

Abstract
Background
Mutations in the gene coding for the transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI) are found in 8% to 10% of subjects with common variable immunodeficiency (CVID). Although heterozygous mutations may coincide with immunodeficiency in a few families, most mutation-bearing relatives are not hypogammaglobulinemic. Thus, the role of TACI mutations in producing the immune defect remains unclear.
Objective
This study examined the expression and function of TACI mutations in healthy heterozygous relatives.
Methods
We examined the surface and intracellular expression of TACI protein in EBV-transformed B cells of patients and relatives with mutations in 7 families, binding of a proliferation-inducing ligand, and secretion of IgG and IgA by ligand-activated B cells. We tested whether Toll-like receptor 9 agonists increased TACI expression and whether an agonistic anti-TACI antibody could induce activation-induced cytidine deaminase mRNA in those with mutations.
Results
Intracellular and extracellular TACI expression was defective for B cells of all subjects with mutations, including subjects with CVID and relatives. Although Toll-like receptor 9 triggering normally up-regulates B-cell TACI expression, this was defective for all subjects with mutations. Triggering TACI by an agonistic antibody showed loss of activation-induced cytidine deaminase mRNA induction in all mutation-bearing B cells. However, ligand-induced IgG and IgA production was normal for healthy relatives but not for subjects with CVID.
Conclusion
Thus, B cells of relatives of subjects with CVID who have mutations in TACI but normal immune globulin levels still have detectable in vitro B-cell defects.
Keywords: CVID, TACI, hypogammaglobulinemia, activation-induced cytidine deaminase, Toll-like receptor 9
Common variable immunodeficiency (CVID) is a heterogeneous primary immunodeficiency disease complex characterized by low levels of serum IgG and IgA and/or IgM coupled with antibody deficiency.1 The syndrome includes impaired B-cell maturation, impaired somatic hypermutation, reduced numbers of circulating memory and isotype-switched memory B cells, and diminished maturation of plasma cells.2–4 That the CVID syndrome is caused by mutations in a number of genes potentially contributing to the diverse phenotypes5–7 is supported by the identification of autosomal recessive mutations in the inducible costimulator, CD19, CD81, CD20, CD21, and B cell-activating factor (BAFF) in a few cases.8–12 However, more complex from the viewpoint of pathogenesis are mutations in the transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI, TNFRSF13B), identified in about 8% to 10% of patients but sometimes found in healthy subjects who are not hypogammaglobulinemic.13–17
TACI is a cell membrane receptor for 2 ligands, a proliferation-inducing ligand or APRIL (TNFSF13) and BAFF (TNFSF13B). BAFF also binds with high affinity to the BAFF receptor but poorly to a third B-cell receptor, B-cell maturation antigen (BCMA). By contrast, APRIL binds only to TACI but much less well to BCMA.18–20 TACI protein is expressed on the surface of B cells, in particular marginal zone and CD27+ memory B cells and is upregulated on activation by Toll-like receptor 9 (TLR9).21 Appropriate activation of TACI leads to B-cell differentiation, up-regulation of activation-induced cytidine deaminase (AID) mRNA, isotype switch, and maturation to immunoglobulin production, leading especially to T cell-independent antibody production.18,20,22–25 As for other TNF family members, TACI functions as a trimer; the first cysteine-rich domain (CDR1) has been suggested as important for the preassembly of TACI into trimers or larger multimers, independently of ligand interactions.23 The second CRD is essential for the binding of both BAFF or APRIL to TACI.19 Downstream, TACI signaling proteins include TNF-R–associated factors and calcium-modulator and cyclophilin ligand, subsequently activating nuclear factor-κB via the canonical pathway and ultimately intersecting with myeloid differentiation antigen (MYD88), which is also required for signaling.21
Subjects with CVID have both homozygous and heterozygous mutations in the extracellular, transmembrane, and intracellular regions of TACI.13–17 The extracellular mutation (C104R) leads to a disruption of the second CRD, impairing BAFF and APRIL binding.19 The C104R and A181E mutations lead to signaling impairments and defective antibody production in vivo in homologous murine models.24,25 These and other TACI transmembrane or intracytoplasmic mutations in CVID are presumed to lead to impaired BAFF and APRIL signaling, loss of class switch, and ultimately hypogammaglobulinemia. Although TACI mutations are significantly associated with CVID17 and occasionally with immune deficiency in first-degree relatives of patients with CVID,13–16 family members with heterozygous mutations are much more commonly healthy and not hypogammaglobulinemic, leading to questions about the function of this receptor. However, the loss of 1 functional allele might lead to degree of haploinsufficiency,26 potentially relevant for a trimeric receptor that has stringent binding requirements for a multimerized ligand.27 This hypothesis does not however explain why relatives are more commonly not immune deficient and suggests the presence of additional defects in patients. We have shown that subjects with Smith-Magenis syndrome, who commonly have only 1 copy of TACI because of deletions on chromosome 17, have impaired TACI expression, loss of ligand binding, and poorer anticarbohydrate anybody production.28 In the present study we show that family members with the same mutations in heterozygous or homozygous form, although not hypogammaglobulinemic, still have impaired B-cell TACI expression, reduced ligand binding, and markedly defective upregulation of AID mRNA, showing a dominantly inherited, selective in vitro activation defect.
METHODS
Patients, families, B-cell lines
Peripheral blood B cells of patients with CVID with heterozygous, compound heterozygous, or homozygous mutations in TACI in 7 families and their first-degree relatives were studied. Clinical histories and serum immune globulins for all subjects with antibody evaluations for patients were obtained (Tables I and II). Genomic DNAwas examined for TACI mutations as described.16 Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized peripheral blood by Ficoll-Hypaque (Pharmacia, Uppsala, Sweden) density gradient centrifugation. EBV-transformed B cell lines were produced from peripheral blood B cells of these subjects and healthy controls and cultured at 37°C in RPMI 1640 medium (Gibco, Carlsbad, Calif) with l-glutamine and 10% heat-inactivated fetal calf serum (FCS). Family F, containing both parents with C104R heterozygous mutations and 3 children with homozygous C104R defects, only 2 of whom have hypogammaglobulinemia, has previously been described (Table I).29
TABLE I.
Inheritance of TACI mutations in patients and families
| Family and member | Relation | Sex | Age at time of study (y) | Mutation | Clinical manifestation | IgG (mg/dL) | IgA (mg/dL) | IgM (mg/dL) | IgG1 (mg/dL) | IgG2 (mg/dL) | IgG3 (mg/dL) | IgG4 (mg/dL) | Percent T cells | Percent B cells |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A.I | Patient | F | 25 | A181E | Gastrointestinal tract infections, arthritis; ITP; AHA; uveitis; granulomatous skin lesions; pulmonary infiltrates; splenectomy; Coombs test positive | 164 | 5 | 20 | 84 | 7 | 34 | 5 | 59 | 25 |
| A.II | Sister | F | 29 | A181E | Ulcerative colitis; anticardiolipin syndrome; fetal loss | 989 | 83 | 127 | 515 | 333 | 89 | 27 | — | — |
| A.III | Mother | F | 55 | A181E | None | 889 | 178 | 252 | 519 | 345 | 47 | 21 | — | — |
| B.I | Patient | F | 19 | A181E | Chronic sinusitis | 371 | 14 | 4 | 182 | 47 | 15 | 21 | 67 | 24 |
| B.II | Mother | F | 48 | A181E | None | 876 | 78 | 92 | — | — | — | — | — | — |
| C.I | Patient | F | 42 | L171R/A181E | Pneumonia; URIs, arthritis, Sweet syndrome; aseptic meningitis | 390 | 77 | 25 | 204 | 92 | 20 | 19 | 74 | 8 |
| C.II | Father | M | 76 | L171R | None | 759 | 229 | 97 | 325 | 394 | 72 | 122 | — | — |
| C.III | Sister | F | 46 | L171R | None | 927 | 97 | 71 | 474 | 383 | 65 | 18 | — | — |
| D.I | Patient | F | 39 | C104R | ITP, splenectomy; lymphadenopathy | 436 | 47 | 54 | 295 | 99 | 60 | <0.2 | 38 | 42 |
| D.II | Father | M | 75 | C104R | None | 823 | 228 | 80 | 460 | 207 | 66 | <0.2 | — | — |
| D.III | Son | M | 10 | C104R | None | 743 | 84 | 95 | 437 | 120 | 15 | <0.2 | — | — |
| E.I | Patient | F | 47 | C104R/S194X | Pneumonia; AHA × 3; ITP × 1; pulmonary infiltrates; splenectomy | 200 | <6 | 24 | 162 | 9 | 63 | <1 | 65 | 28 |
| E.II | Brother | M | 79 | C104R | None | 919 | 85 | 90 | — | — | — | — | — | — |
| E.III | Brother | M | 57 | S194X | None | 612 | 94 | 22 | — | — | — | — | — | — |
| E.IV | Sister | F | 50 | C104R | Died of pseudomonas sepsis | 892 | 85 | 58 | 620 | 158 | 11 | <7 | — | — |
| E.V | Sister | F | 80 | None | None | 807 | 108 | 34 | 522 | 135 | 20 | <7 | — | — |
| E.VI | Brother | M | 49 | S194X | None | 546 | 22 | <5 | — | — | — | — | — | — |
| F.I | Patient | F | 33 | C104R/C104R | Diarrhea; chronic otitis; pneumonia | 310 | 34 | 15 | 184 | 114 | 35 | — | 66 | 18 |
| F.II | Patient | F | 35 | C104R/C104R | Diarrhea; Crohn disease | 476 | 88 | 62 | 263 | 145 | 37 | 2 | 50 | 11 |
| F.III | Brother | M | 28 | C104R/C104R | None | 820 | 52 | 104 | 488 | 234 | 43 | 26 | 46 | 23 |
| F.IV | Mother | F | 80 | C104R | None | 1070 | 243 | 69 | 600 | 287 | 36 | 46 | 59 | 15 |
| F.V | Father | M | 80 | C104R | None | 1300 | 104 | 174 | — | — | — | — | 60 | 7 |
| G.I | Patient | F | 10 | A181E | ITP; AIHA | 400 | 21 | 32 | 262 | 70 | 63 | <1 | 71 | 14 |
| GII | Mother | F | 38 | A181E | None | 822 | 248 | 91 | — | — | — | — | — | — |
| G.III | Father | N | 41 | None | None | Nl | Nl | Nl | — | — | — | — | — | — |
Normal ranges: IgG, 639–1349 mg/dL; IgA, 70–312 mg/dL; IgM, 56–352 mg/dL; IgG1, 280–1020 mg/dL; IgG2, 60–790 mg/dL; IgG3, 14–240 mg/dL; IgG4, 11–330 mg/dL; percent T cells, 55%–83%; percent B cells, 6%–19%.
AIHA, Autoimmune hemolytic anemia; F, female; ITP, idiopathic thrombocytopenia; M, male; Nl, normal; URI, upper respiratory infection; –, data not available.
TABLE II.
Antibody evaluations
| Family member | Tetanus | Diphtheria | Measles | Mumps | Rubella | Varicella | Pneumococci | Hemophilus |
|---|---|---|---|---|---|---|---|---|
| B.I | Positive | Positive | Positive | Positive | Positive | Positive | 5 of 12 | Positive |
| C.I | Positive | Positive | — | Positive | Positive | — | 2 of 12 | — |
| D.I | Positive | — | — | — | — | — | 1 of 12 | Negative |
| E.I | Positive | Positive | Positive | Positive | Negative | Positive | 7 of 14 | Negative |
| G.I | Positive | Positive | — | — | — | — | — | Negative |
Antibodies to vaccine antigens before immune globulin replacement; the laboratory lowest level of protective was used to indicate positive; if less, antibody was called negative. For pneumococci, after immunization with a 23-valent vaccine, tests to determine protective titers are indicated, considering 1.3 mg/mL to be protective.
Extracellular and intracellular TACI expression; APRIL binding
Surface expression of TACI on B cells of EBV-transformed B-cell lines from subjects with CVID, family members, and healthy controls was determined by flow cytometry with the use of biotinylated polyclonal goat anti-TACI antibody or a control biotinylated goat-IgG (R&D Systems, Minneapolis, Minn) and secondary staining with streptavidin-phycoerythrin (BD Pharmingen, San Diego, Calif). Intracellular TACI expression was determined with goat anti-TACI antibody, after treatment with 1% saponin in complete medium with FCS for permeabilization, followed by streptavidin-phycoerythrin staining. To assess TACI binding of the ligand APRIL, EBV B cells were incubated with 250 ng/mL FLAG-tagged MegaAPRIL (Axxora, San Diego, Calif) on ice in the presence of heparin (1000 U/mL); 1 μg/mL biotin-anti-FLAG monoclonal M2 antibody (Sigma, St Louis, Mo) was then added. The cells were washed and examined with streptavidin- phycoerythrin. Flow cytometry (fluorescence-activated cell sorting or FACS) was performed with a FACSCalibur (Becton Dickinson, Mountain View, Calif.)
TLR9 activation and TACI expression
Stimulation of B cells by TRL-9 ligation normally enhances the expression of TACI on normal B cells.21 To determine whether upregulation of TACI occurs for B cells of subjects with CVID with mutations, we incubated EBV cells or peripheral blood B cells with the cytosine phosphate guanine (CpG)–DNA TLR9 activator, ODN2006 (Invivogen, San Diego, Calif) at 0, 0.6, 1.5, and 3.0 μg/mL for 48 hours and examined the surface expression of TACI on B cells by FACS as described in the Extracellular and intracellular TACI expression; APRIL binding section.
TACI-induced AID mRNA
Triggering TACI initiates the upregulation of AID mRNA in both normal B cells and EBV-transformed B-cell lines.23 To examine the effects of TACI mutations, we tested EBV B cells of subjects with CVID with compound heterozygous mutations from families C, E, and F, as well as B cells from relatives with the same mutations in heterozygous form, compared with EBV cells of healthy controls. For this, 5 × 104 B cells were cultured in 48-well plates with a 1 μg/mL of an agonistic biotin-conjugated TACI mAb (11H3; eBioscience, San Diego, Calif) or isotype control, with or without 100 ng/mL IL-4 or IL-10 (R&D Systems) for 48 hours. To further crosslink TACI receptors, 5 mL of antibiotin microbeads IgG microbeads (Miltenyi Biotec, Cambridge, Mass) was added. AID mRNA was isolated (RNeasy Mini Kit; Qiagen, Valencia, Calif) and reverse transcribed (SuperScript III First-Strand cDNA synthesis kit; Invitrogen, Carlsbad, Calif). Quantitative real-time PCR was performed with LightCycler Real-Time PCR system and FastStart DNA Master SYBR Green I kit (Roche Diagnostics, Indianapolis, Ind), using β-actin mRNA as control for copy number. The following primers were used for AID mRNA: (5′-TGCTCTTCCTCGGCTACATCTC-3′; 5′-AACCTCATACAGGGGCAAAAGG-3′) and β-actin (5′-CTGAACCC CAAGGCCAACAG-3′; 5′-CCAGAGAAGAGGAGGATGCG-3′).16
IgG and IgA production
Peripheral mononuclear cells of patients and family members or healthy controls were incubated with MegaAPRIL (200 ng/mL; Axxora), with or without IL-10 (10 ng/mL; R&D Systems) or CD40-ligand (300 ng/mL; Axxora) plus IL-10 for 12 days in complete medium containing 10% FCS. The cell supernatant fluids of these cultures were then tested for IgG and IgA content with the use of sandwich ELISA reagents (Bethyl Laboratories, Montgomery, Tex).
Statistical analyses
The Spearman correlation test was used to compare intracellular and extracellular expressions of TACI in lymphoblastoid cell lines, using 95% confidence intervals. To compare TACI expression for B cells of groups of subjects or to compare the ability of lymphoblastoid B cells of patients, relatives, and controls to bind the ligand APRIL, the nonparametric 2-tailed Mann-Whitney test was used. This test was also used in comparisons of mRNA induction in B cells of subjects with mutations and controls and to examine differences for IgG and IgA production for subjects with CVID, relatives, and controls. To consider the effects of multiple comparisons, some data sets were also examined with one-way ANOVA. All data were analyzed with GraphPad Prism software, considering P ≤.05 to be significant.
RESULTS
Patients and mutational analyses
The baseline serum immunoglobulins and clinical histories of patients with CVID with heterozygous, compound heterozygous, or homozygous TACI mutations and their first-degree relatives in 7 families are shown on Table I. Table II provides further data, when available, on antibody production for the subjects with CVID before immunoglobulin replacement. As previously reported, the predominant mutations were C104R and A181E, but other mutations were also found. Subjects with CVID in each family are identified by the low serum levels of IgG, IgA, and IgM. Family members also with TACI mutations had normal serum immune globulins, with the exceptions of both brothers E.III and E.IV (each with S194X stop codons) who had reduced serum IgG and especially low serum levels of IgM. Neither had any significant clinical history of infections or other illnesses. Although autoimmunity was common in the patients,16 most family members were healthy; however, the sister of patient A.I had ulcerative colitis and was known to have anticardiolipin syndrome that led to fetal loss, and the sister of E.I. (E.IV) died of an overwhelming pseudomonas infection of unknown origin.
Mutations lead to impaired expression of intracellular and extracellular TACI
B cells from EBV cell lines from patients and their family members were examined to compare the surface and intracellular protein expression of TACI with control EBV B cells from healthy donors. Fig 1 shows that cells of all patients (open circles) identified in their respective families had significantly less TACI expression (surface or intracellular) than cells of their family members with the same mutations (shaded open circles) compared with the healthy controls (Mann-Whitney test: patients to controls, P = .01; relatives to controls, P = .04). In addition, for all cells examined, extracellular and intracellular expressions of TACI were highly significantly related (P < .0001, Spearman r = 0.86.) However, B cells of both family members and patients had less intracellular and extracellular expressions of TACI than B cells of the healthy controls (filled circles) (P =.01 and .008).
FIG 1.
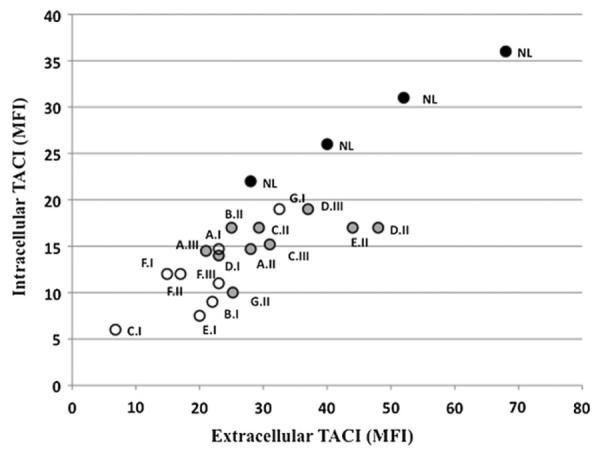
Impaired expression of surface and intracellular TACI. The mean fluorescence intensity for surface TACI expression (MFI; x-axis) on EBV-transformed B-cell lines of subjects and relatives with mutations was determined and compared with the same cells examined for intracellular TACI expression (y-axis). Subjects with CVID are shown as open circles, relatives with mutations are shown as shaded circles, and controls are shown as black circles. These results were compared with normal control (NL) B-cell lines with no mutations (black circles). The subject numbers from Table I are indicated. The expression of intracellular and extracellular TACI for all cell lines, shown for 1 experiment that was representative of 3 performed, was highly correlated (Spearman test r = 0.8553, P = .0001).
Heterozygous and homozygous mutations impair APRIL binding
The capacity of mutated TACI receptors on lymphoblastoid B cells of both patients and family members to bind the ligand APRIL was then compared with that of B cells of healthy controls (Fig 2). Although the heterozygous C104R mutation would be likely to impair ligand binding, B cells of both patients and relatives with all the other heterozygous mutations were also impaired, suggesting that, as for TACI expression, reduced ligand binding is another general feature of the heterozygous state. The ability of normal EBV cells to bind APRIL was significantly greater than either CVID B cells or B cells of relatives (Mann Whitney test P =.024, P =.001). However, patients and relatives with mutations had similar and reduced ligand-binding capacities (P=.104).
FIG 2.
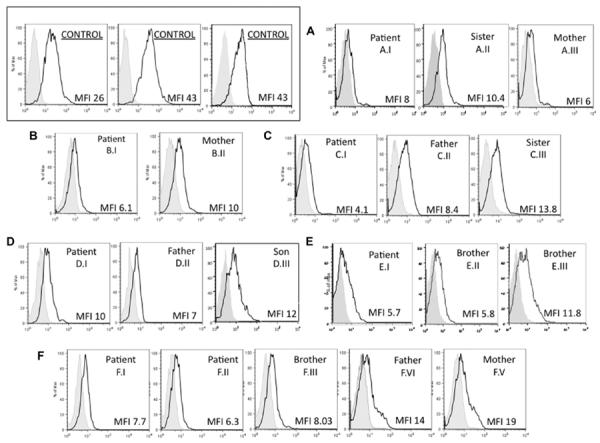
Heterozygous mutations impair APRIL binding. B-cell lines of patients with CVID and relatives with TACI mutations (as identified on each panel from Table I) were tested to determine the surface binding of Mega-FLAG-APRIL, using monoclonal anti-FLAG-biotin and phycoerythrin streptavidin, in comparison with B cells of 3 healthy controls (upper left). These subjects are from 6 families (A–F) and also identified on each panel by the same numbers as given in Table I. For each cell line, mean fluorescence intensity (MFI; x-axis) for FLAG-APRIL binding is indicated in each panel for each B-cell population. Cell counts are on the y-axis. FLAG-APRIL binding fluorescence is indicated in the open areas; controls (biotin anti-FLAG) are marked in the shaded areas of each panel. Data shown are from 1 experiment, representative of 3 performed on these cell lines. Differences between normal B cells (6 tested) and CVID B cells, or B cells of relatives were both significant (Mann Whitney test P = .024, P = .001.) Patients and relatives were not different (P = .104). Note: MFI for the 6 controls were 20, 23, 34, 26, 43, and 25.
Impaired TACI upregulation after TLR9 activation
TLR9 activation normally upregulates TACI expression on normal B cells or B-cell lines. Examining the EBV B cells of the large Family E allowed us to compare the effect of both heterozygous and compound heterozygous S194X and C104R mutations with cells of healthy controls. Fig 3, A, shows that the subject with CVID with compound heterozygous mutations C104R/S194X and family members with heterozygous mutations C104R or S194X had defective expression of TACI after incubation with the TLR9 agonist. At CpG concentrations of either 1.5 or 3.0 μg/mL, controls had greater TACI expression than the subject with CVID or her family members with mutations (Mann Whitney test P=.03.) By contrast, the one sister with no TACI defect (E.V) had normally increased B-cell TACI expression on TLR on activation, which was similar to the other control B cells examined.
FIG 3.
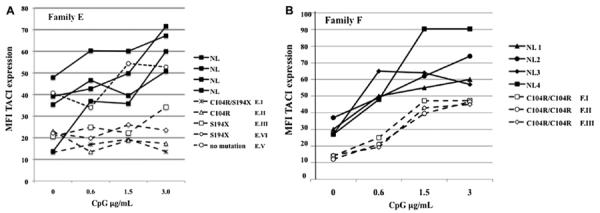
A, TACI upregulation by TLR9 activation. EBV cells from 5 subjects from Family E were cultured with the TLR9 agonist CpG oligodeoxynucleotide (ODN) at the indicated concentrations for 48 hours; B-cell surface TACI expression was determined by FACS, using biotin-labeled anti-TACI followed by streptavidinphycoerythrin. The patient with CVID and her family members have the indicated mutations; one sister (E.V.) has no mutation. B cells of 4 unrelated controls (NL) are included for comparison. B, Similarly, PBMCs of subjects from 3 subjects of Family F were cultured with CpG ODN 2006 to examine upregulation of TACI expression on peripheral blood B cells. TACI expression was determined by FACS as for subjects in Fig 3, A. Similarly treated peripheral blood B cells of 4 unrelated controls (NL) were included for comparison. MFI, Mean fluorescence intensity.
Peripheral blood B lymphocytes of other subjects with mutations also showed impaired baseline surface TACI. As an example, for 3 siblings, all with homozygous C104R mutations (F.I, F.II, and F.III; only 2 of whom have CVID29), TLR9 activation increased the lower baseline of TACI expression on the peripheral blood B cells of these subjects, but still not into the range noted for control peripheral B cells (Fig 3, B).
TACI-induced expression of AID mRNA
Activating TACI on human B cells leads to isotype switch, initiated by the expression of AID mRNA. We examined this aspect of TACI function in B cells of EBV cell lines from patients and controls in Families C, E, and F, in which there were subjects with CVID with homozygous and relatives with heterozygous mutations (Fig 4, A–C). After stimulation of B cells with an agonist anti-TACI antibody, with or without the addition of cytokines IL-4 or IL-10, AID mRNA was quantitated in comparison with similarly treated B cells from healthy controls. By showing each patient with CVID (black bars) and their heterozygous relatives (shaded bars) compared with B cells of 3 healthy controls (open bars), the induction of AID mRNA was found significantly impaired for B cells of all subjects with either heterozygous or homozygous mutations in all 3 families. Although for unclear reasons, the B cells of subjects with mutations in Family E displayed significantly reduced AID mRNA induction when activated by cytokines IL-4 or IL-10 alone, this was not observed for the B cells of subjects with mutations in Families C and F. However, for experiments in which anti-TACI antibody was used to activate, the differences between controls and subjects with mutations were even greater, with P values ranging from .02 and .04, as marked on each figure. Data for each family were also found significantly different with the use of one-way ANOVA, P = .002. (Note that in Family 4b, B cells of the normal sister [also shown as an open bar] had results similar to the healthy controls.) Thus, directly triggering TACI in each case shows a dominant class switch defect, not apparent from the clinical phenotype (Fig 4).
FIG 4.
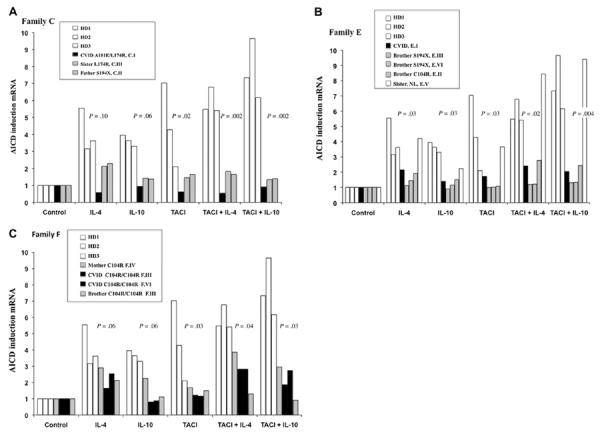
TACI-induced expression of AID mRNA. To examine whether single or compound mutations in TACI affect TACI activation that lead to isotype switch, the induction of AID mRNA by an agonist anti-TACI antibody, with or without added IL-4 or IL-10, was examined for B cells of subjects from Families C, E, and F compared with similarly treated cells from a healthy control. The index patients with CVID (C1, E1, and F1) have 2 mutations in TACI, whereas other members are either normal or have 1 mutation. For this B cells were cultured with biotin-labeled anti-TACI on antibiotin beads for 24 hours, and AID mRNA was quantitated by real-time PCR. Results for AID mRNA for Family C is given in A, Family E in B, and Family F in C. The significant differences (Mann-Whitney test) for induction of AID mRNA for controls compared with subjects for each condition are indicated on the figure.
APRIL-induced immunoglobulin production
To compare IgG and IgA production for subjects with CVID, their family members with mutations, and healthy controls, PBMCs were cultured with MegaAPRIL, with or without IL-10, or with CD40L with IL-10 as a control activator, for 13 days. Supernatant fluids were examined for IgG (Fig 5, A) or IgA (Fig 5, B). Although variability in secretion of both isotypes was observed, CVID relatives with TACI mutations had statistically similar amounts of IgG and IgA production compared with control B cells (Mann-Whitney test P = .2 and .5; ANOVA P = .16), whereas, as usual, B cells of all subjects with CVID in comparison with controls had impaired IgG (Mann-Whitney test P = .04) and especially IgA secretion (Mann-Whitney test P=.001). These data were also significantly different by ANOVA (IgG, P=.038; IgA, P=.001). (Note that CVID B cells activated by CD40L+IL-10 also did not have IgG or IgA secretion comparable with normal controls, P =.01 and P =.002.)
FIG 5.
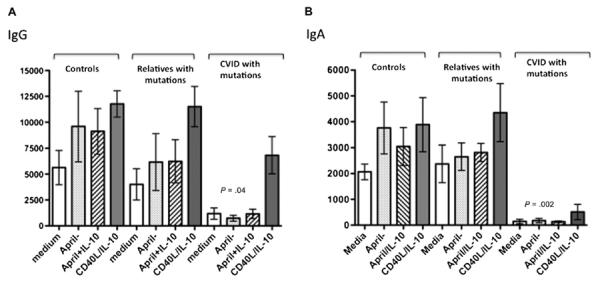
Immune globulin production in vitro. PBMCs of subjects with CVID (n = 6) or family members (n = 6) with the same TACI mutations (C104R, A181E, S194X) or cells of healthy controls (n 5 7) were incubated with the indicated activators for 12 days, and IgG (A) or IgA (B) was determined. Relatives with TACI mutations had similar amounts of IgG and IgA production in comparison with control B cells (Mann-Whitney test P = .2 and .5, and ANOVA P = .1638), but subjects with CVID had impaired IgG (P = .04) and especially IgA secretion (P = .001) compared with controls.
DISCUSSION
Although CVID is a relatively more common immune defect, it differs from many of the other genetic defects of humans in that it often has a later onset, usually between the ages of 20 and 40 years, and an unusually heterogeneous phenotype. These distinctions have suggested that a number of genes could contribute to the loss of B-cell function.5,6 To unravel the complexity of this syndrome, previous studies have concentrated on multiplex families with CVID and/or selective IgA deficiency in various members30,31 and consanguineous families with several members with CVID.32 These studies have yielded mutations in the T-cell activation receptor inducible costimulator and a handful of interesting autosomal recessive defects that involve the B-cell receptor signaling pathways or the BAFF receptor, all of which are important for B-cell survival and signaling. Concentrating on the more numerous patients with sporadic defects, genomic studies have shown that CVID is a complex syndrome but with a unique genetic signature, suggesting the involvement of many genes to support numerous aspects of human immunity.7
Defects of the TACI receptor, found in 8% to 10% of patients with CVID, have provided confirmation that TACI signaling is likely to be important in B-cell function in humans; however, because family members uncommonly have any clinically or laboratory evident immune defects, the role of this receptor in B-cell immunity has been unclear. However, we show that the expression of TACI protein is reduced in B cells of both subjects with CVID with homozygous or heterozygous mutations and their apparently healthy mutation-bearing relatives. Perhaps as a consequence of reduced TACI expression, binding of the ligand APRIL is also impaired. We also find that TACI expression on either lymphoblastoid B cells or peripheral blood B cells cannot be fully enhanced by activation by TLR9 activation in amounts comparable with control B cells. Direct triggering of the TACI receptor by an agonistic antibody in vitro shows that all the mutations examined, even in heterozygous form, lead to markedly impaired upregulation of AID mRNA, a requirement for isotype switch. However, in concert with the normal in vivo serum immune globulin levels in most of these relatives, culturing PMBCs with APRIL, with or without cytokines, leads to secretion of normal amounts of both IgG and IgA, unlike the B cells of subjects with CVID with mutations, which are quite impaired. These observations lead to the conclusion that with sufficient B-cell activation, perhaps because of the presence of additional normal lymphoid cells and cytokines in the germinal center environment, mutation-bearing B cells from the healthy relatives are compensated for loss of full TACI function. These factors overall presumably lead to normal immunoglobulin production. APRIL, used as an activator in these cultures, is known to have other binding partners, including cell surface proteoglycans33 and BCMA, which may also allow immunoglobulin secretion.
Although one sister in one family (A.II) had severe autoimmunity, the brothers E.III and E.VI with intracytoplasmic stop codons (S194X) had reduced serum IgM levels, and another sister (E.IV) died at age 50 of an overwhelming septic infection, the relatives of the subjects with CVID examined here were healthy and not overtly immune deficient. However, specific antibody production was not examined and could be suboptimal. In this regard, pneumococcal vaccination responses might be pertinent because TACI may exert a role in promoting T-cell–independent antibody. Impaired B-cell expression of TACI in the neonatal state could be related to delayed anticarbohydrate antibody responses.34 We also noted that the loss of 1 allele of TACI in the Smith-Magenis syndrome was associated with poorer vaccine responses to pneumococci.28
Our observations suggest that, although the role of the TACI receptor in human B-cell biology is weak and, when impaired, cannot be said to singly cause the CVID syndrome, selected mutations, even in heterozygous form, lead to dominantly inherited B defects that are demonstrable in vitro.
Clinical implications: Subjects with CVID have mutations in TACI, but family members with the same mutations are generally not hypogammaglobulinemic. Here, we show that these relatives still have detectable and selective in vitro B-cell defects.
Acknowledgments
We thank the patients and their families.
Supported by the National Institutes of Health AI-101093, AI-467320, and AI-48693 and National Institute of Allergy and Infectious Diseases Contract 03-22 (C.C.-R.). M.M.-G. was recipient of a fellowship (CM05/00145) from the Ministerio de Sanidad y Consumo of Spain.
Abbreviations used
- AID
Activation induced cytidine deaminase
- APRIL
A proliferation-inducing ligand
- BAFF
B-cell activating factor
- BCMA
B-cell maturation antigen
- CpG
Cytosine phosphate guanine
- CRD1
First cysteine-rich domain
- CVID
Common variable immunodeficiency
- FACS
Fluorescence-activated cell sorting
- FCS
Fetal calf serum
- PBMC
Peripheral blood mononuclear cell
- TACI
Transmembrane activator and calcium-modulating cyclophilin ligand interactor
- TLR
Toll-like receptor
Footnotes
Disclosure of potential conflict of interest: C. Cunningham-Rundles has received grants from the National Institutes of Health and has received royalties from UpToDate. The rest of the authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Cunningham-Rundles C. Common variable immunodeficiency. Curr Allergy Asthma Rep. 2001;1:421–9. doi: 10.1007/s11882-001-0027-1. [DOI] [PubMed] [Google Scholar]
- 2.Warnatz K, Denz A, Drager R, Braun M, Groth C, Wolff-Vorbeck G, et al. Severe deficiency of switched memory B cells (CD27(+)IgM(−)IgD(−)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. 2002;99:1544–51. doi: 10.1182/blood.v99.5.1544. [DOI] [PubMed] [Google Scholar]
- 3.Bonhomme D, Hammarstrom L, Webster D, Chapel H, Hermine O, Le Deist F, et al. Impaired antibody affinity maturation process characterizes a subset of patients with common variable immunodeficiency. J Immunol. 2000;165:4725–30. doi: 10.4049/jimmunol.165.8.4725. [DOI] [PubMed] [Google Scholar]
- 4.Ochtrop ML, Goldacker S, May AM, Rizzi M, Draeger R, Hauschke D, et al. T and B lymphocyte abnormalities in bone marrow biopsies of common variable immunodeficiency. Blood. 2011;118:309–18. doi: 10.1182/blood-2010-11-321695. [DOI] [PubMed] [Google Scholar]
- 5.Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–86. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 6.Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119:1650–7. doi: 10.1182/blood-2011-09-377945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orange JS, Glessner JT, Resnick E, Sullivan KE, Lucas M, Ferry B, et al. Genome-wide association identifies diverse causes of common variable immunodeficiency. J Allergy Clin Immunol. 2011;127:1360–7.e6. doi: 10.1016/j.jaci.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Zelm MC, Reisli I, van der Burg M, Castano D, van Noesel CJ, van Tol MJ, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354:1901–12. doi: 10.1056/NEJMoa051568. [DOI] [PubMed] [Google Scholar]
- 9.Warnatz K, Salzer U, Rizzi M, Fischer B, Gutenberger S, Bohm J, et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci U S A. 2009;106:13945–50. doi: 10.1073/pnas.0903543106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuijpers TW, Bende RJ, Baars PA, Grummels A, Derks IA, Dolman KM, et al. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J Clin Invest. 2010;120:214–22. doi: 10.1172/JCI40231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Zelm MC, Smet J, Adams B, Mascart F, Schandene L, Janssen F, et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest. 2010;120:1265–74. doi: 10.1172/JCI39748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thiel J, Kimmig L, Salzer U, Grudzien M, Lebrecht D, Hagena T, et al. Genetic CD21 deficiency is associated with hypogammaglobulinemia. J Allergy Clin Immunol. 2012;129:801–10.e6. doi: 10.1016/j.jaci.2011.09.027. [DOI] [PubMed] [Google Scholar]
- 13.Salzer U, Chapel HM, Webster AD, Pan-Hammarstrom Q, Schmitt-Graeff A, Schlesier M, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. 2005;37:820–8. doi: 10.1038/ng1600. [DOI] [PubMed] [Google Scholar]
- 14.Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005;37:829–34. doi: 10.1038/ng1601. [DOI] [PubMed] [Google Scholar]
- 15.Goldacker S, Warnatz K. Tackling the heterogeneity of CVID. Curr Opin Allergy Clin Immunol. 2005;5:504–9. doi: 10.1097/01.all.0000191888.97397.b3. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Radigan L, Salzer U, Behrens TW, Grimbacher B, Diaz G, et al. Trans-membrane activator and calcium-modulating cyclophilin ligand interactor mutations in common variable immunodeficiency: clinical and immunologic outcomes in heterozygotes. J Allergy Clin Immunol. 2007;120:1178–85. doi: 10.1016/j.jaci.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan-Hammarström Q, Salzer U, Du L, Björkander J, Cunningham-Rundles C, Nelson DL, et al. Mutations in TNFRSF13B encoding TACI and selective IgA deficiency. Nat Genet. 2007;39:429–30. doi: 10.1038/ng0407-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mackay F, Schneider P, Rennert P, Browning J. BAFF and APRIL: a tutorial on B cell survival. Annu Rev Immunol. 2003;21:231–64. doi: 10.1146/annurev.immunol.21.120601.141152. [DOI] [PubMed] [Google Scholar]
- 19.Hymowitz SG, Patel DR, Wallweber HJ, Runyon S, Yan M, Yin J, et al. Structures of APRIL-receptor complexes: like BCMA, TACI employs only a single cysteine-rich domain for high affinity ligand binding. J Biol Chem. 2005;280:7218–27. doi: 10.1074/jbc.M411714200. [DOI] [PubMed] [Google Scholar]
- 20.Schneider P. The role of APRIL and BAFF in lymphocyte activation. Curr Opin Immunol. 2005;17:282–9. doi: 10.1016/j.coi.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 21.He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, et al. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat Immunol. 2010;11:836–45. doi: 10.1038/ni.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castigli E, Wilson SA, Scott S, Dedeoglu F, Xu S, Lam KP, et al. TACI and BAFFR mediate isotype switching in B cells. J Exp Med. 2005;201:35–9. doi: 10.1084/jem.20032000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garibyan L, Lobito AA, Siegel RM, Call ME, Wucherpfennig KW, Geha RS. Dominant-negative effect of the heterozygous C104R TACI mutation in common variable immunodeficiency (CVID) J Clin Invest. 2007;117:1550–7. doi: 10.1172/JCI31023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JJ, Jabara HH, Garibyan L, Rauter I, Sannikova T, Dillon SR, et al. The C104R mutant impairs the function of transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI) through haploinsufficiency. J Allergy Clin Immunol. 2010;126:1234–41.e2. doi: 10.1016/j.jaci.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JJ, Rauter I, Garibyan L, Ozcan E, Sannikova T, Dillon SR, et al. The murine equivalent of the A181E TACI mutation associated with common variable immunodeficiency severely impairs B-cell function. Blood. 2009;114:2254–62. doi: 10.1182/blood-2008-11-189720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salzer U, Bacchelli C, Buckridge S, Pan-Hammarstrom Q, Jennings S, Lougaris V, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. 2009;113:1967–76. doi: 10.1182/blood-2008-02-141937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bossen C, Cachero TG, Tardivel A, Ingold K, Willen L, Dobles M, et al. TACI, unlike BAFF-R, is solely activated by oligomeric BAFF and APRIL to support survival of activated B cells and plasmablasts. Blood. 2008;111:1004–12. doi: 10.1182/blood-2007-09-110874. [DOI] [PubMed] [Google Scholar]
- 28.Chinen J, Martinez-Gallo M, Gu W, Cols M, Cerutti A, Radigan L, et al. Trans-membrane activator and CAML interactor (TACI) haploinsufficiency results in B-cell dysfunction in patients with Smith-Magenis syndrome. J Allergy Clin Immunol. 2011;127:1579–86. doi: 10.1016/j.jaci.2011.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Pomar N, Detkova D, Arostegui JI, Alvarez A, Soler-Palacin P, Vidaller A, et al. Role of TNFRSF13B variants in patients with common variable immunodeficiency. Blood. 2009;114:2846–8. doi: 10.1182/blood-2009-05-213025. [DOI] [PubMed] [Google Scholar]
- 30.Vorechovsky I, Zetterquist H, Paganelli R, Koskinen S, Webster AD, Bjorkander J, et al. Family and linkage study of selective IgA deficiency and common variable immunodeficiency. Clin Immunol Immunopathol. 1995;77:185–92. doi: 10.1006/clin.1995.1142. [DOI] [PubMed] [Google Scholar]
- 31.Schroeder HW, Jr, Schroeder HW, III, Sheikh SM. The complex genetics of common variable immunodeficiency. J Investig Med. 2004;52:90–103. doi: 10.1136/jim-52-02-17. [DOI] [PubMed] [Google Scholar]
- 32.Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Drager R, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. 2003;4:261–8. doi: 10.1038/ni902. [DOI] [PubMed] [Google Scholar]
- 33.Ingold K, Zumsteg A, Tardivel A, Huard B, Steiner QG, Cachero TG, et al. Identification of proteoglycans as the APRIL-specific binding partners. J Exp Med. 2005;201:1375–83. doi: 10.1084/jem.20042309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaur K, Chowdhury S, Greenspan NS, Schreiber JR. Decreased expression of tumor necrosis factor family receptors involved in humoral immune responses in preterm neonates. Blood. 2007;110:2948–54. doi: 10.1182/blood-2007-01-069245. [DOI] [PubMed] [Google Scholar]