

Abstract
Helicobacter pylori infects over 3 billion people worldwide and is the primary risk factor for gastric cancer. Most individuals infected with H. pylori develop only asymptomatic gastritis; however, some develop ulcers or gastric adenocarcinoma. We demonstrate that one previously unappreciated parameter influencing H. pylori disease outcome is variation in the preinfection host microbiota. Utilizing a mouse model, we altered the microbiota by antibiotic treatment and found that these alterations resulted in significantly lowered H. pylori-triggered inflammation. Specifically, antibiotic pretreatment reduced CD4+ T-helper cells and Ifnγ transcript levels in gastric tissue after H. pylori infection. The bacterial communities in mice with a reduced response to H. pylori displayed many differences from those in untreated mice, including significantly more cluster IV and XIVa Clostridium spp., bacteria known to influence inflammation via regulatory T cell populations. Our findings suggest that microbiota composition, perhaps Clostridium spp., contributes to the variable disease outcome of H. pylori infection by altering the recruitment of CD4+ T cells to the gastric compartment. Our results suggest that gastric microbiota could be used as a diagnostic tool to determine which patients are at risk for developing severe disease.
INTRODUCTION
The bacterial gastric pathogen Helicobacter pylori colonizes more than half of the world's population (1, 2). Most infected people remain asymptomatic; however, ∼10% develop either peptic ulcers, gastric adenocarcinoma, or mucosa-associated lymphoid tumors (1–3). It is not yet possible to predict who will develop disease and what form it will take (3). Additionally, H. pylori infections protect against diseases such as esophageal cancer and asthma (1, 4, 5). As a result, most H. pylori infections are not treated unless the infected individual displays symptoms. However, it would be desirable to cure H. pylori infections that will progress to gastric cancer as this disease has few treatment options and high mortality (6). Several variables that determine H. pylori-associated disease outcome have been identified, but they do not fully explain disease risk. These past studies focused on variation in H. pylori genetic composition (2, 3, 7), host genetics (1–3), and environmental factors (2), but there has been no examination of the role played by the host microbiota.
Microbiotas have been implicated in aspects of immune system regulation and development (8–10), and altered microbiota communities have been implicated in both ameliorating (8, 11) and enhancing (12–14) disease symptoms. Specifically, dysbiosis of microbiota has been shown to influence inflammatory bowel disease (IBD) (12), obesity (13), and immune responses to Citrobacter rodentium (8) and Helicobacter hepaticus (15). Gastric microbial communities from people infected with H. pylori are somewhat different from those of uninfected people (16, 17), suggesting an interaction between H. pylori and the gastric microbial community. Whether specific communities of the microbiota make an individual more susceptible to H. pylori infection or disease is unknown.
In this study, we investigate how the microbiota affects disease that develops from H. pylori infection using the well-established H. pylori mouse model. Our studies were motivated by initial observations that identical mouse strains from different vendors responded differently to H. pylori infection. We report that these mice have variations in their normal gastric microbiota, akin to what has been seen in mouse intestinal microbiota (8). More in-depth studies found that antibiotic-induced alterations in the normal mouse microbiota shaped the immune response to H. pylori in a manner that suggested that specific microbiota members can reduce H. pylori-initiated inflammation. Mice that displayed low H. pylori-initiated inflammation had high gastric abundances of Clostridia species. These data thus suggest that variations in specific microbiota members can have a dramatic effect on H. pylori-associated disease and suggest new avenues for curbing H. pylori inflammation-related diseases such as ulcers and gastric cancer.
MATERIALS AND METHODS
H. pylori strains and growth conditions.
Helicobacter pylori strain SS1 (18), a gift of Jani O'Rourke (University of New South Wales), was cultured on Columbia blood agar (Difco) with 5% defibrinated horse blood (Hemostat Labs, Davis, CA), 50 μg/ml cycloheximide, 10 μg/ml vancomycin, 5 μg/ml cefsulodin, 2.5 units/ml polymyxin B, and 0.2% β-cyclodextrin. Mouse stomach samples were plated on the same medium plus 5 μg/ml trimethoprim, 8 μg/ml amphotericin B, 10 μg/ml nalidixic acid, and 200 μg/ml bacitracin. For mouse infection, H. pylori was grown with shaking in brucella broth (Difco) with 10% fetal bovine serum (FBS; Gibco) and incubated at 37°C with 7 to 10% O2, 10% CO2, and 80 to 83% N2 overnight. We inoculated mice orally intragastrically via a 20-gauge by 1.5-in. feeding needle with 500 μl containing ∼1 × 107 CFU/ml H. pylori bacteria.
Animal infections.
The University of California, Santa Cruz (UCSC), Institutional Animal Care and Use Committee approved all animal protocols and experiments. Female C57BL/6N mice (Helicobacter-free; Taconic Farms [TF], Germantown, NY, or Charles River Laboratories [CRL], Hollister, CA) were housed at the animal facility of UCSC under standard conditions in sterile cages, with sterile bedding and water and with a microisolator cage top. Cages were changed under laminar-flow sterile conditions. For experiments, 0.6 g/liter of penicillin/streptomycin (Omega Scientific) was administered ad libitum in the water bottle for 8 days; the antibiotic was replenished every 2 days. Two days after completing antibiotic treatment, mice slated for reconstitution were orally intragastrically fed 200 μl of stomach homogenates from non-antibiotic-treated C57BL/6N mice. Ten days after receiving the stomach homogenates, mice were inoculated orally intragastrically with H. pylori. Mice were 7 weeks old at the time of H. pylori inoculation; age-matched uninfected mice were included in all experiments. Four weeks postinoculation the animals were sacrificed via CO2 narcosis; the stomachs were dissected, opened along the lesser curvature, and divided into longitudinal strips. The tissue pieces were treated as follows: (i) homogenized using a Bullet Blender (Next Advance) with 1.0-mm zirconium silicate beads and plated to determine the number of H. pylori CFU/gram of stomach or used for DNA isolation for determining microbial profiles; (ii) frozen in liquid nitrogen and stored at −80°C for quantitative reverse transcription-PCR (RT-PCR); or (iii) stored in cold Hanks balanced salt solution (HBSS; Lonza) to be used in flow cytometry experiments.
Flow cytometric characterization of cells.
To prepare single-cell suspensions, mouse stomachs were dissected, opened longitudinally along the lesser curvature, and placed in cold HBSS (Lonza). The stomach was cut with a razor blade into 2-cm slices and incubated for 45 min at 37°C in HBSS supplemented with 10% fetal bovine serum (FBS), 15 mM HEPES, 5 mM EDTA, and 0.014% dithiothreitol (DTT). Subsequently, the tissue was rinsed with cold HBSS and incubated for 1 h at 37°C in 10 ml of RPMI medium with 10% FBS plus 1 mg/ml Dispase (Roche) and 0.25 mg/ml collagenase (Roche). The tissue was then vortexed and filtered through a 40-μm-pore-size cell strainer (BD Biosciences). The strainer was rinsed with 10 ml of cold HBSS. After samples were filtered, the cells were collected by centrifugation and washed two times in cold phosphate-buffered saline (PBS). The cells were then incubated on ice with IgG (US Biological) for 10 min, followed by a 20-min incubation with the test antibody. Optimal concentrations for the antibodies were determined in prior experiments to be 2 μg/ml for Alexa Fluor 488-CD3 (eBioscience), phycoerythrin (PE)-Cy7-CD4 (eBioscience), allophycocyanin (APC)/Cy7-CD8 (Biolegend), and Alexa Fluor 647-CD45 (Biolegend). An isotype control was included. Subsequently, the cells were centrifuged and washed twice in PBS plus 2.0% FBS and then fixed in 2% paraformaldehyde for 10 min on ice. A total of 50,000 cells were counted on a BD LSR II instrument (BD Biosciences) and analyzed using FlowJo (BD Biosciences) software; results are presented as a percentage of the 50,000 cells counted. Two-way analysis of variance (ANOVA) with Tukey's range test was used for statistical analysis.
qPCR.
Quantitative analysis of Ifnγ, Il17, Il4, Foxp3, and Il10 in the mouse stomach was performed by real-time quantitative PCR (qPCR). RNA was isolated from mouse stomach samples using TRIzol (Gibco). Stomachs were homogenized with a Polytron (Kinematica, Switzerland) in 1 ml of TRIzol per 100 mg of stomach tissue. Following homogenization, a standard phenol-chloroform extraction was used. A total of 1 to 3 μg of RNA was converted to cDNA using an RT2 First Strand Kit (Superarray Bioscience Corporation). The cDNA was measured in qPCRs with SYBR green master mix (Bioline). The data were normalized to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and are presented as fold increase over mock treatment using the ΔΔCT (where CT is threshold cycle) method (19). Samples were analyzed in triplicate along with controls lacking reverse transcriptase. qPCR for bacterial species in the mouse stomach was performed on DNA samples isolated from homogenized mouse stomach using a DNEasy Kit (Qiagen), and bacteria were quantified using a standard curve of plasmids containing the bacterial 16S rRNA gene. Two-way ANOVA in conjunction with Tukey's range test was used for statistical analysis.
Pathology.
Gastric tissue preserved in 22-oxacalcitriol (OCT) was sectioned, stained with hematoxylin and eosin, and evaluated in a blind fashion by a pathologist (J. E. Carter). Each slide was evaluated by two grading methods as outlined by Eaton et al. (Fig. 1A and B) (20, 21). Each slide was assessed twice per method to ensure reproducibility. For the experiment shown in Fig. 1A, lymphocytic infiltration was scored as outlined by Eaton et al. (20), as follows: 0, no infiltrate; 1, mild multifocal infiltration; 2 mild widespread infiltration; 3, mild widespread and moderate multifocal infiltration; 4, moderate widespread infiltration; and 5, moderate widespread and severe multifocal infiltration. For the experiment shown in Fig. 1B, the method of Eaton et al. (21) was used to determine the percentage of fields that showed evidence of the following: (i) polymorphonuclear leukocytes (PMN), defined as a field with several clusters of three or more neutrophils; (ii) gastritis, defined as inflammatory cell infiltrate of any cell type that was sufficient to displace the gastric glands; and (iii) metaplasia, defined as loss of parietal cells with concomitant replacement by mucus-type cells.
Fig 1.

Female C57BL/6N mice from different vendors mount different inflammatory responses to H. pylori and possess various amounts of key Lactobacillus spp. (A) Inflammation grade scored on a scale of 0 (no lymphocytic infiltration) to 5 (moderate, widespread and severe multifocal lymphocytic infiltration) as described by Eaton et al. (20). (B) Inflammation grade of the same tissue as in panel A, scored by determining the percentage of fields that contain PMN, gastritis, or metaplasia, as outlined by Eaton et al. (21). Gastric tissue was from mice from either Charles River Laboratories (CRL) or Taconic Farms (TF) infected with wild-type H. pylori strain SS1 (WT) for 6 months or from uninfected (UI) controls. PMN, polymorphonuclear leukocytes (neutrophils). (C) H. pylori colonization levels in CFU counts per gram of stomach tissue at 2 and 6 months postinoculation. (D) Quantitative PCR on the 16S gene for Lactobacillus species ASF360 and ASF361 in 2-month-old uninfected mice from CRL or TF. For all panels, n ≥ 6 mice per group infected with H. pylori, and n = 3 for uninfected groups. For panels B and D, data are presented as averages ± standard errors of the means. *, P < 0.05; **, P < 0.01, Student's t test.
Phylochip analysis.
Bacterial DNA was isolated using a DNeasy Blood and Tissue kit (Qiagen), including pretreatment for Gram-positive bacteria. The isolated DNA was sent to Second Genome (San Francisco, CA) for bacterial 16S DNA amplification. Microbial profiles for each sample were generated by hybridizing the bacterial 16S rRNA gene amplicons to the Phylochip (Second Genome). The Phylochip has been validated to detect 90% of cloned subfamilies at a 2.5-fold higher diversity than cloning (22). The Phylochip array contains 1,016,064 probes. For analysis, the probe gene sequences are clustered into groups known as operational taxonomic units (OTUs). Each OTU summarizes an average of 0.5% sequence divergence between probes and includes an average of 37 probe pairs, each of which includes a perfectly matching probe and a control, mismatching probe. In total, the Phylochip array contains 59,959 OTUs, which represent 147 phyla, 1,123 classes, and 1,219 orders within the Archaea and Bacteria (22, 23). An Adonis test was utilized for finding significant differences among discrete categorical or continuous variables. A two-sided t test was used to determine the subfamilies with highly significant populations between treatment groups.
RESULTS
Mice from Taconic and Charles River Laboratories have different inflammatory responses to H. pylori.
While studying the mouse inflammatory response to H. pylori infection, we noted significant differences in the degrees of inflammation that H. pylori strain SS1 triggered in C57BL/6N mice from Charles River Laboratories (CRL) or Taconic Farms, Inc. (TF) (Fig. 1A and B). Mice from both vendors reacted with increased inflammation compared to the uninfected controls. However, two methods of inflammation grading (20, 21) demonstrated that CRL mice had significantly higher inflammation grades than TF mice (Fig. 1A and B) even though H. pylori colonized mice from each group equally (Fig. 1C). This outcome suggested a difference between CRL and TF mice that could not be attributed to the H. pylori infecting strain, length of infection, or bacterial load or to mouse genetics, husbandry, gender, or age at infection. Studies have found differences in intestinal microbiota between C57BL/6 mice from different vendors (8). Thus, we examined differences in the gastric microbial communities in CRL versus TF C57BL/6N mice by comparing the amounts of Lactobacillus species ASF360 and ASF361, two resident bacteria of the mouse gastric microbiota (24). We noticed that there was significantly more ASF360 bacteria in the TF mice than in the CRL mice, while ASF361 bacteria were found in significantly higher numbers in the CRL mice than in the TF mice (Fig. 1D). These data are consistent with the idea that mice from different vendors have different resident stomach microbial populations.
The murine stomach microbiota is refractory to H. pylori-triggered perturbations and similar to the human stomach microbiota.
The differing levels of Lactobacillus ASF360 and ASF361 bacteria in CRL and TF mice suggest that different gastric microbial constituents may influence the degree of H. pylori-induced inflammation in the mouse model. The normal mouse stomach microbiota had not been characterized, so as a first step we identified the constituents of the normal mouse stomach microbiota. We focused our work on TF mice to eliminate complications from possible unknown variables and isolated total DNA from the stomachs of five specific-pathogen-free C57BL/6N mice (Helicobacter-free TF). We amplified the 16S rRNA genes and used this DNA in hybridization studies with the Phylochip microbial profiling system, a microarray-based method that identifies and measures the relative abundances of more than 59,000 individual microbial taxa (22). This analysis found that there were 10,207 species groups, termed operational taxonomic units (OTU), in any mouse stomach, with 2,056 of those OTUs found in all five mice (see Table S1 in the supplemental material). The majority of these isolates came from members of the Firmicutes phylum, with members from the class Clostridia being the most common (Fig. 2A). Members of the Bacteroidetes and Verrucomicrobia phyla were the second and third most highly represented phyla. The 2,056 OTUs represented 110 different families (see Fig. S1 in the supplemental material). The stomach phylotypes with the most members were the Bacteroidetes, Firmicutes, Proteobacteria, and Actinobacteria, a composition which is similar to what has been observed in humans except that in human samples Fusobacteria are also dominant (16, 17, 25).
Fig 2.
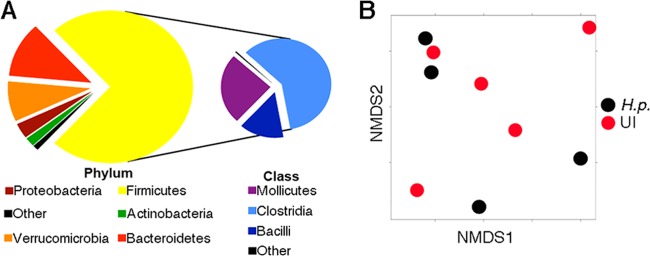
The murine stomach microbiota is refractory to H. pylori-triggered perturbations and similar to the human stomach microbiota. (A) Pie charts depicting phylum and class level distribution of 2,056 taxa that were present in all noninfected TF mice; 74% of the 2,056 taxa are Firmicutes, and, specifically, 44% of the total are members of the class Clostridia. A total of 12,032 taxa were present in at least one of five mouse stomachs. (B) Significant differences in microbiotas are not apparent between H. pylori-infected and noninfected samples, as indicated by nonmetric multidimensional scaling (NMDS) based on the Bray-Curtis distance between samples given the presence/absence of 12,032 taxa present in at least one of nine mouse samples; each dot represents one mouse in the study. H.p., 4-week H. pylori infection; UI, uninfected.
We next used the Phylochip analysis described above to determine whether 4 weeks of infection with H. pylori strain SS1 changed the core gastric microbiota. We compared the microbiota differences using both abundance of bacterial species and their presence/absence. The microbiota communities from H. pylori-positive or -negative mice were not overall significantly different from each other (Fig. 2B; see also Fig. S2 in the supplemental material) but did display a few substantial differences in specific taxa (see Fig. S3 and Table S2). The OTUs affected by H. pylori colonization included decreased Firmicutes (class Bacilli), Bacteroidetes, and Proteobacteria and increased Firmicutes (class Clostridia), Proteobacteria (genus Helicobacter), and Verrucomicrobia (see Fig. S3).
Fewer CD4+ T cells infiltrate into the stomach in response to H. pylori when mice have been pretreated with antibiotics.
We hypothesized that different preinfection stomach microbial populations might affect the inflammatory response to H. pylori infection based on the inflammatory differences we observed between the TF and CRL C57BL/6N mice (Fig. 1). Thus, we performed an experiment in which we altered the starting microbial populations by antibiotic treatment of mice from a common source, TF. We included three treatment groups in this analysis: the first group was normal (not antibiotic treated), the second group received antibiotics for 8 days, and the third group received antibiotics for 8 days followed by gavage with stomach homogenates from normal mice to reconstitute the normal mouse stomach microbiota (Fig. 3A). For each treatment group, five mice were infected with H. pylori, and five were left uninfected. To determine whether the pretreatment altered the total number of bacteria in the stomach, we performed qPCR for bacterial 16S rRNA genes and demonstrated no significant difference in levels between any of the groups (see Fig. S4A in the supplemental material). Furthermore, H. pylori colonized each experimental group similarly (see Fig. S4B), suggesting that any inflammatory differences are not due to differences in H. pylori colonization levels.
Fig 3.

Fewer CD4+ T cells infiltrate the stomach in response to H. pylori when mice have been pretreated with antibiotics. (A) Experimental setup for altering the microbiota and infecting with H. pylori. (B) Representative flow cytometry plots showing the percentages of CD4+ and CD8+ cells of the CD45+ CD3+ gastric lymphocytes from tissue 4 weeks postinoculation. Numbers in quadrants indicate the percentages of positively stained cells. Quadrants were determined based on isotype controls (data not shown). (C) CD45+ cell percentage is presented as the percentage of CD45+-stained cells out of 50,000 cells counted (left panel). CD4+ and CD8+ cell percentages are presented as the percentages of positively stained cells out of 50,000 cells counted (right panel). Data were obtained from two independent experiments with similar results (n ≥ 7 mice for each group). (D to F) Total gastric mRNA expression of Ifnγ, Il17, and Il4. mRNA expression levels were normalized to the level of the housekeeping gene, Gapdh, and are expressed as fold change from mock treatment, using the ΔΔCT method (19). H.p., H. pylori-infected; Ab, antibiotic treatment; RC, reconstituted with gastric microbiota; WT, wild type; PI, postinfection; wks, weeks. Data in panels C to F are presented as averages ± standard errors of the means. N.S., not significant; *, P < 0.05, by two-way ANOVA and Tukey's range test.
To determine whether there were inflammatory differences between the groups, we analyzed the mouse stomachs by flow cytometry for CD45+ CD3+ CD4+ T-helper cells and CD45+ CD3+ CD8+ T-cytotoxic cells (Fig. 3B and C). We chose these cells as they are known to arrive relatively early in response to H. pylori infection and also are reflective of the host response at later time points (26, 35). As expected, normal mice developed a CD4+ response to H. pylori infection that was significantly above that of uninfected mice (Fig. 3B and C). Interestingly, mice treated with antibiotics did not respond immunologically to an H. pylori infection, displaying levels of CD4+ T-helper cells that were comparable to those of uninfected controls (Fig. 3B and C). In contrast, mice with reconstituted gastric microbiota had a robust influx of CD4+ T-helper cells that was comparable to the normal mouse response to H. pylori (Fig. 3B and C). Thus, preinfection antibiotic treatment affected the outcome of H. pylori-host interactions and specifically dampened the H. pylori immune response.
To determine what type of immune response was inhibited in the antibiotic-treated mice, we examined transcripts encoding inflammatory cytokines for T-helper type 1 (Th1) (Ifnγ), T-helper type 17 (Th17) (Il17), and T-helper type 2 (Th2) (Il4) responses in the mouse stomach (Fig. 3D to F). We found that the Ifnγ level was much lower in H. pylori-infected antibiotic-treated mice than in normal mice or reconstituted mice (Fig. 3D), similar to the pattern we observed with the infiltrating CD4+ T cells (Fig. 3C). In contrast, the other cytokines did not display marked differences (Fig. 3E and F).
Antibiotic treatment causes significant changes to the stomach microbiota.
Our finding that antibiotic treatment dampens the host Th1 response to H. pylori is consistent with the notion that preinfection microbial populations affect the extent of H. pylori-triggered immune cell infiltration. We hypothesized that there were significant differences in the microbiotas between antibiotic-treated and normal mice and thus compared the stomach microbiotas of these two groups using the Phylochip microbial profiling system. We found that the microbial populations were significantly different from each other, both in the presence/absence of specific species and in their abundances (Fig. 4A; see also Fig. S5 in the supplemental material). Specifically, over 4,400 OTUs were different between the two groups, with 55% significantly decreased in the antibiotic-treated mice and 45% significantly increased, compared to levels in normal mice (Fig. 4B and C, respectively; see also Tables S3 and S4).
Fig 4.

The microbiota of antibiotic-treated mice is significantly different from that of the untreated mice and is dominated by members of the Clostridiales. (A) Significant differences in microbiotas are apparent between antibiotic-treated and untreated H. pylori-infected samples, as indicated by nonmetric multidimensional scaling (NMDS) based on the Bray-Curtis distance between samples given the presence/absence of 12,765 taxa present in at least one of 10 mouse samples; each dot represents one mouse in the study. A total of 4,400 OTUs were different between the antibiotic-treated H. pylori-infected mice and the H. pylori-infected untreated mice. Of those OTUs, 55% were significantly decreased in the antibiotic-treated mice, and 45% were significantly increased. (B and C) Pie charts illustrate phylum and class level distributions between bacterial taxa that either decreased (B) or increased (C) in the antibiotic-treated mice compared to levels in the nontreated mice. The majority of the taxa that were increased in the antibiotic treated mice were members of the Clostridia. (D) Ninety-four percent of the Clostridia OTUs that were increased in the antibiotic-treated mice were in the order Clostridiales; the chart details those members of the Clostridiales at the family and genus level. (E and F) Total gastric mRNA expression of Foxp3 and Il10. mRNA expression levels were normalized to the level of the housekeeping gene, Gapdh, and are expressed as fold change from mock treatment, using the ΔΔCT method. H.p., H. pylori-infected; Ab, antibiotic treatment; RC, reconstituted with gastric microbiota (n = 5 for each group). For panels E and F, data are displayed as averages ± standard errors of the means. *, P < 0.05, by two-way ANOVA and Tukey's range test.
Members of the Firmicutes phylum of Gram-positive bacteria stood out as having substantial changes between the antibiotic-treated and normal mice. Some members of the Firmicutes were decreased in the antibiotic-treated group, mainly members of the Mollicutes class (41% of all the decreased OTUs) (Fig. 4B). Other members of the Firmicutes were increased in the antibiotic-treated group, including members of the class Clostridia (46% of all the increased OTUs) (Fig. 4C); most of these fell within the order Clostridiales. Faecalibacterium species and Clostridium species genera with Clostridiales have been found to affect immune responses (8, 10, 27). We focused additional analysis on the Clostridium genus because a large number of these species were increased in antibiotic-treated mice (Fig. 4D). The genus Clostridium is subdivided into 19 clusters; members of clusters IV and XIVa are known to induce regulatory T cells (Tregs) in the colon (10). Analysis of the Clostridium spp. that increased in our antibiotic-treated mice determined that they fell into clusters IV and XIVa (see Fig. S6 in the supplemental material). Tregs are known to influence H. pylori pathogenesis. Specifically, Treg responses that arose either as a result of a Helicobacter bilis infection or in neonatal mice resulted in a reduced H. pylori-triggered pathology (28, 29). We did not, however, detect any markers of Treg-associated transcripts (Foxp3 or Il10) in the antibiotic-treated mice, suggesting that elevated Treg cell levels do not underlie the inflammatory differences in our model (Fig. 4E and F).
DISCUSSION
In this study, we employed antibiotic treatment and gastric reconstitution to demonstrate that antibiotic-induced alterations in the microbiota before H. pylori infection reduce the number of CD4+ T cells that develop in response to H. pylori infection. Our studies were built from the observation that C57BL/6N mice from different vendors had different levels of inflammatory responses to H. pylori infection and possessed different amounts of two Lactobacillus species known to inhabit the stomach. Microbiota differences are common between mice from different vendors. For example, other studies have reported that there are intestinal microbiota differences in C57BL/6 mice from different vendors (8).
Our results demonstrate that H. pylori infection does not substantially change the overall stomach microbiota composition but that it does affect some stomach microbiota members. Comparable findings were reported from a small-scale terminal restriction fragment length polymorphism analysis in H. pylori-infected C57BL/6 mice (30). Other studies similarly found that the H. pylori-induced changes were restricted two phylotypes—a reduction in Bacteroidetes and an increase in Firmicutes (31)—in transgenic insulin-gastrin (INS-GAS) mice. Humans are reported to show slightly more significant changes in gastric microbiota in response to H. pylori, with some similarities to the phylotypes that change in mice (16, 17, 25). These human studies, as well as the INS-GAS mouse studies, were all done after longer periods of H. pylori infection. Long-term infections may lead to more substantial changes in the microbiota over time due to accompanying immune or physiological changes, such as loss of parietal cells and increased gastric glandular atrophy. These studies are consistent with the idea, however, that the introduction of H. pylori into a stomach does not initially impact the microbiota.
Subsequently, we were interested in determining how the initial microbiota composition influences the immune response to H. pylori. Our studies found that antibiotic-treated mice were recalcitrant to H. pylori-triggered inflammation and lacked Th1 cytokines after 4 weeks of infection. Microbiota members can drive many different immune responses, including Th1, as recently described for Bilophila wadsworthia in Il10−/− mice (32). We did not detect B. wadsworthia in the gastric samples from our mice, but it is plausible that the microbiota of the antibiotic-treated mice had less of a Th1-promoting microbe or more of a Th1-inhibiting species. Such species remain to be discovered, however. It is tempting to speculate that the reduced inflammatory response we observe early postinoculation would drive conditions that would result in reduced inflammation long term; other work has demonstrated that the CD4 response at 4 weeks accurately reflects the long-term H. pylori inflammatory response (26). However, future experiments will need to examine the enduring effects on the inflammatory state.
One caveat to our studies is that the antibiotic treatment disturbed the microbiota outside the gastric compartment, which in turn might have affected the gastric immune response. Several studies have demonstrated that the microbiota in one organ can affect the immune response in another, including studies that showed that normal intestinal Helicobacter bilis or Helicobacter muridarum coinfection decreased H. pylori pathology (28, 33). We attempted to diminish these effects by employing mice that were Helicobacter free and thus lacked intestinal Helicobacter bacteria, including H. bilis and H. muridarum. Additionally, we reconstituted our mice with stomach microbiota, aiming to isolate the effect of the gastric microbiota on the immune response to H. pylori.
It is also difficult to be certain that all of our gastric microbial species are truly autochthonous members of the gastric microbiota. Mice are coprophagic, and so at least some fecal bacteria will retransit through the stomach. As suggested by Savage, we attempted to rule out nonautochthonous species by focusing on species found only in all five mouse stomachs (34). Further studies will be needed to verify the stability of our species over a longer time course.
Our results suggest that differing preinfection gastric microbial populations affect the subsequent immune response to H. pylori, thus contributing to the diverse outcomes of H. pylori infection. Our work suggests that microbial composition could be used as a marker to predict the disease outcome of H. pylori infection and that manipulating the stomach microbiota may be an avenue to pursue to prevent disease in response to H. pylori infection.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for the technical support received from Bari Holm Nazario, UCSC California Institute for Regenerative Medicine Shared Stem Cell Facility, and we thank Martin Polz (MIT) for supplying the p360 and p361 plasmids for Lactobacillus quantification. We acknowledge Donald R. Smith (UCSC) for assistance with statistical analysis. We are also grateful to the late David Schauer (MIT) for informative and encouraging discussions about stomach microbiota early in this work.
The described project was supported by grant number AI050000 (to K.M.O.) from the National Institutes of Allergy and Infectious Diseases (NIAID) at the National Institutes of Health and the University of California Cancer Research Coordinating Committee (to K.M.O.).
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Published ahead of print 19 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00044-13.
REFERENCES
- 1. Polk DB, Peek RM. 2010. Helicobacter pylori: gastric cancer and beyond. Nat. Rev. Cancer 10:403–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wroblewski LE, Peek RM, Wilson KT. 2010. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin. Microbiol. Rev. 23:713–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Atherton JC. 2006. The pathogenesis of Helicobacter pylori-induced gastro-duodenal diseases. Annu. Rev. Pathol. 1:63–96 [DOI] [PubMed] [Google Scholar]
- 4. Cover TL, Blaser MJ. 2009. Helicobacter pylori in health and disease. Gastroenterology 136:1863–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, Reuter S, Maxeiner J, Hansson M, Taube C, Quiding-Järbrink M, Müller A. 2012. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori--specific immune tolerance, and asthma protection. J. Clin. Invest. 122:1082–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peek RM, Jr, Blaser MJ. 2002. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2:28–37 [DOI] [PubMed] [Google Scholar]
- 7. Weel JF, van der Hulst RW, Gerrits Y, Roorda P, Feller M, Dankert J, Tytgat GN, van der Ende A. 1996. The interrelationship between cytotoxin-associated gene A, vacuolating cytotoxin, and Helicobacter pylori-related diseases. J. Infect. Dis. 173:1171–1175 [DOI] [PubMed] [Google Scholar]
- 8. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Littman DR. 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139:485–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio C-W, Santacruz N, Peterson Da, Stappenbeck TS, Hsieh C-S. 2011. Peripheral education of the immune system by colonic commensal microbiota. Nature 478:250–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov II, Umesaki Y, Itoh K, Honda K. 2011. Induction of colonic regulatory t cells by indigenous clostridium species. Science 331:337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mazmanian SK, Round JL, Kasper DL. 2008. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453:620–625 [DOI] [PubMed] [Google Scholar]
- 12. Frank DN, St Amand AL, Feldman Ra, Boedeker EC, Harpaz N, Pace NR. 2007. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. U. S. A. 104:13780–13785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe Ba, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. 2009. A core gut microbiome in obese and lean twins. Nature 457:480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garrett WS, Gallini Ca, Yatsunenko T, Michaud M, DuBois A, Delaney ML, Punit S, Karlsson M, Bry L, Glickman JN, Gordon JI, Onderdonk AB, Glimcher LH. 2010. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8:292–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Whary MT, Taylor NS, Feng Y, Ge Z, Muthupalani S, Versalovic J, Fox JG. 2011. Lactobacillus reuteri promotes Helicobacter hepaticus-associated typhlocolitis in gnotobiotic B6.129P2-IL-10tm1Cgn (IL-10−/−) mice. Immunology 133:165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. 2008. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One 3:e2836 doi:10.1371/journal.pone.0002836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maldonado-Contreras A, Goldfarb KC, Godoy-Vitorino F, Karaoz U, Contreras M, Blaser MJ, Brodie EL, Dominguez-Bello MG. 2011. Structure of the human gastric bacterial community in relation to Helicobacter pylori status. ISME J. 5:574–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee A, O'Rourke J, De Ungria MC, Robertson B, Daskalopoulos G, Dixon MF. 1997. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology 112:1386–1397 [DOI] [PubMed] [Google Scholar]
- 19. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 20. Eaton KA, Radin MJ, Krakowka S. 1995. An animal model of gastric ulcer due to bacterial gastritis in mice. Vet. Pathol. 32:489–497 [DOI] [PubMed] [Google Scholar]
- 21. Eaton KA, Danon SJ, Krakowka S, Weisbrode SE. 2007. A reproducible scoring system for quantification of histologic lesions of inflammatory disease in mouse gastric epithelium. Comp. Med. 57:57–65 [PubMed] [Google Scholar]
- 22. DeSantis TZ, Brodie EL, Moberg JP, Zubieta IX, Piceno YM, Andersen GL. 2007. High-density universal 16S rRNA microarray analysis reveals broader diversity than typical clone library when sampling the environment. Microb. Ecol. 53:371–383 [DOI] [PubMed] [Google Scholar]
- 23. Hazen TC, Dubinsky Ea, DeSantis TZ, Andersen GL, Piceno YM, Singh N, Jansson JK, Probst A, Borglin SE, Fortney JL, Stringfellow WT, Bill M, Conrad ME, Tom LM, Chavarria KL, Alusi TR, Lamendella R, Joyner DC, Spier C, Baelum J, Auer M, Zemla ML, Chakraborty R, Sonnenthal EL, D'Haeseleer P, Holman H-YN, Osman S, Lu Z, Van Nostrand JD, Deng Y, Zhou J, Mason OU. 2010. Deep-sea oil plume enriches indigenous oil-degrading bacteria. Science 330:204–208 [DOI] [PubMed] [Google Scholar]
- 24. Sarma-Rupavtarm RB, Ge Z, Schauer DB, Fox JG, Polz MF. 2004. Spatial distribution and stability of the eight microbial species of the altered Schaedler flora in the mouse gastrointestinal tract. Appl. Environ. Microbiol. 70:2791–2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, Perez-Perez G, Blaser MJ, Relman DA. 2006. Molecular analysis of the bacterial microbiota in the human stomach. Proc. Natl. Acad. Sci. U. S. A. 103:732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rolig AS, Carter JE, Ottemann KM. 2011. Bacterial chemotaxis modulates host cell apoptosis to establish a T-helper cell, type 17 (Th17)-dominant immune response in Helicobacter pylori infection. Proc. Natl. Acad. Sci. U. S. A. 108:19749–19754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux J-J, Blugeon S, Bridonneau C, Furet J-P, Corthier G, Grangette C, Vasquez N, Pochart P, Trugnan G, Thomas G, Blottière HM, Doré J, Marteau P, Seksik P, Langella P. 2008. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. U. S. A. 105:16731–16736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lemke LB, Ge Z, Whary MT, Feng Y, Rogers AB, Muthupalani S, Fox JG. 2009. Concurrent Helicobacter bilis infection in C57BL/6 mice attenuates pro-inflammatory H. pylori-induced gastric pathology. Infect. Immun. 77:2147–2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Arnold IC, Lee JY, Amieva MR, Roers A, Flavell Ra, Sparwasser T, Müller A. 2011. Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 140:199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tan MP, Kaparakis M, Galic M, Pedersen J, Pearse M, Wijburg OL, Janssen PH, Strugnell RA. 2007. Chronic Helicobacter pylori infection does not significantly alter the microbiota of the murine stomach. Appl. Environ. Microbiol. 73:1010–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lofgren JL, Whary MT, Ge Z, Muthupalani S, Taylor NS, Mobley M, Potter A, Varro A, Eibach D, Suerbaum S, Wang TC, Fox JG. 2011. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology 140:210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos Da, Jabri B, Chang EB. 2012. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 487:104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ge Z, Feng Y, Muthupalani S, Eurell LL, Taylor NS, Whary MT, Fox JG. 2011. Coinfection with enterohepatic Helicobacter species can ameliorate or promote Helicobacter pylori-induced gastric pathology in C57BL/6 mice. Infect. Immun. 79:3861–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Savage DC. 1972. Associations and physiological interactions of indigenous microorganisms and gastrointestinal epithelia. Am. J. Clin. Nutr. 25:1372–1379 [DOI] [PubMed] [Google Scholar]
- 35. Bamford K, Fan X, Crowe S, Leary J, Gourley W. 1998. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology 114:482–492 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.