
Figure.
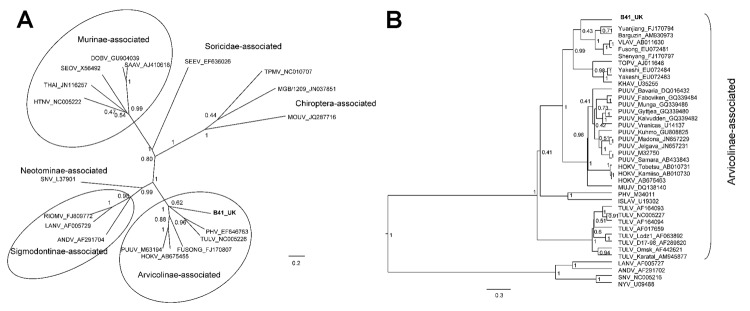
Bayesian phylogenetic trees constructed by using the models HKY+gamma for partial large segment sequences (n = 19) (A) and GTR+gamma for partial small segment sequences (n = 39) (B) within BEAST software (6) with Markov chain Monte Carlo chain lengths of 10 million and strict clock. Optimum substitution models were estimated by using MEGA5 (7). The trees are drawn to scale; branch lengths are measured in the number of substitutions per site. The numbers at each node are posterior probabilities. All effective sample size values exceeded 150 for partial L and 1,600 for partial S sequences. The phylogenetic position of virus isolated from field vole B41 (in boldface) is shown in relation to representative hantaviruses (A) and more closely related Arvicolinae-associated hantaviruses (B). GenBank accession numbers are shown next to taxonomic names. Scale bars indicate nucleotide substitutions per site. VLAV, Vladivostok virus; TOPV, Topografov virus; KHAV, Khabarovsk virus; PUUV, Puumala virus; HOKV, Hokkaido virus; MUJV, Muju virus; PHV, Prospect Hill virus; ISLAV, Isla Vista virus; TULV, Tula virus; LANV, Laguna Negra virus; ANDV, Andes virus; SNV, Sin Nombre virus; NYV, New York virus.