83% (11/23) in FTLD–ALS patients. In sporadic patients, the overall frequency decreased to 4.68%, with frequencies of 3.63% (13/358) in FTLD, and 18.52% (5/27) in FTLD–ALS.
To evaluate the distribution of the G4C2 expansion, we calculated, overall and per clinical phenotype, mutation frequencies per country (Supp. Table S2; Table 3). In Belgium, Portugal, and Italy, the pathological G4C2 expansion mutation showed a comparable overall frequency ranging between 6.09% and 7.86%, and close to the average overall European frequency of 8.38%. However, a marked enrichment was observed in the Spanish (25.49%) and Swedish (21.33%) patient cohorts. In contrast, in the German patients, only 3.52% were carriers.
Table 3.
Meta-analysis of C9orf72 Prevalence Studies in Western Europe
| Total | Familial | Sporadic | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Country | Total n | Carriers n | Carriers (%) | Total n | Carriers n | Carriers (%) | Total n | Carriers n | Carriers (%) | Studies represented in the meta-analysis |
| Totala | 2636 | 263 | 9.98 | 756 | 140 | 18.52 | 1804 | 113 | 6.26 | |
| Belgium | 369 | 29 | 7.86 | 115 | 19 | 16.52 | 254 | 10 | 3.94 | Gijselinck et al. (2012), European EOD consortium |
| Denmark | 82 | 10 | 12.20 | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | Lindquist et al. (2012) |
| Finland | 75 | 22 | 29.33 | 27 | 13 | 48.15 | 48 | 9 | 18.75 | Majounie et al. (2012) |
| France | 200 | 36 | 18.00 | 50 | 22.00 | 44 | 150 | 14 | 9.33 | Majounie et al. (2012) |
| Germany | 228 | 11 | 4.82 | 66 | 5 | 7.58 | 162 | 6 | 3.7 | European EOD consortium, Majounie et al. (2012) |
| Italy | 345 | 21 | 6.09 | 86 | 5 | 5.81 | 259 | 16 | 6.18 | European EOD consortium |
| The Netherlands | 340 | 35 | 10.29 | 116 | 30 | 25.86 | 224 | 5 | 2.23 | Majounie et al. (2012) |
| Portugal | 151 | 10 | 6.62 | 92 | 4 | 4.35 | 59 | 6 | 10.17 | European EOD consortium |
| Spain | 51 | 13 | 25.49 | 20 | 6 | 30 | 31 | 7 | 22.58 | European EOD consortium |
| Sweden | 82 | 17 | 20.73 | 14 | 8 | 57.14 | 74 | 9 | 12.16 | European EOD consortium, Majounie et al. (2012) |
| UK | 713 | 59 | 8.27 | 170 | 28 | 16.47 | 543 | 31 | 5.71 | Majounie et al. (2012) |
Meta-analysis combined prevalences from the European EOD consortium study, the Gijselinck et al., Lancet Neurol 2012 study [Gijselinck et al., 2012], the Majounie et al., Lancet Neurol 2012 study [Majounie et al., 2012], and the Lindquist et al., Clin Neurol study [Lindquist et al., 2012].
Patient samples from the Czech Republic, Bulgaria, and Austria from the European EOD consortium study, and from Sardinia from the Majounie study, contained less than 20 patients and were therefore excluded from the meta-analysis. N.A.: information not available.
C9orf72-Associated Clinical and Pathological Phenotype
Of 73 G4C2 expansion carriers in the European cohort, 50 received a clinical diagnosis of FTLD and 23 of FTLD–ALS. The average onset age in all carriers was 58.0 ± 7.5 years with an onset age range of 40–75 years and was comparable in the FTLD and FTLD–ALS subgroups (Supp. Table S3). The average disease duration in 31 deceased carriers was 5.3 ± 3.5 years (range 1–14 years) but differed in the clinical subgroups. Survival was on average 1.4 years shorter for the FTLD–ALS carriers with 4.7 ± 3.5 years (n = 17, range 1–14 years), compared with the FTLD patients with 6.1 ± 3.6 years (n = 14, range 1–14 years). Of 23 G4C2 expansion carriers, more extensive clinical information was available to allow subclassification to the different FTLD phenotypes. In 22, clinical presentation was conform bvFTD (95.65%) and one presented with PNFA. Autopsied brain was available for 11 European G4C2 expansion carriers (two Portuguese, five Spanish, one Austrian, one Czech, and two Swedish patients). In all 11 cases, we found TDP-43 pathology in the frontal and temporal neocortex, in the hippocampus and neostriatum, which was compatible with type B TDP proteinopathy [Mackenzie et al., 2010]. Accordingly, TDP-43 immunoreactive neuronal cytoplasmic inclusions (NCI) and dystrophic neurites were widespread over the entire cortical thickness, but NCI were more pronounced in the pyramidal cells of layer 2 compared with the deeper cortical layers. In addition to the TDP-43 positive pathology, p62 immunoreactive NCI were present in the granular layer of the dentate gyrus of the hippocampus, and in the granular layer of the cerebellar cortex. Further, p62 positive irregular granular NCI were observed in pyramidal neurons of the CA4 and CA3 region of the hippocampus.
Genomic Complexity in the C9orf72 Region
The C9orf72 G4C2 repeat is contiguous with a GC-rich, LCS, comprising exon 1 of the C9orf72 transcript NM_018325.3 (Fig. 1). We sequenced the GC-rich LCS in 317 unrelated patients of the Flanders-Belgian cohort and in 752 controls (Fig. 1). We observed heterozygous deletions of 5–23 base pairs (bp) in a total of 19 individuals of which 10 patients carrying a G4C2 expansion (10/27 = 37.04%), five noncarrier patients (5/290 = 1.72%), and four control persons (4/752 = 0.53%; Table 4). These variable deletions were significantly more frequently observed in carriers of a G4C2 expansion compared with the group of noncarrier patients (OR = 33.53; 95% CI 10.31–109.09; P < 0.001) and controls (OR = 93.50; 95% CI 28.53–306.39; P < 0.001). Remarkably, nine of 10 (90.00%) expansion carriers presented with the same heterozygous 10-bp GTGGTCGGGG deletion (g.26747_26756 delGTGGTCGGGG; Supp. Fig. S1), which was not observed in patient noncarriers and controls (Table 4). This 10-bp deletion is contiguous with the G4C2 repeat and joins two 100% GC sequences, thereby extending the GC-rich motif of the G4C2 repeat with imperfect repeats (Fig. 1). In this context, it is striking that deletion of the GTGGT motif (Fig. 1) was seen in all 10 deletion carriers of the 27 unrelated patients carrying an expanded G4C2 repeat (37.04%), only once in the noncarrier patients (1/290 = 0.34%) and once in control individuals (1/752 = 0.13%; Table 4). To replicate these findings, we successfully sequenced the LCS in 57 unrelated patient carriers and 114 patient noncarriers from the European cohort. We observed a comparable high frequency of deletions and insertions (indels) in the patient carriers (14/57 = 24.56%) and no indels in the noncarriers (0/114, <0.88%; OR = 36.79; 95% CI 4.69–288.35; P = 0.001; Table 4). Seven of 14 (50.00%) patient carriers presented the same 10-bp deletion and in 10 of 14 (71.43%) patient carriers GTGGT was deleted (Table 4). Of note, children of the FTLD patient with the g.26747_26751delGTGGT deletion but without expansion did not show de novo expansions. This deletion is located on an allele of 5 repeat units.
Table 4.
Low Complexity Sequence (LCS) Indels in Carriers of a Pathological G4C2 Expansion and in Noncarriers of the Flanders-Belgian and European cohorts
| Diagnosis | Number of individuals | Indel genomic mutation namea,b |
|---|---|---|
| Flanders-Belgian cohort | ||
| Patients expansion carriers (10/27) | ||
| FTLD | 7 | g.26747_26756delGTGGTCGGGG |
| FTLD–ALS | 2 | g.26747_26756delGTGGTCGGGG |
| FTLD–ALS | 1 | g.26742_26764delGGGGCGTGGTCGGGGCGGGCCCG |
| Patients without expansion (5/290) | ||
| FTLD | 1 | g.26747_26751delGTGGT |
| FTLD | 3 | g.26752_26762delCGGGGCGGGCC |
| FTLD—ALS | 1 | g.26752_26774delCGGGGCGGGCCCGGGGGCGGGCC |
| Controls (4/752) | ||
| Control | 3 | g.26752_26762delCGGGGCGGGCC |
| Control | 1 | g.26746_26761delCGTGGTCGGGGCGGGC |
| European cohort | ||
| Patients expansion carriers (14/57) | ||
| FTLD | 6 | g.26747_26756delGTGGTCGGGG |
| FTLD | 1 | g.26747_26751delGTGGT |
| FTLD | 1 | g.26747_26768delGTGGTCGGGGCGGGCCCGGGGG |
| FTLD | 1 | g.26753_26764delGGGGCGGGCCCG |
| FTLD | 1 | g.26775_26776insG |
| FTLD | 1 | g.26775_26776insGGGGCGGGCCCG |
| FTLD–ALS | 1 | g.26747_26756delGTGGTCGGGG |
| FTLD–ALS | 1 | g.26746_26773del CGTGGTCGGGGCGGGCCCGGGGGCGGGC |
| FTLD–ALS | 1 | g.26752_26762delCGGGGCGGGCC |
The nucleotide sequence GTGGT is most frequently deleted in the LCS adjacent to the G4C2 repeat in C9orf72 (indicated in bold).
gDNA numbering relative to reverse complement of contig AL451123.12 and starting at nucleotide 1.
The LCS is comprised in the PCR fragments produced by the forward RP-PCR assay to identify G4C2 expansion carriers (Fig. 1) [Gijselinck et al., 2012]. To eliminate influences of LCS variability in G4C2 expansion detection and for sizing normal repeat alleles, we developed a reverse RP-PCR assay on the sense strand (Fig. 1). This assay confirmed the presence of the G4C2 expansion in patients of both cohorts.
Sizing of Normal C9orf72 G4C2 Repeat Lengths
We used a STR genotyping assay (STR-PCR; Fig. 1), to size both normal alleles in noncarriers and the wild-type allele in G4C2 expansion carriers. In addition, we used the reverse RP-PCR assay (Fig. 1) to validate the size of the longest allele in nonexpansion carriers. When we compared the allele lengths between the two assays, we obtained 99% concordance. Discordant allele scoring could be explained by the presence of a LCS deletion. This implies that the STR-PCR can be used for correct sizing of normal repeat alleles. The observed lengths of the G4C2 repeat ranged from 2 to 24 units in nonexpansion carriers of the Flanders-Belgian cohort (g.26724GGGGCC[2_24]; relative to reverse complement of AL451123.12; Fig. 2A) and between 2 and 21 units in the European cohort. The shortest allele contained one unit less than the reference genome (NCBIbuild37–hg19; Fig. 1). Frequency distribution of normal repeat alleles showed a trimodal allele distribution (Fig. 2A). Using an estimation–maximization algorithm implemented in the R Package MCLUST, the three groups could be defined by a cutoff at 4 and 7 units (2–3 units, 4–6 units, and 7–24 units), with a probability of good classification of 0.9944.
Figure 2.
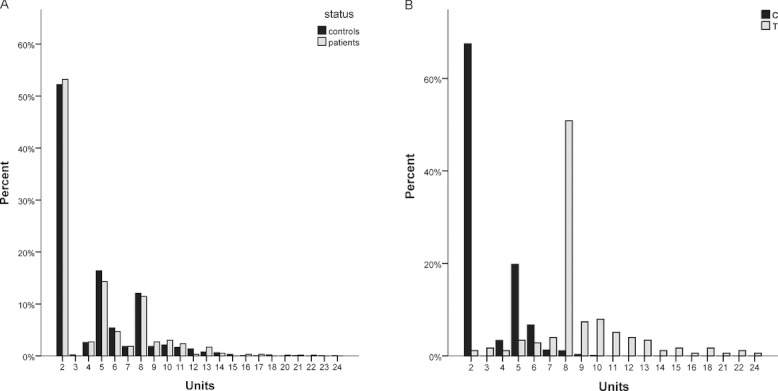
A: Distribution of normal repeat lengths in the Flanders-Belgian patients and control individuals. Histograms of G4C2 repeat units sized <60 repeats in Flanders-Belgian patients, excluding patients with mutations in known causal genes or with a pathological G4C2 expansion, compared with control individuals. B: Correlation of normal repeat lengths with rs2814707 alleles. Histograms of G4C2 repeat units in 610 control individuals homozygous for the rs2814707 C-allele and 53 homozygous for the rs2814707 T-allele.
Intermediate Repeat Length and the Chromosome 9 Risk Haplotype
In the Flanders-Belgian cohort, we previously had calculated a significant association of disease with the risk T-allele of rs2814707 (P = 0.008) [Gijselinck et al., 2012] tagging the chromosome 9p risk haplotype. Repeat expansion carriers contributed the largest fraction of the attributed risk but residual association signal remained in the nonexpansion carriers homozygous for the T-allele (OR = 1.75; 95% CI 1.02–3.01; P = 0.042; Supp. Table S4) [Gijselinck et al., 2012]. We used three different PCR genotyping assays to test for pathological G4C2 expansions, making it unlikely that the residual association could be explained by missed mutation carriers. Also, we analyzed C9orf72 for other mutations by sequencing all exons in 493 patients and by exon-based analysis of putative deletions/duplications in 413 patients. Except for one patient-specific missense mutations (c.196A>T in NM_018325.3 [p.Thr66Ser]) without a clear in silico deleterious effect, no other pathogenic mutations were identified. When we compared the distribution of normal repeat lengths between the rare T-allele and the common C-allele, we observed that alleles of at least 7 repeat units were strongly correlated with the T-allele (P < 0.001; Fig. 2B), corresponding with allele group 3 of the trimodal frequency distribution. We defined this group (7–24 units) as intermediate repeat alleles. When we recalculated genetic association of rs2814707 with disease after excluding individuals homozygous for intermediate alleles, the residual association disappeared (P = 0.121; Supp. Table S4). However, we could not demonstrate direct association of these intermediate alleles with disease (Supp. Table S5). Although a slight increase in frequency of intermediate alleles (25.1%) was observed as compared with the control individuals (22.6%; Supp. Table S5). Also homozygous intermediate repeat carriers were slightly more frequent in the patient group (6.1% vs. 4.6%).
We could not observe evidence in favor of the hypothesis that intermediate repeats are unstable and might trigger pathological G4C2 expansion, as the younger generation in 20 families of index patients with an intermediate repeat allele up to 22 units, did not show longer intermediate repeat alleles or pathological expansions.
Reporter Gene Analysis
To evaluate the effect of repeat length on C9orf72 promoter activity, we measured the RLA of constructs containing a C9orf72 promoter fragment with 2, 9, 17, and 24 units. We demonstrated a highly significant decrease of the RLA between fragments with 9, 17, and 24 units compared with the wild-type allele comprising 2 units (P < 0.001) with a maximum decrease of 52% in the 24 units containing promoter (Fig. 3). These data show that intermediate repeat alleles result in a significantly reduced C9orf72 promoter activity compared with the wild-type alleles.
Figure 3.

Transcriptional activity of C9orf72 promoter with alleles of different repeat length. Bars represent relative Gaussia/Cypridina luciferase activities (RLA) for the different C9orf72 constructs compared with the wild-type allele of 2 units, for an increasing amount of repeat units. Values represent the mean (±SDEV) of 36 independent measurements relative to the 2 units wild-type allele. The significance of differences in expression was calculated using the Mann–Whitney U test. P values are presented above the bars.
Discussion
In the present study, we have assessed the geographical distribution of the pathological G4C2 expansions in an extended pan-European patient cohort of FTLD patients, originating from Italy, Germany, Portugal, Sweden, Spain, Czech Republic, Bulgaria, Austria, and Belgium, ascertained within a newly formed, European EOD consortium. The prevalence of pathological G4C2 expansions in the total European cohort (73/845, 8.64%) as well as in the FTLD-only (50/781, 6.40%) and FTLD–ALS (23/64, 35.94%) subgroups, was comparable to frequencies others and we published in the three original gene identification reports on C9orf72 [Dejesus-Hernandez et al., 2011; Gijselinck et al., 2012; Renton et al., 2011].
The FTLD syndrome in the European G4C2 expansion carriers was most often characterized by behavioral disturbances (95.7%), which was in line with our observations from an in-depth genotype–phenotype correlation study in the Belgian C9orf72 carriers [Van Langenhove et al., 2012b]. Thirty-two percent of expansion carriers also developed concomitant ALS symptomatology. Further, we observed a wide range in onset age and disease duration, suggestive of genetic and/or environmental (epigenetic) disease modifiers [Van Langenhove et al., 2012b]. Of 13 G4C2 expansion carriers, post-mortem neuropathological diagnosis confirmed brain deposition of TDP-43 inclusions, explaining 30.23% (13/43) of TDP-43 positive patients. Because up to 70% of the TDP pathology patients remained unresolved after C9orf72 analysis, we performed mutation profiling of the other TDP-43-associated genes GRN, VCP, and TARDBP. This revealed one additional TARDBP mutation, p.Ile383Val, in a patient from the Barcelona Brain Bank. Interestingly, this mutation located in the C-terminal of the protein was reported once before in one US ALS family [Rutherford et al., 2008], yet in this Spanish patient—diagnosis SD, age at onset 60 years, age at death 75 years, father with dementia—no clinical nor pathological signs of MND were recorded. Taken together, of the 43 TDP-43 pathology patients 67.44% (29/43) remained unresolved by the known genes. In 17 familial TDP patients, this represented 58.82% (10/17) of patients without a known mutation, pointing to at least one, but likely more, other TDP-43-associated genes yet to be discovered. Eleven C9orf72 positive TDP-43 pathology carriers were submitted to further in-depth histopathological evaluation. Although the neuropathological signature of C9orf72 has proven to be more diverse than initially anticipated (reports ranged from strong TDP proteinopathy reminiscent of FTLD-TDP type A [Murray et al., 2011; Snowden et al., 2012; Stewart et al., 2012], to complete absence of TDP-43 pathology [Gijselinck et al., 2012; Murray et al., 2011] (reviewed in Cruts et al., Trends Neurosci, under revision), patients in our study all fitted the criteria of TDP proteinopathy type B [Mackenzie et al., 2010], albeit with relatively low lesion load. Eight of the 11 patients displayed concomitant AD-pathology, yet, AD stages were relatively mild (Braak stage I–III for neurofibrillary tangle pathology and Stage B for β-amyloid pathology) and probably this finding was age related more than related to the C9orf72 expansion. In the five cases stained with p62 antibody, 100% showed immunoreactive NCI in the dentate gyrus of the hippocampus, in the CA4 and CA23 pyramidal neurons of the hippocampus, and the granular cortical layer of the cerebellum. As this lesion load is far more pronounced than the TDP-43 lesions, it can be assumed that the presence of p62 pathology is a highly distinctive feature of C9orf72-associated FTLD [Al-Sarraj et al., 2011]. In another recent large-scale screening of the C9orf72 repeat expansion by Majounie et al. (2012), frequencies were reported for a total of 4,448 ALS patients and 1,425 FTLD patients from European and US Caucasian populations, and compared with smaller sets of patients from other ethnic backgrounds. Of interest with respect to the present study, the Majounie study included FTLD patients from the UK, the Netherlands, France, Finland, Germany, Sardinia, and Sweden. Except for a small number of German (n = 29) and Swedish (n = 7) patients, these countries were not represented in our study and vice versa. Further, a study conducted in Denmark identified 10 C9orf72 expansion carriers in a cohort of 82 Danish FTLD patients [Lindquist et al., 2012]. To get an even more representative picture of the prevalence and distribution of the C9orf72 repeat expansion in Western Europe, we performed a meta-analysis of the FTLD cohorts from our European EOD consortium study (n = 845), our previously reported Flanders-Belgian cohort (n = 360) [Gijselinck et al., 2012], the Majounie study (n = 1381) [Majounie et al., 2012], and the Lindquist study (n = 82) [Lindquist et al., 2012]. This resulted in prevalence numbers on 2,668 FTLD patients from 15 European countries (Table 3, countries with patients groups smaller than 20 were not included in the meta-analysis). The overall frequency of the C9orf72 pathological G4C2 expansion in Western Europe was 9.98% in total FTLD, 18.52% in familial FTLD, and 6.26% in sporadic FTLD. Looking at the per country distribution, Belgium, Italy, The Netherlands, Portugal, and the UK showed an average total prevalence ranging from 6.09% to 10.29%. Denmark showed a slightly increased prevalence of 12.20%, and in France the prevalence was about double the average prevalence with 18.00%. In line with the hypothesis of a Scandinavian founder effect for the chromosome 9—C9orf72 haplotype [Mok et al., 2011], frequencies were highest in Finland (29.33%) and high in Sweden (20.73%). Yet, in contrast with this hypothesis and the expected North–South axis for the founder haplotype prevalence, frequencies for C9orf72 reached 25.49% in Spain. To the other extreme, prevalence in Germany was just 4.82%. Of note, Ireland, Luxembourg, Switzerland, Poland, Norway, and Greece are not yet represented in this meta-analysis of Western Europe.
We exactly sized the normal range of repeat alleles in both the Flanders-Belgian and European cohorts using two different assays and observed a range between 2 and 24 units. The allele frequencies showed a trimodal distribution with a long tail containing the rare longer alleles. Alleles in group 3 of this trimodal distribution (intermediate alleles; 7–24 units) are almost solely present on the chromosome 9 risk haplotype tagged by the rs2814707 T-allele. We asked the question whether these alleles might act as a risk factor on FTLD disease susceptibility or could have a mild effect on gene expression. Although intermediate repeat alleles were not significantly associated with disease in the Flanders-Belgian cohort, the slight increase in frequency in patients compared with control individuals explained the residual association we had observed for homozygous carriers of the rs2814707 T-allele (OR = 1.75; 95% CI 1.02–3.01; P = 0.042). A group of 16 FTLD–ALS patients without pathological expansion was included in this cohort. Although this is a small group, it is remarkable that seven of 32 alleles (21.9%) have at least 10 units, which is significantly higher than 7.2% in control individuals (OR = 3.685; 95% CI 1.560–8.706; P = 0.003). This suggests that intermediate alleles might rather have a risk effect in diseases involving ALS. Because the European cohort was collected for a frequency study of the repeat expansion, we did not have ethnicity matched control individuals for an association study.
Flanders-Belgian and European patient carriers of intermediate repeats (7–24) did not show an earlier onset age compared with carriers of shorter repeats (2–6; data not shown). Also, the variable onset age in patients with a pathological G4C2 expansion [Gijselinck et al., 2012], could not be explained by a modifying effect of number of repeat units in the wild-type allele (data not shown), in contrast to what has been shown for patients with Huntington's disease [Aziz et al., 2009].
The mechanism by which the G4C2 intermediate repeats might contribute to disease pathogenesis remained elusive. In this study, we performed reporter gene expression studies to evaluate the effect of intermediate repeats according to their size on C9orf72 promoter activity. We provided compelling evidence that transcriptional activity of the C9orf72 promoter significantly decreases with an increasing amount of repeat units with a maximal reduction of 52% of a 24 units containing promoter compared with 2 units on the wild-type allele (Fig. 3). To better discriminate the cutoff from which decreased transcriptional activity is apparent, more different length alleles should be investigated, although from these data it is clear that intermediate alleles affect the normal transcriptional activity of the C9orf72 promoter. With the cloned intermediate alleles, we did not reach an expression level of virtually 0 as expected in pathogenic expansion alleles. Therefore, individuals with the number of units higher than 24 are needed to better interpret the grey zone between normal and expanded alleles. These in vitro data confirm that the G4C2 repeat is located in the promoter region of C9orf72 as we previously suggested [Gijselinck et al., 2012]. It also favors the loss-of-function hypothesis that was based on 50% reduction of C9orf72 levels in brain of pathological expansion carriers preventing expression of the mutant allele [Gijselinck et al., 2012]. However, in this study, we did not identify other simple sequence mutations or deletions/duplications pointing to a loss-of-function mechanism, except for one patient-specific missense mutation (p.Thr66Ser) without clear pathogenic nature. Therefore, other molecular mechanisms might also contribute to disease including sequestration of RNA-binding proteins and RNA toxicity [Dejesus-Hernandez et al., 2011], which can also lead to a decrease of the mRNA level. These two potential disease mechanisms are not mutually exclusive, and might also lead together to degeneration of neuronal populations in either the frontal cortex (FTLD) or the spinal cord (ALS).
By what mechanisms the G4C2 repeat expands to a pathological size range remains to be discovered. In our first C9orf72 study in the Flanders-Belgian cohort [Gijselinck et al., 2012], we showed that the majority of the pathological G4C2 expansions were located on the same risk haplotype tagged by the rare T-allele of SNP rs2814707, and strongly associated with FTLD and ALS. These observations were confirmed in the European study with 100% of European carriers of a pathological G4C2 expansion carrying at least one T-allele. It has been proposed that this tight genetic association might be explained by a single founder mutation on this risk haplotype [Mok et al., 2011; Majounie et al., 2012]. An alternative hypothesis is that a specific genomic context on this risk haplotype is rendering the G4C2 repeat less stable and making it more prone to expansion into the pathological size range. In this context, we and others [Dejesus-Hernandez et al., 2011] showed that the G4C2 intermediate repeats are also highly significantly overrepresented on the same risk haplotype, suggesting that these intermediate repeats are more prone to replication slippage and unstable inheritance and thus triggering pathological expansions. The observation that intermediate alleles have the same linkage disequilibrium with a specific SNP allele as the expanded alleles is also previously made in other repeat expansion disorders, for example, Fragile X Syndrome [Gunter et al., 1998]. The appearance of pathological G4C2 expansions in apparently sporadic patients in this and previous studies [Dejesus-Hernandez et al., 2011; Gijselinck et al., 2012; Renton et al., 2011], seems to support this hypothesis. However, we could not observe increasing G4C2 repeat length or de novo expansions in the younger generations of families of index patients with intermediate repeat length up to 22 units. Therefore, intermediate alleles might rather be considered as predisposing alleles for further stepwise expansion over probably many generations instead of pre-mutations. A study using sporadic ALS trios did also not show evidence of repeat instability between two generations [Pamphlett et al., 2012].
Further, we observed in patients of both the Flanders-Belgian and European cohorts with a pathological G4C2 expansion, a significantly higher frequency of short indels in the GC-rich LCS adjacent to the G4C2 repeat (24/84, 28.57%) compared with patient noncarriers (5/404, 1.24%; P < 0.001) and control persons (4/752, 0.53%; P < 0.001). Remarkably, in 23.81% (20/84) of repeat expansion carriers, a deletion of the GTGGT motif contiguous with the G4C2 repeat was comprised within the indel (Fig. 1), compared with 0.25% in the noncarrier patients and 0.13% in control individuals (Table 1). This GTGGT deletion joins the two GC-rich sequences, increasing the overall GC content of the LCS. This has already been reported for CAG expansion diseases, where the expandability of the repeat increases with a higher GC-content of the surrounding DNA sequence [Brock et al., 1999; Nestor and Monckton, 2011]. Also, the GTGGT deletion creates one long contiguous imperfect G4C2 repeat, which is likely more prone to replication slippage. Therefore, the GTGGT sequence is potentially an important stabilizer of the G4C2 repeat. This resembles the loss of AGG interspersion in unstable Fragile X alleles [Kunst and Warren, 1994; Larsen et al., 2000]. Moreover, all carriers of a GTGGT deletion in the LCS had at least one rs2814707 T-allele and the 10-bp deletion was cotransmitted with the G4C2 expansion in relatives of two expansion carriers, which indicates that it is likely located on the risk haplotype and might trigger the expansion. Nevertheless, it should be noted that not all carriers of a pathological G4C2 expansion also have an LCS indel and thus we cannot exclude that the indels are rather a consequence of the close neighborhood of the expansion destabilizing the genomic region. Of note, the indels found in nonexpansion carriers are mostly not affecting the GTGGT sequence but rather stabilizing the repeat by deleting 100% GC-rich sequence. The GTGGT deletion in one FTLD patient without repeat expansion is located on a short allele with 5 units, which is probably not unstable enough to expand. A GTGGT deletion on an intermediate allele will most likely be not stable and hence not observed.
A better understanding of the underlying mechanism causing the G4C2 instability will be essential to assess disease risk and improve clinical benefits. Further, elucidating how G4C2 expansions lead to disease will be crucial to unveil biological pathways and key molecules in the disease process as targets for future therapies.
Acknowledgments
The authors are grateful to the participants contributing to this study from within the European EOD consortium and the BELNEU consortium. Also, we thank the personnel of the Genetic Service Facility of VIB (http://www.vibgeneticservicefacility.be) for their support of the genetic analyses. The IWT provided a PhD fellowship to T.V.L. and the FWO to S.P., postdoctoral fellowships were granted by the FWO to I.G., and J.v.d.Z.
Disclosure Statement: The authors report no conflict of interest with regard to the reported findings.
Appendix
European EOD Consortium members that contributed to the data in this article:
Department of Molecular Genetics, VIB, Antwerp, Belgium, Neurodegenerative Brain Diseases Group: Julie van der Zee, Ilse Gijselinck, Lubina Dillen, Tim Van Langenhove, Jessie Theuns, Stéphanie Philtjens, Kristel Sleegers, Veerle Bäumer, Githa Maes, Ellen Corsmit, Marc Cruts, Christine Van Broeckhoven; Neurogenomics Group: Albena Jordanova
Institute Born-Bunge, University of Antwerp, Antwerp, Belgium, Laboratory of Neurogenetics: Julie van der Zee, Ilse Gijselinck, Lubina Dillen, Tim Van Langenhove, Stéphanie Philtjens, Jessie Theuns, Kristel Sleegers, Veerle Bäumer, Githa Maes, Marc Cruts, Christine Van Broeckhoven; Laboratory of Neurochemistry and Behavior: Sebastiaan Engelborghs, Peter P. De Deyn; Laboratory of Neurobiology: Patrick Cras
Department of Neurology and Memory Clinic, Hospital Network Antwerp, Middelheim and Hoge Beuken, Antwerp, Belgium: Sebastiaan Engelborghs, Peter P. De Deyn
University of Leuven, Leuven, Belgium, Brain and Emotion Laboratory: Mathieu Vandenbulcke; Laboratory for Cognitive Neurology: Rik Vandenberghe
University Hospitals Leuven Gasthuisberg, Leuven, Belgium, Department of Psychiatry: Mathieu Vandenbulcke; Department of Neurology: Rik Vandenberghe
Centre for Ageing Brain and Neurodegenerative Disorders, Neurology Unit, University of Brescia, Brescia, Italy: Barbara Borroni, Alessandro Padovani, Silvana Archetti
Department of Psychiatry and Psychotherapy, Technische Universität München, Munich, Germany: Robert Perneczky, Janine Diehl-Schmid
Department of Neurodegeneration, Hertie Institute for Clinical Brain Research and Centre of Neurology, Tuebingen, Germany: Matthis Synofzik, Walter Maetzler, Jennifer Müller vom Hagen, Ludger Schöls
German Research Center for Neurodegenerative Diseases (DZNE), University of Tuebingen, Germany: Matthis Synofzik, Walter Maetzler, Jennifer Müller vom Hagen, Ludger Schöls; University of Bonn, Germany: Michael T. Heneka, Frank Jessen, Alfredo Ramirez, Delia Kurzwelly, Carmen Sachtleben, Wolfgang Mairer
Institute of Molecular Medicine, Faculty of Medicine, University of Lisbon, Lisbon, Portugal: Alexandre de Mendonça, Gabriel Miltenberger-Miltenyi, Sónia Pereira, Clara Firmo
Faculty of Medicine, University of Lisbon, Lisbon, Portugal: José Pimentel
Alzheimer's disease and Other cognitive disorders unit, Department of Neurology, Hospital Clinic, Barcelona, Spain: Raquel Sanchez-Valle, Albert Llado, Anna Antonell, Jose Molinuevo
Biobanc, Hospital Clinic, Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain: Ellen Gelpi
KI'Alzheimer Disease Research Center, Karolinska Institutet, Stockholm, Sweden, Department of Neurobiology, Care Sciences and Society: Caroline Graff, Huei-Hsin Chiang, Marie Westerlund; Department of NVS: Charlotte Forsell, Lena Lillius
Karolinska University Hospital, Stockholm, Sweden, Department of Geriatric Medicine, Genetics unit: Caroline Graff, Anne Kinhult Ståhlbom, Håkan Thonberg; Department clinical cytology and pathology: Inger Nennesmo
Department of Psychiatry and Neurochemistry, Institute of Neuroscience and Physiology, Sahlgrenska Academy, University of Gothenburg: Anne Börjesson-Hanson
Department of Neurological and Psychiatric Sciences, University of Florence, Florence, Italy: Benedetta Nacmias, Silvia Bagnoli, Sandro Sorbi, Valentina Bessi, Irene Piaceri
Neurology Department, Centro Hospitalar Universitário de Coimbra, Coimbra, Portugal: Isabel Santana, Beatriz Santiago
University of Coimbra, Coimbra, Portugal, Faculty of Medicine: Isabel Santana, Maria Helena Ribeiro; Center for Neuroscience and Cell Biology: Maria Rosário Almeida, Catarina Oliveira
Department of Neurology Centro Hospitalar de São João, and Department of Clinical Neuroscience and Mental Health, Faculty of Medicine University of Porto, Porto, Portugal: João Massano, Carolina Garret
Hospital de Angra do Heroísmo, Azores, Portugal: Paula Pires
Laboratory of Epidemiology Neuroimaging & Telemedicine & National Centre for Alzheimer's and Mental Diseases, IRCCS Centro San Giovanni di Dio, FBF, Brescia, Italy: Giovanni Frisoni, Orazio Zanetti, Cristian Bonvicini
Medical University Sofia, Sofia, Bulgaria, Department of Neurology: Stayko Sarafov, Ivailo Tournev; Molecular Medicine Center, Department of Biochemistry: Albena Jordanova
Department of Cognitive Science and Psychology, New Bulgarian University, Sofia, Bulgaria: Ivailo Tournev
Institute of Neurology, Medical University Vienna, Vienna, Austria: Gabor G. Kovacs, Thomas Ströbel
University of Bonn, Germany, Department of Neurology, Clinical Neuroscience Unit: Michael T. Heneka, Frank Jessen, Alfredo Ramirez, Delia Kurzwelly, Carmen Sachtleben, Wolfgang Mairer; Department of Psychiatry: Frank Jessen
Department of Pathology and Molecular Medicine, Thomayer Hospital, Prague, Czech Republic: Radoslav Matej, Eva Parobkova
Ludwig-Maximilians-Universität Munich, Germany, Neurologische Klinik: Adrian Danel; Zentrum für Neuropathologie und Prionforschung: Thomas Arzberger
Section of Neuropathology, Department of Neurological, Neuropsychological, Morphological and Movement Sciences, University of Verona, Italy: Gian Maria Fabrizi, Silvia Testi
Unità Operativa Complessa di Neuropatologia d'U, Azienda Ospedaliera Universitaria Integrata di Verona: Sergio Ferrari, Tiziana Cavallaro
Cyclotron Research Centre and Department of Neurology, University of Liège, Liège, Belgium: Eric Salmon
Department of Neurology, University Hospital Ghent and University of Ghent, Ghent, Belgium: Patrick Santens
Department of Neurology, Antwerp University Hospital, Edegem, Belgium: Patrick Cras
Supplementary material
Disclaimer: Supplementary materials have been peer-reviewed but not copyedited.
{kind=link}
References
- Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobagyi T, Shaw CE. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MNDALS. Acta Neuropathol. 2011;122:691–702. doi: 10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- Arighi A, Fumagalli GG, Jacini F, Fenoglio C, Ghezzi L, Pietroboni AM, De RM, Serpente M, Ridolfi E, Bonsi R, Bresolin N, Scarpini E, et al. Early onset behavioral variant frontotemporal dementia due to the C9ORF72 hexanucleotide repeat expansion: psychiatric clinical presentations. J Alzheimers Dis. 2012;31:447–452. doi: 10.3233/JAD-2012-120523. [DOI] [PubMed] [Google Scholar]
- Aziz NA, Jurgens CK, Landwehrmeyer GB, van Roon-Mom WM, van Ommen GJ, Stijnen T, Roos RA. Normal and mutant HTT interact to affect clinical severity and progression in Huntington disease. Neurology. 2009;73:1280–1285. doi: 10.1212/WNL.0b013e3181bd1121. [DOI] [PubMed] [Google Scholar]
- Bigio EH. Motor neuron disease: the C9orf72 hexanucleotide repeat expansion in FTD and ALS. Nat Rev Neurol. 2012;8:249–250. doi: 10.1038/nrneurol.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeve BF, Boylan KB, Graff-Radford NR, Dejesus-Hernandez M, Knopman DS, Pedraza O, Vemuri P, Jones D, Lowe V, Murray ME, Dickson DW, Josephs KA, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. 2012;135:765–783. doi: 10.1093/brain/aws004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer AL, Mackenzie IR, Boeve BF, Baker M, Seeley WW, Crook R, Feldman H, Hsiung GY, Rutherford N, Laluz V, Whitwell J, Foti D, et al. Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTDALS family. J Neurol Neurosurg Psychiatry. 2011;82:196–203. doi: 10.1136/jnnp.2009.204081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock GJ, Anderson NH, Monckton DG. Cis-acting modifiers of expanded CAGCTG triplet repeat expandability: associations with flanking GC content and proximity to CpG islands. Hum Mol Genet. 1999;8:1061–1067. doi: 10.1093/hmg/8.6.1061. [DOI] [PubMed] [Google Scholar]
- Chio A, Borghero G, Restagno G, Mora G, Drepper C, Traynor BJ, Sendtner M, Brunetti M, Ossola I, Calvo A, Pugliatti M, Sotgiu MA, et al. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain. 2012;135:784–793. doi: 10.1093/brain/awr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejesus-Hernandez M, Mackenzie IR, Boeve C, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari R, Mok K, Moreno JH, Cosentino S, Goldman J, Pietrini P, Mayeux R, Tierney MC, Kapogiannis D, Jicha GA, Murrell JR, Ghetti B, et al. Screening for C9ORF72 repeat expansion in FTLD. Neurobiol Aging. 2012;33:1850. doi: 10.1016/j.neurobiolaging.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijselinck I, Engelborghs S, Maes G, Cuijt I, Peeters K, Mattheijssens M, Joris G, Cras P, Martin JJ, De Deyn PP, Kumar-Singh S, Van Broeckhoven C, et al. Identification of 2 Loci at chromosomes 9 and 14 in a multiplex family with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol. 2010;67:606–616. doi: 10.1001/archneurol.2010.82. [DOI] [PubMed] [Google Scholar]
- Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, Engelborghs S, Sieben A, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11:54–65. doi: 10.1016/S1474-4422(11)70261-7. [DOI] [PubMed] [Google Scholar]
- Gunter C, Paradee W, Crawford DC, Meadows KA, Newman J, Kunst CB, Nelson DL, Schwartz C, Murray A, Macpherson JN, Sherman SL, Warren ST. Re-examination of factors associated with expansion of CGG repeats using a single nucleotide polymorphism in FMR1. Hum Mol Genet. 1998;7:1935–1946. doi: 10.1093/hmg/7.12.1935. [DOI] [PubMed] [Google Scholar]
- Hsiung GY, Dejesus-Hernandez M, Feldman HH, Sengdy P, Bouchard-Kerr P, Dwosh E, Butler R, Leung B, Fok A, Rutherford NJ, Baker M, Rademakers R, et al. Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain. 2012;135:709–722. doi: 10.1093/brain/awr354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumps C, Van RN, Heyrman L, Goossens D, Del-Favero J, Noguera R, Vandesompele J, Speleman F, De PK. Multiplex Amplicon Quantification (MAQ), a fast and efficient method for the simultaneous detection of copy number alterations in neuroblastoma. BMC Genomics. 2010;11:298. doi: 10.1186/1471-2164-11-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunst CB, Warren ST. Cryptic and polar variation of the fragile X repeat could result in predisposing normal alleles. Cell. 1994;77:853–861. doi: 10.1016/0092-8674(94)90134-1. [DOI] [PubMed] [Google Scholar]
- Laaksovirta H, Peuralinna T, Schymick JC, Scholz SW, Lai SL, Myllykangas L, Sulkava R, Jansson L, Hernandez DG, Gibbs JR, Nalls MA, Heckerman D, et al. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol. 2010;9:978–985. doi: 10.1016/S1474-4422(10)70184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen LA, Armstrong JS, Gronskov K, Hjalgrim H, Macpherson JN, Brondum-Nielsen K, Hasholt L, Norgaard-Pedersen B, Vuust J. Haplotype and AGG-interspersion analysis of FMR1 (CGG)(n) alleles in the Danish population: implications for multiple mutational pathways towards fragile X alleles. Am J Med Genet. 2000;93:99–106. doi: 10.1002/1096-8628(20000717)93:2<99::aid-ajmg4>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Le Ber I, Camuzat A, Berger E, Hannequin D, Laquerriere A, Golfier V, Seilhean D, Viennet G, Couratier P, Verpillat P, Heath S, Camu W, et al. Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology. 2009;72:1669–1676. doi: 10.1212/WNL.0b013e3181a55f1c. [DOI] [PubMed] [Google Scholar]
- Lindquist S, Duno M, Batbayli M, Puschmann A, Braendgaard H, Mardosiene S, Svenstrup K, Pinborg L, Vestergaard K, Hjermind L, Stokholm J, Andersen B, et al. Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clin Genet. 2012;9999 doi: 10.1111/j.1399-0004.2012.01903.x. [DOI] [PubMed] [Google Scholar]
- Luty AA, Kwok JB, Thompson EM, Blumbergs P, Brooks WS, Loy CT, Dobson-Stone C, Panegyres PK, Hecker J, Nicholson GA, Halliday GM, Schofield PR. Pedigree with frontotemporal lobar degeneration–motor neuron disease and Tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9. BMC Neurol. 2008;8:32. doi: 10.1186/1471-2377-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119:1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney CJ, Beck J, Rohrer JD, Lashley T, Mok K, Shakespeare T, Yeatman T, Warrington EK, Schott JM, Fox NC, Rossor MN, Hardy J, et al. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain. 2012;135:736–750. doi: 10.1093/brain/awr361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chio A, Restagno G, Nicolaou N, Simon-Sanchez J, Van Swieten JC, Abramzon Y, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323–330. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok K, Traynor BJ, Schymick J, Tienari PJ, Laaksovirta H, Peuralinna T, Myllykangas L, Chio A, Shatunov A, Boeve BF, Boxer AL, Dejesus-Hernandez M, et al. The chromosome 9 ALS and FTD locus is probably derived from a single founder. Neurobiol Aging. 2011;33:e3–e8. doi: 10.1016/j.neurobiolaging.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Al Chalabi A, Andersen PM, Hosler B, Sapp P, Englund E, Mitchell JE, Habgood JJ, de Belleroche J, Xi J, Brown RH. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66:839–844. doi: 10.1212/01.wnl.0000200048.53766.b4. [DOI] [PubMed] [Google Scholar]
- Murray ME, Dejesus-Hernandez M, Rutherford NJ, Baker M, Duara R, Graff-Radford NR, Wszolek ZK, Ferman TJ, Josephs KA, Boylan KB, Rademakers R, Dickson DW. Clinical and neuropathologic heterogeneity of c9FTDALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 2011;122:673–690. doi: 10.1007/s00401-011-0907-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- Nestor CE, Monckton DG. Correlation of inter-locus polyglutamine toxicity with CAG*CTG triplet repeat expandability and flanking genomic DNA GC content. PLoS One. 2011;6:e28260. doi: 10.1371/journal.pone.0028260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamphlett R, Cheong PL, Trent RJ, Yu B. Transmission of C9orf72 hexanucleotide repeat expansions in sporadic amyotrophic lateral sclerosis: an Australian trio study. Neuroreport. 2012;23:556–559. doi: 10.1097/WNR.0b013e3283544718. [DOI] [PubMed] [Google Scholar]
- Pearson JP, Williams NM, Majounie E, Waite A, Stott J, Newsway V, Murray A, Hernandez D, Guerreiro R, Singleton AB, Neal J, Morris HR. Familial frontotemporal dementia with amyotrophic lateral sclerosis and a shared haplotype on chromosome 9p. J Neurol. 2011;258:647–655. doi: 10.1007/s00415-010-5815-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, et al. A hexanucleotide repeat expansion in C9ORF72 Is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, Sreedharan J, Vance C, et al. Novel mutations in TARDBPTDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatunov A, Mok K, Newhouse S, Weale ME, Smith B, Vance C, Johnson L, Veldink JH, van Es MA, van den Berg LH, Robberecht W, Van DP, et al. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 2010;9:986–994. doi: 10.1016/S1474-4422(10)70197-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Sanchez J, Dopper EG, Cohn-Hokke PE, Hukema RK, Nicolaou N, Seelaar H, de Graaf JR, de KI, van Schoor NM, Deeg DJ, Smits M, Raaphorst J, et al. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain. 2012;135:723–735. doi: 10.1093/brain/awr353. [DOI] [PubMed] [Google Scholar]
- Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM, Jones M, Gerhard A, Davidson YS, Robinson A, Gibbons L, Hu Q, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012;135:693–708. doi: 10.1093/brain/awr355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart H, Rutherford NJ, Briemberg H, Krieger C, Cashman N, Fabros M, Baker M, Fok A, Dejesus-Hernandez M, Eisen A, Rademakers R, Mackenzie IR. Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol. 2012;123:409–417. doi: 10.1007/s00401-011-0937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troakes C, Maekawa S, Wijesekera L, Rogelj B, Siklos L, Bell C, Smith B, Newhouse S, Vance C, Johnson L, Hortobagyi T, Shatunov A, et al. An MND/ALS phenotype associated with C9orf72 repeat expansion: abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology. 2011;32:505–514. doi: 10.1111/j.1440-1789.2011.01286.x. [DOI] [PubMed] [Google Scholar]
- Valdmanis PN, Kabashi E, Dion PA, Rouleau GA. ALS predisposition modifiers: knock NOX, who's there? SOD1 mice still are. Eur J Hum Genet. 2008;16:140–142. doi: 10.1038/sj.ejhg.5201961. [DOI] [PubMed] [Google Scholar]
- Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M, Arnold SE, Mann DM, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234–239. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Es MA, Veldink JH, Saris CG, Blauw HM, van Vught PW, Birve A, Lemmens R, Schelhaas HJ, Groen EJ, Huisman MH, Van Der Kooi AJ, De VM, et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009;41:1083–1087. doi: 10.1038/ng.442. [DOI] [PubMed] [Google Scholar]
- Van Langenhove T, Van Broeckhoven C, van der Zee J. The molecular basis of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum. Ann Med. 2012a doi: 10.3109/07853890.2012.665471. [Epub ahead of print.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Langenhove T, van der Zee J, Gijselinck I, Engelborghs S, Vandenberghe R, Vandenbulcke M, De Bleeckere J, Sieben A, Versijpt J, Ivanoiu A, Deryck O, Willems C, et al. 2012b. Distinct clinical characteristics of C9orf72 expansion carriers compared to GRNMAPT and non-mutation carriers in a Flanders-Belgian FTLD cohort. Arch Neurol [In press]
- Vance C, Al Chalabi A, Ruddy D, Smith BN, Hu X, Sreedharan J, Siddique T, Schelhaas HJ, Kusters B, Troost D, Baas F, de Jong V, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2–21.3. Brain. 2006;129:868–876. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- Weckx S, Del Favero J, Rademakers R, Claes L, Cruts M, De Jonghe P, Van Broeckhoven C, De Rijk P. novoSNP, a novel computational tool for sequence variation discovery. Genome Res. 2005;15:436–442. doi: 10.1101/gr.2754005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Weigand SD, Boeve BF, Senjem ML, Gunter JL, Dejesus-Hernandez M, Rutherford NJ, Baker M, Knopman DS, Wszolek ZK, Parisi JE, Dickson DW, et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain. 2012;135:794–806. doi: 10.1093/brain/aws001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.