

Abstract
Continuous exposure of cultured cortical neurons to moderate hypoxia (1% O2) elevates cellular accumulation of hypoxia-inducible factor-1α (HIF-1α) and improves basal survival of cultured cortical neurons. We examined the effects of adaptation to moderate hypoxia on the vulnerability of cultured neurons to the acute injury of simulated ischemia-reperfusion. Cortical neurons cultured continuously in 1% O2 were markedly protected against simulated ischemia-reperfusion, with protection persisting through 72 hours after ischemia. Neurons from 1% O2 conditions were also highly resistant to glutamate-induced NMDA receptor-dependent excitotoxic injury, despite expression of NMDA receptors at levels not significantly changed from controls. Inhibition of prolyl hydroxylase, mimicking cellular signaling effects of hypoxia including HIF-1α stabilization, also protected neurons against simulated ischemia-reperfusion injury. Nevertheless, genetic deletion of HIF-1α expression did not diminish the protection of neurons adapted to 1% O2 from excitotoxicity or ischemia-reperfusion injury, nor did it prevent the protective effect of prolyl hydroxylase inhibition. We conclude that chronic exposure to moderate hypoxia, through HIF-1α-independent mechanisms, produces strong protective effects against excitotoxic and ischemia-reperfusion related injury.
Keywords: hypoxia, adaptation, ischemia, reperfusion, neuroprotection, hypoxia-inducible factor, prolyl hydroxylase, excitotoxicity, glutamate
Introduction
On the microscopic scale, local tissue partial pressure of oxygen (pO2) measurements within brain gray matter are found to be remarkably heterogeneous, probably reflecting spatial O2 gradients between capillaries. These values range from an upper limit of 35–45 Torr to as low as 5–11 Torr (~1–2% O2 fraction, ‘moderate hypoxia’) (Grote, et al., 1996, Metzger and Heuber, 1977). Spatially averaged values lie in the range of 30–35 Torr (~4– 5% O2 fraction). We have recently observed the effects of culturing cortical neurons in moderately hypoxic atmospheres of 1% O2, simulating conditions in these minima of physiological O2 gradients. Although abrupt exposure to 1% O2 is lethal for neurons after prior incubation in ambient (20% O2) atmospheres (Banasiak, et al., 2004, Wick, et al., 2002), continuous exposure to 1% O2 from the time of plating actually results in improved survival, through a mechanism associated with HIF-1α accumulation and dependent in part on extracellular VEGF signaling (Li, et al., 2005). As the changes induced by culturing in 1% O2 appear to depend on chronic exposure and to persist after removal from the low O2 atmosphere, the response can be considered an adaptation to the moderate hypoxia.
Despite the evident links between O2 levels and ischemia, there has been little investigation of effects of chronic adaptation to hypoxia in studies of neuronal ischemic injury. However, a great deal of recent work has focused on the related topic of ‘ischemic preconditioning’ (or ‘hypoxic preconditioning’) of neurons, whereby a brief exposure to a non-lethal insult can produce marked protection against subsequent ischemic injury, usually after 24–48 hours (Chen and Simon, 1997, Gidday, et al., 1994, Hakim and Simon, 2004). Ischemic preconditioning has been shown to occur through mechanisms including NO release, erythropoietin or VEGF release, and Akt activation (Gidday, et al., 1999, Gonzalez-Zulueta, et al., 2000, Prass, et al., 2003, Wick, et al., 2002), and an association with upregulation of HIF-1 and its target genes has been reported (Bergeron, et al., 2000, Bernaudin, et al., 2002). We hypothesized that the adaptive changes in neurons occurring in chronic exposure to moderate hypoxia might also provide protection against ischemia-reperfusion injury through HIF-1-dependent mechanisms. We therefore explored the vulnerability of neurons adapted to moderate hypoxia (1% O2) to ischemia-reperfusion injury, and to glutamate receptor-mediated excitotoxic injury, which is known to underlie much of the injury in neuronal ischemia. Here we report that hypoxic adaptation produces remarkable protection against both forms of injury. Unexpectedly, although the neuronal protection produced by hypoxic adaptation was recapitulated by prolyl hydroxylase inhibition resulting in HIF-1α accumulation, it was not diminished by genetic deletion of HIF-1α, suggesting that protection by hypoxic adaptation occurs by mechanisms independent of HIF-1α expression. These results point to a novel form of neuroprotection against ischemia-reperfusion and excitotoxic injury, through chronic adaptation to moderate hypoxia.
Methods
Primary cultures of cortical neurons
All animal procedures were approved by the University of Chicago Institutional Animal Care and Use Committee. Dissociated cultures of cortical neurons were prepared from C57BL/10 mice, embryonic day E17.5 were plated on poly L-lysine-coated cover slips (at 8 × 104 cells/cm2) or in poly-L-lysine coated 96 well plates (at 5 × 104 cells per well) and maintained in serum-free medium as previously described (Li, et al., 2005). Neuronal cultures were maintained continuously from plating in a conventional humidified 37°C culture incubator with standard gaseous conditions (5% CO2 mixed with room air, producing an O2 fraction of 20%), or at reduced O2 tensions of 5% O2, or 1% O2. These latter were produced within a hypoxic chamber consisting of a humidified, 37°C, CO2- and O2-regulated work station (Coy Laboratory Products, Grass Lake, MI, USA) maintained at settings of 5% CO2 and 5% O2, containing nested within it a second independently regulated 37°C humidified hypoxia chamber set at 1% O2 (Pro-Ox 110 culture chamber and meter, Biospherix, Redfield, NY). O2 electrode calibrations were regularly readjusted using 100% dry N2 and room air as 0% O2 and 21% O2 calibration standards. Neurons were utilized for simulated ischemia experiments at 8 – 10 days in vitro.
HIF-1α genetic deletion
Cortical neuronal cultures, similar to those made from wild-type mice, were also generated from transgenic mice, homozygous for loxP sites flanking the essential exon 2 of the HIF-1α gene (designated HIF-1α fl/fl) (Ryan, et al., 2000). These mice, generated on the C57BL/6 strain background, were provided as the kind gift of Dr. Randall Johnson. Recombinant replication-deficient cre-recombinase –expressing adenoviral vector (Stec, 1999), adenovirus carrying cre recombinase linked to green fluorescent protein, or corresponding empty viral vector Bgl II, purchased from the University of Iowa Gene Transfer Vector Core facility, were obtained as CsCl gradient-purified virus at concentration 1010 pfu/ml, aliquoted, and stored at −80°C. Coverslips of cortical neurons at DIV 1 (density approximately 80,000 cells/cm2) were placed in culture dishes and covered with medium containing adenovirus at a multiplicity of infection (M.O.I.) of 50–100 pfu/cell. After 2 hour incubation at 37°C, cells were washed and returned to incubation in fresh medium.
Ischemia-reperfusion simulation in vitro
Real-time monitoring of effects of simulated ischemia-reperfusion injury was conducted as previously described (Li, et al., 2007), employing continuous superfusion of cultured neurons at 37°C with oxygenated saline or with an ‘ischemia solution’ containing (in mM): 0 glucose, 114 NaCl, 21.4 NaHCO3, 8.0 KCl, 0.8 MgSO4, 1.0 NaH2PO4, 1.2 CaCl2, and equilibrated for at least 3 hours with a regulated gaseous environment of 0.5% O2, 20% CO2, and 79.5% N2. Delayed survival at 24–72 hours following simulated ischemia was assayed by applying the same pre-equilibrated ischemia solution to coverslips of cultured neurons in a culture dish. After return to standard oxygenated culture medium for 24 or 72 hours, coverslips were removed and subjected to survival assays or fixation for staining. For microtubule-associated protein-2 (MAP-2) immunostaining, coverslips were rapidly removed from the perfusion chamber during simulated ischemia or reperfusion and immediately submersed in 4% paraformaldehyde solution at 37°C for fixation (15 min), followed by immunostaining using MAP-2 monoclonal antibody (Chemicon; 1:200) and an Alexa-488-linked goat anti-mouse secondary antibody (Molecular Probes; 1:300), and imaging by epifluorescence microscopy.
Survival assays
Survival assays of neurons plated on glass coverslips were conducted using a fluorescein diacetate/propidium iodide live/dead assay and automated counting of living and dead cells using CellProfiler™ based software (Carpenter, et al., 2006), as previously described (Li, et al., 2007). Relative survivals were calculated as those normalized to survival in parallel control cover slips at the same time point and cultured in the same O2 level prior to the experiment. Survival assays in 96 well plate format were conducted by wash of cells in HEPES-buffered physiological saline, followed by exposure to 10 μM Calcein-AM (from 10 mM stock in dimethylsulfoxide) for 10 min at 37°C, followed by washing and lysis of cells with 0.1% Triton X-100. Fluorescence was read in a Synergy fluorescence microplate reader at 485 nm excitation and 528 nm emission. Each experiment utilized all 8 wells in a column for each condition, with the average fluorescence reading taken after rejection of any outlying values more than 3 standard deviations from the column’s mean. Relative survival was taken as the average fluorescence divided by that in a normalization control condition.
Whole cell patch clamp recordings
Whole-cell voltage clamp recordings of ligand-gated currents were performed as previously described, using a CsCl-based intracellular solution (Brorson, et al., 2004). N-methyl-D-aspartate (NMDA) receptor responses and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor responses were measured and analyzed as previously described (Brorson, et al., 2004, Brorson, et al., 1999). Fractional desensitization of NMDA receptor responses was calculated as the difference between the peak current and the current remaining at 900 msec after peak, divided by the peak current. For determination of fractional Mg2+ block, peak currents evoked in Mg2+-containing saline were compared to those evoked in Mg2+-free saline.
Western blotting
Western blots for glutamate receptor subunits were performed largely as previously described (Brorson, et al., 2004), with loading of membrane protein preparations from cultured neurons (20 μg per lane), separation by 8% SDS-polyacrylamide gel electrophoresis, and transfer to polyvinylidene fluoride (PVDF) microporous membranes (Amersham Pharmacia Biotech). Primary antibodies applied were: anti-NR1 (rabbit polyclonal anti NMDAε1 (H-54) sc-9056, 1:2000, Santa Cruz Biotechnology); anti-NR2A (rabbit monoclonal IgG clone A12W, 1:1000, Upstate); anti-NR2B (monoclonal clone BWJHL, 1:1000, Upstate). Secondary antibodies (horseradish peroxidase-conjugated goat anti–rabbit or anti-mouse IgG, Bio-Rad Laboratories, Hercules, CA) were applied at 1:5000 dilutions for 1 hr at R.T. Blots were developed with ECL Plus (Amersham), and digital images were acquired on the Syngene Chemgenius2 Bioimaging system, with care to avoid signal saturation. Band intensity was quantified as peak area (net of background signal) in line scans of each lane, using ImageJ software. To assess for equal loading of these membrane protein preps (lacking actin or other soluble control proteins), total protein in each lane of the blots was quantified by Coomassie Blue staining. These measures of total protein loading were used to normalize quantified immunoreactive band strengths.
Western blots for HIF-1α and HIF-2α were performed on whole cell lysates, prepared as previously described (Li, et al., 2005), and loaded at 50 μg per lane onto 6%-8% SDS-PAGE gels. HIF-1α immunoblotting utilized an antibody generously provided by Dr. Regina Young and Dr. Celeste Simon of the University of Pennsylvania (Mansfield, et al., 2005), applied at 1:2000 dilution to proteins transferred to PVDF membrane and detected by the digital methods described above. HIF-2α immunoblotting utilized anti-HIF-2α polyclonal antibody (1:1000, Novus Biologicals, Littleton, CO, USA). However, protein bands for HIF-2α were not detectable by digital imaging of PVDF blots. Instead, for HIF-2α blots, proteins were transferred to nitrocellulose membranes (Protran; Whatman GmbH, Germany) and visualized by film-based detection using an enhanced chemiluminescence detection kit (Thermo Scientific Pierce). For these whole-cell lysates, anti-β actin (Sigma) was used as a loading control.
Data analysis
Pooled results are reported in text and graphed as mean values ± standard deviation (S.D.). Tests of significance were carried out by one-way or two-way ANOVA testing, as stated in figure legends. Where testing for normality failed, testing with one-way ANOVA on Ranks was substituted. ANOVA was followed by Student-Newman-Keuls post-hoc pairwise comparisons, with p < 0.05 considered significant (Sigma-Stat version 1.0, Jandel Scientific Corp.).
Materials
Calcein-AM was purchased from Biotium (Hayward, CA). GYKI 53655 was the kind gift of Dr. D. Bleakman, Lily Research Laboratories. Other chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
Results
Hypoxia-adapted neurons are protected from ischemia-reperfusion injury
We initially asked whether culturing neurons in 1% O2 might alter neuronal sensitivity to ischemia-reperfusion injury. As previously reported (Li, et al., 2005), cortical neurons cultured continuously in atmospheres of 1% O2 at baseline were morphologically intact and well-differentiated, expressing the neuron-specific somato-dendritic marker MAP-2 (Figure 1). One hour of exposure to simulated ischemia (O2 and glucose deprivation, pH 6.8, and moderately elevated K+, CO2, and glutamate), followed by return to oxygenated medium, produced rapid morphological degradation and early elimination of the majority of neurons cultured under conventional conditions (20% O2 and 5% CO2 partial pressures), as previously described (Li, et al., 2007). In striking contrast, such simulated ischemia-reperfusion produced little evidence of injury in neurons cultured from the time of plating in 1% O2 (Figure 1). Evaluation of cytoskeletal integrity by MAP-2 immunostaining detected only minor degrees of dendritic beading and pruning in these 1% O2-adapted neurons, contrasting with the severe morphological degradation within 2 hours after simulated ischemia in 20% O2-adapted neurons.
Figure 1. Morphological changes during ischemia-reperfusion in cortical neurons.
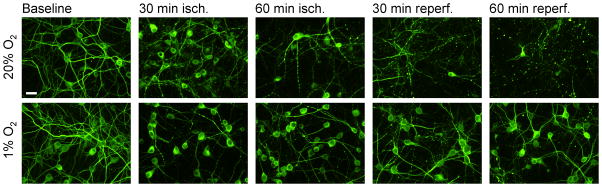
Representative images of cortical neurons, 10 days in vitro, fixed and immunostained for MAP-2 at intervals during simulated ischemia –reperfusion. Neurons cultured in 20% O2 and 1% O2 conditions were fixed at baseline (after 30 min equilibration in oxygenated saline), after 30 min or 60 min simulated ischemia, and following 1 or 2 hours of simulated reperfusion. Scale bar, 20 microns for all panels. In neurons from 20% O2 conditions (top row), prominent somatic swelling and dendritic beading was seen during simulated ischemia, followed by widespread dendritic breakdown and loss of cellular MAP-2 immunoreactivity during reperfusion. In neurons cultured in 1% O2 conditions (bottom row), morphological changes seen with ischemia or reperfusion were minimal.
Quantitation of survival after simulated ischemia-reperfusion revealed that relative survival of neurons cultured in 20% O2 conditions was reduced to 0.14 ± 0.08 at 24 hours and 0.13 ± 0.11 at 72 hours (Figure 2Ai). In contrast, survival after ischemia-reperfusion of neurons adapted to 1% O2 was significantly greater than survival in neurons from 20% O2 and nearly the same as in parallel controls, with relative survivals of 0.94 ± 0.11 and 0.92 ± 0.12 at 24 and 72 hours,. Survival of neurons adapted to 1% O2 was also significantly improved as compared to neurons cultured in 5% O2 (similar to average pO2 in mammalian cortex), in which relative survivals were 0.27 ± 0.25 and 0.38 ± 0.30 at 24 and 72 hours of reperfusion, only mildly and non-significantly increased over those in neurons adapted to 20% O2.
In the experiments described above, reperfusion was simulated with medium equilibrated with 20% O2 atmospheres in all groups. When instead, 1% O2-adapted neurons were returned to medium equilibrated with 1% O2 atmospheres, again there was prevention of severe injury at 24 or 72 hours (0.88 ± 0.21 and 0.99 ± 0.01 relative survival at 24 and 72 hours, respectively; compared to 0.20 ± 0.06 and 0.06 ± 0.02 in neurons from 20% O2 with reperfusion in 20% O2; n = 3; not shown). We also examined simulated ischemia without the addition of exogenous glutamate (Figure 2Aii). In this condition, injury was less severe than when 30 μM glutamate was included, but 1% O2 culture conditions again produced significant protection, with relative survivals of 1.02 ± 0.01 and 0.97 ± 0.07 at 24 and 72 hours of reperfusion, as compared with survival rates of 0.79 ± 0.08 and 0.69 ± 0.12 at 24 and 72 hours in neurons from 20% O2 conditions.
To allow demonstration of the timing of neuronal injury during the early minutes of reperfusion following ischemia, we employed a similar model of simulated ischemia-reperfusion injury utilizing continuous saline perfusion of a sealed cell chamber, allowing real-time fluorescence and DIC microscopic imaging (Li, et al., 2007). In this model, the 1% O2-adapted neurons again showed essentially complete protection against reperfusion-induced cell death (Figure 2B, C), contrasting dramatically with the rapid swelling and necrosis within 2 hours produced consistently in this model in neurons adapted to 20% O2 (Li, et al., 2007). Thus, in several versions of simulated ischemic injury, adaptation to moderate hypoxia produced remarkable protection of neurons against reperfusion injury, in both immediate and delayed phases of survival.
Figure 2. Protective effects of hypoxic adaptation against ischemia-reperfusion injury.

A) Relative survival of cortical neurons 24 and 72 hours following 60 min simulated ischemia-reperfusion with 30 μM glutamate (i) and simulated ischemia without added glutamate (ii). Neurons were maintained from time of plating in 20%, 5%, or 1% O2 tensions to DIV 9, when they were subjected to simulated ischemia. Following ischemia, all neurons were returned to conventional 20% O2 conditions for 24 to 72 hours. Neurons adapted to 1% O2 were strongly protected from reperfusion injury (n = 4 and 3, at 24 and 72 hours, respectively; mean ± S.D.; *p<0.05 in two-way ANOVA testing followed by Student-Newman-Keuls post-hoc testing). B) Acute neuronal death (percentage of cells taking up propidium fluorescence) in cells exposed to oxygenated, glucose-containing saline (control) or to simulated ischemia-reperfusion, without or with added glutamate (IR, or IR+ Glu). C) In neurons adapted to conventional 20% O2 conditions, widespread dramatic swelling and acute necrosis resulted within 2 hrs of reperfusion following simulated ischemia (upper panel; arrow indicates an example of an acutely swollen neuron with nuclear propidium fluorescence). In neurons adapted to 1% O2 (lower panel), no measurable cell death occurred (p = 0.66 for comparison of O2 levels, two-way ANOVA).
Hypoxia-adapted neurons are protected from excitotoxic injury
As neuronal injury from ischemia-reperfusion, both in vitro and in vivo, is largely mediated by glutamate-induced excitotoxicity mediated by N-methyl-D-aspartate (NMDA) receptors (Park, et al., 1988, Simon, et al., 1984, Wahlestedt, et al., 1993), we evaluated whether excitotoxic injury underlies neuronal death in this model of ischemia-reperfusion. Treatment during simulated ischemia with the NMDA receptor antagonist MK-801 fully blocked neuronal death induced by simulated ischemia-reperfusion at 24 or 72 hours, even when supplemental glutamate was not added to the simulated ischemic milieu (Figure 3A). This result suggested that the protective effect of hypoxic adaptation of neurons might be fully explained by an amelioration of excitotoxic injury. We therefore examined excitotoxic injury directly, exposing neurons to glutamate for 60 min at a range of concentrations, and determining survival at 24 hours. All glutamate exposures and recovery periods were performed in ambient normoxic conditions. As expected, neurons from 20% O2 or 5% O2 conditions were sensitive to delayed death induced by glutamate of 10–1000 μM. In contrast, neurons adapted to 1% O2 were remarkably resistant to glutamate-induced cell death, even at 1000 μM, a saturating concentration for ionotropic glutamate receptors (Figure 3B). To better characterize the pharmacology of excitotoxic injury in these cells, a fluorescence-based survival assay of neurons cultured in 96 well plates was developed, with survival evaluated at 24 hours following 60 min exposures to glutamate, 0.1 – 1000 μM. By this assay, glutamate produced excitotoxic death of neurons from 20% O2 or from 5% O2 conditions with LD50 values of 10.3 ± 2.4 μM and 12.8 ± 4.2 μM, respectively (n = 6). By visual inspection, at glutamate concentrations of 100 μM or above, near-complete neuronal necrosis occurred within 24 hours, though some background fluorescence was detected. In contrast, in neurons adapted to 1% O2 conditions, 60 min of glutamate exposure produced only small reductions in survival at 24 hours at concentrations of 30 –1000 μM. Survivals across these concentrations were significantly greater than those in either neurons from 20% O2 or 5% O2 conditions, again establishing a remarkable degree of protection by adaptation to 1% O2 against excitotoxic injury (Figure 3C). To determine which glutamate receptor subtype mediated this injury, selective antagonists were applied during exposures to 100 μM glutamate, in neurons taken from 20% O2 conditions. The AMPA receptor antagonist GYKI 53655 (10 μM) produced no significant attenuation of glutamate-induced excitotoxicity, while the NMDA receptor antagonist MK-801 (10 μM) fully blocked neuron death (Figure 3D), confirming that NMDA receptors mediated the excitotoxicity. In summary, adaptation of cultured cortical neurons to 1% O2 produced strong protection against direct NMDA receptor-mediated excitotoxic injury, comparable to that produced against simulated ischemia-reperfusion.
Figure 3. Hypoxic adaptation and glutamate excitotoxicity.

A) Effect of the NMDA antagonist MK-801 (10 μM), on simulated ischemia-reperfusion injury. Relative survival of cortical neurons, all cultured in 20% O2 conditions, at 2, 24, or 72 hours following 60 min of simulated ischemia without (‘IR’) or with added glutamate (30 μM, ‘IR+Glu’). MK-801, added during simulated ischemia, fully prevented subsequent neuronal death, whether exogenous glutamate was added or not (mean ± SD, n = 3; *groups different by two-way ANOVA, p<0.05; effects of reperfusion time were not significant). B) Glutamate excitotoxicity in cultured cortical neurons adapted to 20%, 5%, or 1% O2 conditions (mean ± S.D., n = 4). All neurons were treated with a range of glutamate concentrations for 60 min in physiological saline equilibrated with ambient atmospheric O2, and kept in 20% O2 conditions during the 24 hour recovery period. At each glutamate concentration of 10 μM or above, survival in the 1% O2 condition was significantly greater than in the 5% O2 and 20% O2 conditions (*p < 0.05, two-way ANOVA followed by post-hoc testing). C) Concentration-response curves for glutamate excitotoxicity in neurons cultured in 96 well plate format and adapted to 20%, 5%, or 1% O2 conditions, with survival, measured as uptake of calcein fluorescence, relative to parallel controls. Glutamate exposures were for 60 min in physiological saline in ambient atmospheric (21% O2) conditions (n = 6, mean ± S.E.M.). D) Receptor pharmacology of glutamate excitotoxicity in cortical neurons cultured in multiwell plates. Survival at 24 hrs following exposure to glutamate, 100 μM, applied alone (‘Glu’), in the presence of 10 μM GYKI 53655 (‘GYKI’), with 10 μM MK-801, or with both antagonists. MK-801 protected against excitotoxicity (n = 4, mean ± SD; * p < 0.05 compared to vehicle alone; # p < 0.05 compared to glutamate alone).
Glutamate receptor expression is not altered in hypoxia-adapted neurons
A simple explanation for this robust protection would arise if culturing neurons in 1% O2 could be shown to strongly suppress NMDA receptor expression. We evaluated this possibility in two ways. First, expression of the NMDA receptor subunits NR1, NR2A, and NR2B, the principal NMDA subunits expressed in the forebrain, was evaluated by Western blotting with subunit-specific antibodies. Qualitative expression of each subunit was found in membrane protein preparations from neurons from 20% O2, 5% O2, and 1% O2 conditions (Figure 4A). Appreciable variability in band strengths was attributable in part to variable protein loading. When quantified immunoreactive bands were normalized by total protein staining in each lane, no significant differences between O2 levels were found for NR1, NR2A, or NR2B expression (Figure 4B). The principle AMPA receptor subunits expressed in forebrain neurons, GluR1 and GluR2, were also unaltered in expression (data not shown). These results suggest that no substantial changes were to be found in protein expression of the principle glutamate receptor subunits in neurons adapted to 1% O2 conditions, confirming the normal neuronal phenotype in hypoxia-adapted neurons, and ruling out suppression of glutamate receptor expression as the reason for resistance to ischemia reperfusion and excitotoxic injury.
Figure 4. Glutamate receptor expression and adaptation to hypoxia.

A) Western blotting for principal NMDA receptor subunits NR1, NR2A, and NR2B from membrane preparations from neurons (DIV 8–10) cultured in 20% O2, 5% O2, and 1% O2 environments. B) Quantitation of relative band strength, normalized to protein loading, from neurons from 20%, 5%, and 1% conditions (n = 3 independent samples; mean ± SD; differences not significant by ANOVA). C) Representative whole cell currents elicited by 1 sec applications of 300 μM NMDA and 10 μM glycine (bar) in cortical neurons (DIV 8–10) cultured in 20%, 5%, or 1% O2 atmospheres. D) Whole cell peak currents (top), fractional block by 1 mM Mg2+ (middle) and fractional desensitization over 1 sec (bottom), in cortical neurons cultured in 20% O2 (n = 9), 5% O2 (n = 8), and 1% O2 (n = 9). Each circle represents a measurement from a single cell, with mean values represented by horizontal bars. Distributions were highly overlapping, and no significant differences were found (p = 0.16, p = 0.94, and p = 0.14, respectively, by ANOVA).
However, these immunoblotting results did not fully rule out the possibility that functional, surface-expressed glutamate receptor responses might be somehow altered in neurons adapted to 1% O2 conditions. Thus, we also examined glutamate receptor-mediated electrophysiological responses in these neurons. Whole-cell patch clamp recordings were made in room air within 60 min of removal of cells from their incubation conditions. Evaluation of inward currents elicited by brief applications of NMDA revealed substantial whole-cell inward currents in neurons from each of the culture conditions (Figure 4C). While whole-cell NMDA receptor current amplitudes varied widely from cell to cell, the mean current amplitudes in cells cultured in 20% O2, 5% O2, and 1% O2 were not statistically different (Figure 4D, top). A trend towards a somewhat larger mean NMDA current amplitude in neurons from 20% O2 was largely due to a single outlying cell, and the highly overlapping distributions of current magnitudes did not provide an explanation for the strong protection by hypoxic adaptation based on a reduction in NMDA receptor currents. Similarly, other features of NMDA receptor expression that might explain differences in excitotoxic vulnerability, including fractional block of NMDA currents by 1 mM Mg2+, fractional desensitization of NMDA receptor currents (Figure 4D), and whole-cell current densities (normalized to cell capacitance, not shown), were quite variable from cell to cell but were not significantly different between neurons cultured in 20% O2, 5% O2, or 1% O2 conditions. AMPA receptor responses were also evaluated without detection of significant differences in peak current densities or desensitization rates in neurons adapted to 1% O2 (data not shown). In sum, no substantial differences in expression of NMDA- or AMPA–type glutamate receptors were found that could explain the protective effects of adaptation to 1% O2 against ischemia-reperfusion and excitotoxic injury.
Evaluation of the role of HIF-1 in protection by hypoxic-adaptation
In previous work, we showed that enhanced basal survival of cultured neurons in 1% O2 conditions is associated with accumulation of the transcription factor subunit HIF-1α, depends upon the HIF-1-regulated target gene VEGF, and is diminished by genetic deletion of HIF-1α in neurons (Li, et al., 2005; Li and Brorson, 2006). HIF-1α accumulation, and associated preservation of basal survival, can also be induced in cultured neurons by application of dimethyl oxallyl glycine (DMOG, 25 μM), an inhibitor of the prolyl hydroxylases that regulate the O2-dependent proteasomal degradation of HIF-1α (Li, et al., 2005). If HIF-1α accumulation mediates the protection against simulated ischemia-reperfusion induced by adaptation to 1% O2, prolyl hydroxylase inhibition ought to reproduce this protection. To test this, 25 μM DMOG was applied continuously from the time of initial culture through day 8–10, to neurons in 20% O2. DMOG-treated cells were then subjected to ischemia-reperfusion injury. DMOG treatment produced near-complete protection against ischemia-reperfusion, either without or including added glutamate (Figure 5). Thus prolyl hydroxylase inhibition reproduced protection similar to that found with adaptation to 1% O2.
Figure 5. Effect of prolyl hydroxylase inhibition on ischemia-reperfusion injury.
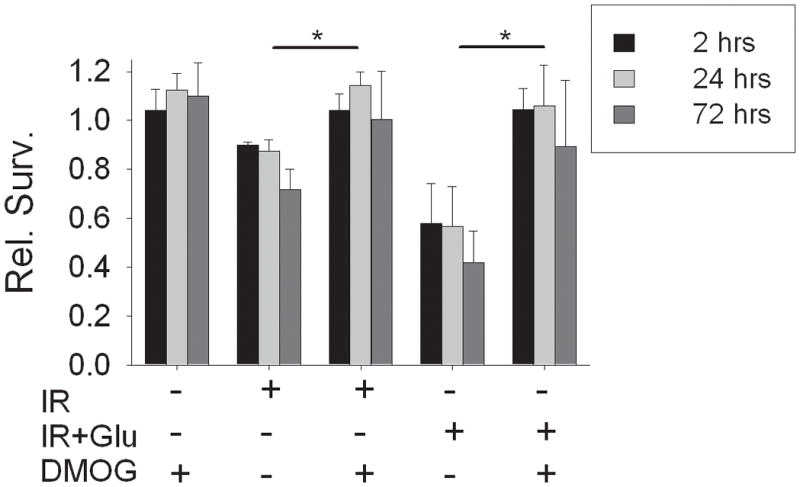
All neurons were cultured under ambient conditions (20% O2), and treated with DMOG (25 μM) from day 0 in culture. Simulated ischemia was applied for 60 min on day 8–10, without (‘IR’) or with added glutamate (‘IR+Glu’), and relative survival compared at 2, 24, and 72 hours. DMOG pre-treatment completely blocked neuronal death during reperfusion. DMOG alone produced no change in relative survival (n = 5, mean ± S.D.; * treatment groups different, p < 0.05 by two-way ANOVA; effects of reperfusion time were not significant).
While prolyl hydroxylase inhibition is known to produce HIF-1α accumulation (Epstein, et al., 2001), other targets of such inhibition may also contribute to neuroprotective effects (Ratan, et al., 2004, Siddiq, et al., 2007). Direct testing of HIF-1 involvement requires blockade or prevention of HIF-1 signaling in neurons cultured in 1% O2. One approach might be pharmacological, applying the DNA–binding peptide antibiotic echinomycin, reported to specifically block the binding of HIF-1 to response elements in target genes (Kong, et al., 2005). However, echinomycin was found to be quite neurotoxic in prolonged applications at sub-nanomolar concentrations (data not shown). Thus we turned to genetic tools to directly test whether HIF-1α itself plays an essential role in the protection by 1% O2 against ischemia-reperfusion injury, making use of neurons cultured from mice with conditional knockout of the HIF-1α gene (Ryan, et al., 2000). These cells were indistinguishable in morphology from wildtype neurons, and as previously observed in wild-type neurons (Li, et al., 2005), showed a strong increase in accumulated HIF-1α levels when exposed chronically to 1% O2 (Figure 6A). Treatment of these HIF-1α fl/fl neurons with an adenoviral vector carrying cre recombinase (AdV cre) produced the expected strong suppression of hypoxia-induced HIF-1α accumulation. As HIF-2α is an alternative candidate for mediating hypoxia-induced protection, we also examined expression of HIF-2α in these cells by Western blotting (Figure 6B). HIF-2α was detected at only faint levels, requiring film-based rather than digital imager-based detection of chemoluminescent signals. Furthermore, HIF-2α accumulation was not upregulated by chronic (7 day) exposures to 1% O2 or to DMOG, nor was it upregulated in a compensatory fashion in cells with HIF-1α knockdown by AdV cre treatment (Figure 6B). Thus no basis could be found to attribute to HIF-2α the protection afforded by either hypoxic adaptation or DMOG, while HIF-1α, upregulated by these treatments, remained a candidate for explaining the protection.
Figure 6. Neuronal expression of hypoxia inducible factors.

Whole-cell lysates were prepared from cortical neurons cultured from HIF-1αfl/fl mice in conditions of 20% O2 or 1% O2. Neurons were treated on day 1 with AdV cre at of 50 or 100 M.O.I., or with the empty vector Bgl II, and harvested at day 8 in vitro. A) Immunoblotting for HIF-1α demonstrated marked hypoxia-induced HIF-1α accumulation, eliminated by AdV cre treatment at 50 or 100 M.O.I. B) Immunoblotting for HIF-2α showed only faint expression in cortical neurons, without upregulation by hypoxia or DMOG treatment (25 μM for 7 days), and only modest upregulation by CoCl2 (300 μM for 4 hours). Knockdown of HIF-1α by AdV cre treatment (100 M.O.I.) did not induce compensatory upregulation of HIF-2α.
HIF-1α fl/fl neurons were therefore cultured in 1% O2 conditions, and treated either with a control viral vector (Bgl II) or with AdV cre. In each culture prep, efficiency of HIF-1α genetic deletion was confirmed by Western blotting for HIF-1α in parallel dishes. When such neurons, adapted to 1% O2, were subjected to simulated ischemia with continuous monitoring of survival in the perfusion model described above, they remained fully protected from acute necrosis over 2 hours of simulated reperfusion, despite HIF-1α deletion (Figure 7).
Figure 7. HIF-1α knockdown did not prevent hypoxia-induced protection against acute ischemia-reperfusion injury.
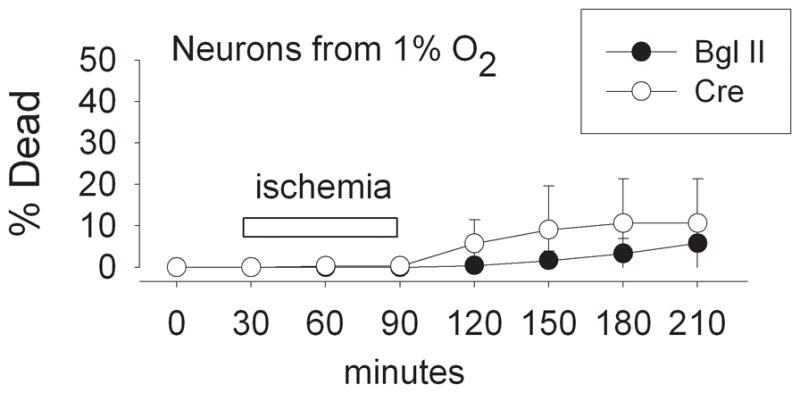
Acute neuronal death during simulated ischemia-reperfusion in cortical neurons from HIF-1α fl/fl mice cultured in 1% O2 and treated with Bgl II viral vector or with AdV cre (100 M.O.I.), monitored during continuous perfusion. HIF-1α knockdown did not abrogate the protective effect of prior adaptation to 1% O2 (n = 4; effect of O2 level not significant by two-way ANOVA).
This result suggests that HIF-1α is not necessary for the neuroprotective effects of hypoxic adaptation in the early phases of reperfusion injury. However, lasting hypoxia-induced protection against later phase neuronal death might still be dependent upon HIF-1α. We therefore examined delayed survival at later time points of 24 and 72 hours following excitotoxic injury or simulated ischemia-reperfusion. For these experiments, an adenoviral vector carrying cre recombinase linked to enhanced green fluorescent protein was utilized, allowing confirmation of effective gene transduction by enumeration of neurons expressing fluorescence. Pilot experiments confirmed that infection with AdV cre-GFP at 100 M.O.I. consistently produced greater than 90% expression of GFP, correlating with strong knockdown of HIF-1α by Western blotting (n = 3, not shown). Delayed survival after simulated ischemia-reperfusion injury was then examined in neurons infected with control or cre recombinase-expressing virus (Figure 8A). Again, the strong protection provided by adaptation to 1% O2 was not diminished at 24 hours or at 72 hours by deletion of HIF-1α expression, whether compared to neurons not exposed to virus or to those infected with control Bgl II virus (n = 5), ruling out a necessary role for HIF-1α in the delayed phase of protection after simulated ischemia-reperfusion as well.
Figure 8. The role of HIF-1α in hypoxia-induced protection against delayed ischemia-reperfusion injury.

All data are from neurons cultured from HIF-1α fl/fl mice, treated at day 1 with 100 M.O.I. empty viral vector Bgl II or with AdV cre-GFP, as indicated. A) Delayed relative survival at 24 hours and 72 hours following simulated ischemia-reperfusion (with glutamate) in neurons cultured for 10 days in 20% O2, 5% O2, or 1% O2 conditions. HIF-1α knockout, in cre-exposed neurons, did not prevent the hypoxia-induced protection (n = 5; N.S., groups not significantly different by two-way ANOVA). B) Excitotoxic injury assayed 24 hours after 60 min exposure to 100 μM glutamate. HIF-1α knockdown with AdV cre-GFP produced no loss of protection against excitotoxic death in neurons from 1% O2 (relative survival, n = 4; cre group compared to Bgl II group not significantly different by two-way ANOVA; effects of O2 level remained significant). C) Effect of prolyl hydroxylase inhibition. Relative survival 24 hours after simulated ischemia-reperfusion in neurons from 20% O2 conditions treated with DMOG (25 μM). Significant protective effects of DMOG were not affected by HIF-1α knockdown in cre-exposed neurons (n = 4; cre group compared to Bgl II group not significantly different by two-way ANOVA; *significant protection by DMOG, p<0.001).
Turning to glutamate-induced excitotoxicity, the complete protective effect of adaptation to 1% O2 against injury from 60 minute exposures to 100 μM glutamate, re-confirmed in neurons infected with control viral vector Bgl II, was also unaltered by HIF-1α deletion produced by treatment with the AdV cre-GFP vector (Figure 8B; n = 4; no significant differences compared to Bgl II-treated neurons). Thus, HIF-1α expression was not required for protection against direct excitotoxic injury by adaptation to 1% O2.
Finally, we also tested whether HIF-1α accumulation is required for the neuroprotection afforded by prolyl hydroxylase inhibition with DMOG. Again, suppression of HIF-1α by cre-mediated gene deletion failed to prevent the protective effect of DMOG pre-treatment of neurons against simulated ischemia-reperfusion injury, with protection similar to that in control neurons (Figure 8C). In sum, these results independently confirmed the protection against excitotoxic or ischemia-reperfusion injury from adaptation to 1% O2, and from prolyl hydroxylase inhibition, and they strongly ruled out a necessary role for HIF-1α in mediating this protection.
Discussion
In the present experiments we show that hypoxic adaptation, known to result in improved basal survival of cultured neurons, also produces profound protection against simulated ischemia-reperfusion injury and against glutamate excitotoxicity. Reperfusion injury following prolonged neuronal ischemia involves multiple mechanisms, with glutamate excitotoxicity mediated by NMDA receptors playing a central role in triggering subsequent injury. Injury following severe ischemia primarily involves rapid, necrotic mechanisms of neuronal death, with contributions from various forms of programmed cell death in areas of less severe ischemia (Bredesen, 2007). In the present model of simulated ischemia applied for 60 min, cell death occurred in association with cell swelling and rapid loss of membrane integrity within 2 hours, with little further cell loss over the subsequent days, suggesting predominance of a necrotic form of cell death. An antagonist of NMDA receptors blocked this ischemia-induced death, confirming the important role for glutamate excitotoxicity, whether or not exogenous glutamate was included in the simulation of ischemia. Prevention of excitotoxic neuronal injury often requires attenuation of NMDA receptor-dependent Ca2+ entry. However, in the present experiments the strong protective effect of prior adaptation to moderate hypoxia against both excitotoxic and ischemia-reperfusion injury occurred without significant reduction of NMDA receptor expression, and without a shift in desensitization or Mg2+-inhibition properties of expressed NMDA receptors that might explain this protection. While the data cannot fully rule out a small contribution to the protection from a modest reduction in NMDA receptor expression in cells cultured in 1% O2, not confirmed statistically, a reduction in average NMDA receptor current amplitude could not plausibly explain the strong protection afforded by hypoxic adaptation across the entire population of cells, with their highly overlapping distributions of NMDA current amplitudes. Further experiments would be required to determine if some difference in membrane targeting of NMDA receptors or in sequestration of receptor-mediated Ca2+ influx might explain the observed protection against excitotoxicity.
Inhibition of prolyl hydroxylase, the enzyme required for O2-induced HIF-1α degradation, resulted in HIF-1α accumulation and in strong protection against ischemia-reperfusion, similar to that seen from hypoxic adaptation. Given this result, and prior work suggesting the importance of HIF-1α to the improvement in basal survival induced by 1% O2 (Li, et al., 2005, Li and Brorson, 2006), we anticipated that HIF-1α would play an important role in protective effects of adaptation to hypoxia against reperfusion and excitotoxic modes of injury. However, genetic deletion of HIF-1α in neurons from HIF-1α fl/fl mice did not reduce any of the protective effects of adaptation to hypoxia, whether against glutamate excitotoxicity or against simulated ischemia-reperfusion injury, nor did it prevent the protection induced by prolyl hydroxylase inhibition. Thus, HIF-1-α independent mechanisms must account for these protective effects.
Prior work has also documented HIF-1α-independent protective effects of prolyl hydroxylase inhibition, and in particular that the homologous molecule HIF-2α may play a protective role in oxidative damage to neurons (Siddiq, et al., 2009). In the present work, HIF-2α was found to be relatively weakly expressed in the neurons, in agreement with a previous report (Chavez, et al., 2006), and was not upregulated by hypoxic adaptation or by DMOG, making it an unlikely target to explain the protective effects of these treatments against ischemia-reperfusion injury. Regardless, the results again highlight the promise of prolyl hydroxylase inhibition as a strategy for neuroprotection (Ratan, et al., 2004).
The near-complete survival of ischemia-reperfusion afforded by hypoxic adaptation suggests that strong neuroprotective mechanisms are involved. Powerful endogenous programs leading to protection against acute and delayed ischemia-triggered death are also well known to be triggered by hypoxic preconditioning, and the present results might be viewed as another version of this well-described phenomenon (Chen and Simon, 1997). However, the stimulus producing protection by hypoxic adaptation, in the present experiments, is a prolonged, moderate lowering of O2 tensions to a level compatible with continued mitochondrial respiration, rather than a brief exposure to functional anoxia or ischemia sufficient to impair respiration. Furthermore, increased levels of phosphorylated Akt, shown to mediate some forms of preconditioning (Wick, et al., 2002), are not found in cortical neurons cultured in 1% O2 (D. Li and J.R. Brorson, unpublished results). Although upregulation of HIF-1 and its gene targets has been shown to be closely associated with neuronal hypoxic preconditioning (Bergeron, et al., 2000, Bernaudin, et al., 2002), specific genetic deletion of neuronal HIF-1α expression failed to prevent hypoxic preconditioning against the injury of focal transient brain ischemia (Baranova, et al., 2007). Similarly, hypoxic preconditioning of photorecepors against phototoxicity was recently found to be independent of HIF-1α (Thiersch, et al., 2009). It is likely that hypoxic adaptation and hypoxic preconditioning involve both overlapping and distinctive pathways of mechanisms promoting neuronal survival in stressful environments.
The role of HIF-1α in neuronal survival of ischemia has been somewhat controversial. In vitro, iron chelators produced HIF-1 upregulation associated with protection against oxidative stress- induced death in cultured cortical neurons (Zaman, et al., 1999), and hypoxia-induced or genetic overexpression of HIF-1α protected sympathetic neurons against NGF-deprivation-induced neuronal death (Xie, et al., 2005), suggesting protective effects of HIF-1α against these stresses. However, in oxygen and glucose deprivation applied to cultured cortical neurons, modeling ischemia, a deleterious effect of HIF-1α on survival was suggested, in that suppression of HIF-1α by viral delivery of a dominant-negative form protected against delayed death (Halterman, et al., 1999). More recently, cell-specific effects were indicated by studies of cortical neuronal survival of a pure hypoxic insult showing that neuronal expression of HIF-1α supported neuronal survival of hypoxia, while astrocytic expression of HIF-1α promoted neuronal death in hypoxia (Vangeison, et al., 2008).
In vivo studies have further examined the role for HIF-1 expression in neuronal tolerance of ischemia. One report demonstrated protection against prolonged (75 min) global forebrain ischemic damage in mice with genetic deletion of brain HIF-1α (Helton, et al., 2005), while another report, also using genetic deletion of forebrain HIF-1α expression, found exacerbation of the damage produced by transient (30 min) MCA occlusion, with the pro-survival effects of HIF-1 appearing at later survival times (Baranova, et al., 2007). Thus, both an unexpected role for HIF-1 in mediating injury in global forebrain ischemia, and a protective role against delayed death following transient focal ischemia, appear to be indicated. Both studies utilized cre recombinase-driven deletion of HIF-1α gene, with cre expression under control of the calcium/calmodulin-dependent kinase CaMK IIα promoter, suggesting that the different outcomes may relate primarily to the different forms of ischemic injury. While all the reasons for this divergence of findings are not clear, these results underscore the potential for HIF-1α’s effects to be either deleterious or beneficial for neuronal survival, depending on the severity and mode of injury, and the likelihood that both the timing and the cellular localization of ischemia-induced HIF-1α expression may determine the net effect on neuronal survival.
The robust neuroprotection by hypoxic adaptation demonstrated in the present experiments points to endogenous cellular responses to the environment that strongly reduce the intrinsic vulnerability of cortical neurons to ischemia-reperfusion injury and excitotoxicity. Elucidation of the mechanisms and mediators underlying this resistance is likely to offer attractive targets for neuroprotection in stroke and brain ischemia.
Acknowledgments
We wish to thank Dr. Randall Johnson for provision of founding breeding pairs of HIF-1αfl/fl mice, Nnana Eleanor Mogongwafor technical assistance, and Dr. Zuohui Shao and Dr. Terry Vanden Hoek for assistance in performing in vitro simulated ischemia-reperfusion experiments. This work was supported by a Grant-in Aid to J.R.B. from the American Heart Association, and by the Valenti Family Foundation.
References
- 1.Banasiak KJ, Burenkova O, Haddad GG. Activation of voltage-sensitive sodium channels during oxygen deprivation leads to apoptotic neuronal death. Neuroscience. 2004;126:31–44. doi: 10.1016/S0306-4522(03)00425-1. [DOI] [PubMed] [Google Scholar]
- 2.Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC. Neuron-specific inactivation of the hypoxia inducible factor 1α increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci. 2007;27:6320–6332. doi: 10.1523/JNEUROSCI.0449-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergeron M, Gidday JM, Yu AY, Semenza GL, Ferriero DM, Sharp FR. Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Annals of Neurology. 2000;48:285–296. [PubMed] [Google Scholar]
- 4.Bernaudin M, Tang Y, Reilly M, Petit E, Sharp FR. Brain genomic response following hypoxia and re-oxygenation in the neonatal rat. Identification of genes that might contribute to hypoxia-induced ischemic tolerance. J Bio Chem. 2002;277:39728–39738. doi: 10.1074/jbc.M204619200. [DOI] [PubMed] [Google Scholar]
- 5.Bredesen DE. Toward a mechanistic taxonomy for cell death programs. Stroke. 2007;38:652–660. doi: 10.1161/01.STR.0000257802.82826.a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brorson JR, Li D, Suzuki T. Selective expression of heteromeric AMPA receptors driven by Flip-flop differences. J Neurosci. 2004;24:3461–3470. doi: 10.1523/JNEUROSCI.5023-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brorson JR, Zhang Z, Vandenberghe W. Ca2+ permeation of AMPA receptors in cerebellar neurons expressing Glu receptor 2. J Neurosci. 1999;19:9149–9159. doi: 10.1523/JNEUROSCI.19-21-09149.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biology. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chavez JC, Baranova O, Lin J, Pichiule P. The transcriptional activator hypoxia inducible factor 2 (HIF-2/EPAS-1) regulates the oxygen-dependent expression of erythropoietin in cortical astrocytes. J Neurosci. 2006;26:9471–9481. doi: 10.1523/JNEUROSCI.2838-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J, Simon R. Ischemic tolerance in the brain. Neurology. 1997;48:306–311. doi: 10.1212/wnl.48.2.306. [DOI] [PubMed] [Google Scholar]
- 11.Epstein ACR, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. Elegans EGL-9 and Mammalian Homologs Define a Family of Dioxygenases that Regulate HIF by Prolyl Hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 12.Gidday JM, Fitzgibbons JC, Shah AR, Park TS. Neuroprotection from ischemic brain injury by hypoxic preconditioning in the neonatal rat. Neurosci Letters. 1994;168:221–224. doi: 10.1016/0304-3940(94)90455-3. [DOI] [PubMed] [Google Scholar]
- 13.Gidday JM, Shah AR, Maceren RG, Wang Q, Pelligrino DA, Holtzman DM, Park TS. Nitric oxide mediates cerebral ischemic tolerance in a neonatal rat model of hypoxic preconditioning. J Cereb Blood Flow Metab. 1999;19:331–340. doi: 10.1097/00004647-199903000-00011. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez-Zulueta M, Feldman AB, Klesse LJ, Kalb RG, Dillman JF, Parada LF, Dawson TM, Dawson VL. Requirement for nitric oxide activation of p21ras/extreacellular regulated kinase in neuronal ischemic preconditioning. Proceedings of the National Academy of Sciences (USA) 2000;97:436–441. doi: 10.1073/pnas.97.1.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grote J, Laue O, Eiring P, Wehler M. Evaluation of brain tissue O2 supply based on results of PO2 measurements with needle and surface microelectrodes. J Autonomic Nervous System. 1996;57:168–172. doi: 10.1016/0165-1838(95)00096-8. [DOI] [PubMed] [Google Scholar]
- 16.Hakim A, Simon R. Ischemic Preconditioning: Introduction. Stroke. 2004;35 (Suppl 1):2675. [Google Scholar]
- 17.Halterman MW, Miller CC, Federoff HJ. Hypoxia-inducible factor-1α mediates hypoxia-induced delayed neuronal death that involves p53. J Neurosci. 1999;19:6818–6824. doi: 10.1523/JNEUROSCI.19-16-06818.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helton R, Cui J, Scheel JR, Ellison JA, Ames C, Gibson C, Blouw B, Ouyang L, Dragatsis I, Zeitlin S, Johnson RS, Lipton SA, Barlow C. Brain-specific knock-out of hypoxia-inducible factor-1α reduces rather than increases hypoxic-ischemic damage. J Neurosci. 2005;25:4099–4107. doi: 10.1523/JNEUROSCI.4555-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kong D, Park EJ, Stephen AG, Calvani M, Cardellina JH, Monks A, Fisher RJ, Shoemaker RH, Melillo G. Echinomycin, a small-molecular inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Research. 2005;65:9047–9055. doi: 10.1158/0008-5472.CAN-05-1235. [DOI] [PubMed] [Google Scholar]
- 20.Li D, Brorson JR. HIF-1α mediates the hypoxic enhancement of in vitro survival of cortical neurons. Society for Neuroscience Abstracts (online), Program no. 186.117 2006 [Google Scholar]
- 21.Li D, Marks JD, Schumacker PT, Young RM, Brorson JR. Physiological hypoxia promotes survival of cultured cortical neurons. European J Neurosci. 2005;22:1319–1326. doi: 10.1111/j.1460-9568.2005.04335.x. [DOI] [PubMed] [Google Scholar]
- 22.Li D, Shao Z, VandenHoek TL, Brorson JR. Reperfusion accelerates acute neuronal death induced by simulated ischemia. Exp Neurol. 2007;206:280–287. doi: 10.1016/j.expneurol.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mansfield KD, Guzy RD, Pan Y, Young RM, Schumacker PT, Simon MC. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-α activation. Cell Metabolism. 2005;1:393–399. doi: 10.1016/j.cmet.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Metzger H, Heuber S. Local oxygen tension and spike activity of the cerebral grey matter of the rat and its response to short intervals of O2 deficiency or CO2 excess. Pflügers Arch. 1977;370:201–209. doi: 10.1007/BF00581695. [DOI] [PubMed] [Google Scholar]
- 25.Park CK, Nehls DG, Graham DI, Teasdale GM, McCulloch J. The glutamate antagonist MK-801 reduces focal ischemic brain damage in the rat. Ann Neurol. 1988;24:543–551. doi: 10.1002/ana.410240411. [DOI] [PubMed] [Google Scholar]
- 26.Prass K, Scharff A, Ruscher K, Löwl D, Muselmann C, Victorov I, Kapinya K, Dirnagl U, Meisel A. Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke. 2003;34:1981–1986. doi: 10.1161/01.STR.0000080381.76409.B2. [DOI] [PubMed] [Google Scholar]
- 27.Ratan RR, Siddiq A, Aminova L, Lange PS, Langley B, Ayoub I, Gensert J, Chavez J. Translation of ischemic preconditioning to the patient. Prolyl hydroxylase inhibition and hypoxia inducible factor-1 as novel targets for stroke therapy. Stroke. 2004;35 (Suppl 1):2687. doi: 10.1161/01.STR.0000143216.85349.9e. [DOI] [PubMed] [Google Scholar]
- 28.Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, Johnson RS. Hypoxia-inducible Factor-1α Is a Positive Factor in Solid Tumor Growth. Cancer Research. 2000;60:4010–4015. [PubMed] [Google Scholar]
- 29.Siddiq A, Aminova LR, Ratan RR. Hypoxia inducible factor prolyl 4-hydroxylase enzymes: Center stage in the battle against hypoxia, metabolic compromise and oxidative stress. Neurochem Res. 2007;32:931–946. doi: 10.1007/s11064-006-9268-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siddiq A, Aminova LR, Troy CM, Suh K, Messer Z, Semenza GL, Ratan RR. Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB- independent pathways. J Neurosci. 2009;29:8828–8838. doi: 10.1523/JNEUROSCI.1779-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simon RP, Swan JH, Meldrum BS. Blockade of N-Methyl-D-Aspartate receptors may protect against ischemic damage in the brain. Science. 1984;226:850–852. doi: 10.1126/science.6093256. [DOI] [PubMed] [Google Scholar]
- 32.Stec DE, Davisson RL, Haskell RE, Davidson BL, Sigmund CD. Efficient liver-specific deletion of a floxed human angiotensinogen transgene by adenoviral delivery of Cre recombinase in vivo. J Biol Chem. 1999;274:21285–90. doi: 10.1074/jbc.274.30.21285. [DOI] [PubMed] [Google Scholar]
- 33.Thiersch M, Lange C, Joly S, Heynen S, Le YZ, Samardzija M, Grimm C. Retinal neuroprotection by hypoxic preconditioning is independent of hypoxia-inducible factor –1α expression in photoreceptors. Eur J Neurosci. 2009;29:2291–2302. doi: 10.1111/j.1460-9568.2009.06781.x. [DOI] [PubMed] [Google Scholar]
- 34.Vangeison G, Carr D, Federoff HJ, Rempe DA. The good, the bad, and the cell type-specific roles of hypoxia inducible factor 1α in neurons and astrocytes. J Neurosci. 2008;28:1988–1993. doi: 10.1523/JNEUROSCI.5323-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wahlestedt C, Golanov E, Yamamoto S, Yee F, Ericson H, Yoo H, Inturrisi CE, Reis DJ. Antisense oligodeoxynucleotides to NMDA-R1 receptor channel protect cortical neurons from ecitotoxicity and reduce focal ischaemic infarctions. Nature. 1993;363:260–263. doi: 10.1038/363260a0. [DOI] [PubMed] [Google Scholar]
- 36.Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J Neurosci. 2002;22:6401–6407. doi: 10.1523/JNEUROSCI.22-15-06401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie L, Johnson RS, Freeman RS. Inhibition of NGF deprivation-induced death by low oxygen involves suppression of BIMEL and activation of HIF-1. J Cell Biol. 2005;168:911–920. doi: 10.1083/jcb.200407079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaman K, Ryu H, Hall D, O’Donovan K, Lin K-I, Miller MP, Marquis JC, Baraban JM, Semenza GL, Ratan RR. Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21waf1/cip1, and erythropoietin. J Neurosci. 1999;19 doi: 10.1523/JNEUROSCI.19-22-09821.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]; J Bio Chem. :9821–9830. [Google Scholar]