

Abstract
Cellular control of protein activities by modulation of their abundance or compartmentalization is not easily measured on a large scale. We developed and applied a method to globally interrogate these processes that is widely useful for systems-level analyses of dynamic cellular responses in many cell types. The approach involves subcellular fractionation followed by comprehensive proteomic analysis of the fractions, which is enabled by a data-independent acquisition mass spectrometry approach that samples every available mass to charge channel systematically to maximize sensitivity. Next, various fraction-enrichment ratios are measured for all detected proteins across different environmental conditions and used to group proteins into clusters reflecting changes in compartmentalization and relative conditional abundance. Application of the approach to characterize the response of yeast proteins to fatty acid exposure revealed dynamics of peroxisomes and novel dynamics of MCC/eisosomes, specialized plasma membrane domains comprised of membrane compartment occupied by Can1 (MCC) and eisosome subdomains. It also led to the identification of Fat3, a fatty acid transport protein of the plasma membrane, previously annotated as Ykl187.
A large number of proteomic studies have focused on profiling or quantifying protein components of specific subcellular compartments or macromolecular structures in a variety of cell types. These include proteomic studies of the nucleus, mitochondrion, Golgi apparatus, cell wall, peroxisome, plasma membrane, nucleolus, lysosome, and vacuole (1). These studies provide valuable insights into the biological functions of the organelles as well as individual protein components. Analysis of organelle protein inventories is complicated by the fact that a given protein may have multiple functional roles (2) and may have multiple subcellular locations as well as spatially and temporally dynamic distributions. For example, our previous study showed that glycerol-3-phosphate dehydrogenase 1 (Gpd1) localizes to not only cytosol but also peroxisomes (3, 4). Localization of Gpd1 to peroxisomes is dependent on both the metabolic status of cells and the phosphorylation of aminoacyl residues adjacent to the peroxisomal targeting signal. In addition, exposure of cells to osmotic stress induces subcellular redistribution of Gpd1 to the cytosol and nucleus. This example reinforces the fact that protein localization is an important and complex aspect of cellular response to environmental stimuli. For this reason, it is important to not only define the proteome of subcellular organelles, but also to measure dynamic distribution of proteins between different compartments.
Shotgun mass spectrometry (MS) approaches have been widely used, especially for comparative and quantitative proteomics. Traditionally, shotgun proteomic analysis has been performed in a data-dependent manner where ion selection for collision induced dissociation relies on a preliminary precursor ion scan, from which peptides are selected and then subjected to collision induced dissociation (5). This general approach has become extremely powerful for determining the protein content of moderately complex mixtures. However, the ability to compare different samples is complicated by the semi-random sampling process of data-dependent acquisition (DDA)1, which under-samples available ions (5, 6). The bias in data-dependent ion selection is generally against ions of low signal to noise ratio leaving a portion of peptide ions detected (as precursor ions) but not identified. Recently, a novel tandem MS based proteome screen termed Precursor Acquisition Independent From Ion Count (PAcIFIC) was shown to identify proteins over an order of magnitude larger dynamic range than standard shotgun proteomic methods in a complex proteome (7, 8). This data-independent acquisition (DIA) MS technique systematically interrogates all available m/z channels for the presence of peptides, regardless of the observation of a precursor ion. PAcIFIC thus provides increased confidence in protein identification because all peptides present, irrespective of abundance, are eventually fragmented, which can dramatically improve protein sequence coverage. Therefore, this approach provides a simplified, direct, and systematic approach for proteomic screening.
The yeast, Saccharomyces cerevisiae, can use a variety of carbon sources; glucose is catabolized mainly by fermentation, whereas nonfermentative carbon sources, such as ethanol, glycerol, and fatty acids are catabolized by respiration (9). In the presence of abundant glucose, fermentation is dominant and glycolysis in the cytosol generates all the necessary energy and biomass, and proteins required for utilization of nonfermentable substrates, including mitochondrial proteins involved in respiration, as well as peroxisomal proteins involved in β-oxidation of fatty acids, are repressed (10). In the absence of a fermentative carbon source, respiration dominates. Use of fatty acids such as oleic acid as the sole carbon source by S. cerevisiae requires the coordinated function of peroxisomes, where fatty acids are catabolized, and mitochondria, where oxidation is completed (11).
We sought to establish a proteomic approach for global analysis of coordinated and conditional protein redistribution. A strategy combining a sensitive and quantitative DIA-MS method with subcellular fractionation was developed to measure changes in protein abundance and subcellular distribution under different conditions. We applied this strategy to globally characterize protein dynamics in response to a switch from glucose- to oleic acid-containing medium. These conditions represent two metabolic conditions of yeast, fermentation and respiration, and were selected because of the wealth of information available in the literature. The analysis generated data for more than 60% of the yeast proteome and identified over 1000 candidate proteins that responded to the stimulus. These data demonstrate that the strategy is capable of measuring condition-specific changes in abundance and redistribution of proteins on a global scale.
EXPERIMENTAL PROCEDURES
Strains and Culture Conditions
For the global analysis of protein abundance and redistribution, yeast cells (strain BY4742) were grown to log phase in YPD (1% yeast extract, 2% peptone, 2% glucose), or SCIM (0.7% yeast nitrogen base, 0.5% yeast extract, 0.5% peptone, 0.5% Tween 40 (w/v), 0.79 g complete synthetic medium/L, 0.5% ammonium sulfate, and 0.15% (v/v) oleic acid). YPD was chosen as the reference condition as it is the standard laboratory growth condition for yeast and thus the data set is compatible with other data sets in the literature for systems-level analyses. SCIM was chosen as it is the standard fatty acid induction medium for yeast. Note that although the switch of carbon source from glucose to oleic acid elicits a very strong cellular response (12), other differences between the growth conditions may also trigger responses; therefore response specificity needs to be ascertained through follow-up experiments (as we have done for Fat3 (see “Results” Section)). For microscopy, diploid cells were grown overnight in YPD and then for 24 h to log phase in either SCIM (with Tween 40 and oleate), or SCIM without Tween 40 and oleate containing either 2% glucose or 3% glycerol. For the cerulenin toxicity assay, all plates were made with YPD medium containing 0.7% KH2PO4 and 2% agar. Some plates also contained 25 μm cerulenin (from 20 mg/ml stock in ethanol; Sigma), or 25 μm cerulenin and 500 μm myristate (C14:0) solubilized in Tween 40 at a final concentration of 0.5% (w/v).
Haploid deletion strains were from the commercially available deletion set (Invitrogen, Carlsbad, CA). Haploid strains tagged with Aequorea Victoria (S65T) GFP are isogenic to BY4741 and are from the commercially available GFP clone collection (Invitrogen) and were converted to diploids by mating to BY4742, and selecting individual zygotes. Strains expressing the Sur7-mCherry chimera were constructed by homologous recombination of the sequences encoding Discosoma sp. red fluorescent protein (13) and hph (hygromycin B resistance gene) amplified together from pBS35 (YRC, University of Washington) in frame and downstream of the SUR7 open reading frame.
Subcellular fractionation and protein digestion
A whole cell lysate (WCL) and an organelle-enriched fraction (20KgP) were prepared from glucose- or oleate-grown cells as described below. For WCL preparations, cells were quickly collected by centrifugation, washed, and immediately lysed in buffer (0.74% β-mercaptoethanol and 0.815 m NaOH) for 10 min at 4 °C. 20KgP fractions were prepared as previously described (14, 15). Briefly, cells were harvested by centrifugation and converted to spheroplasts by incubation with 1 mg Zymolase 100T/g of cells for 1 h at 30 °C. Spheroplasts were lysed by homogenization in 2-(N-morpholino)ethanesulfonic acid buffer (0.65 m sorbitol, 5 mm 2-(N-morpholino)ethanesulfonic acid, pH 5.5) containing 1 mm KCl, 1 mm EDTA, 0.2 mm phenylmethanesulfonyl fluoride, 0.4 μg pepstatin A/ml, and 1 × SIGMAFASTTM Protease inhibitor (Sigma). Cell debris and nuclei were separated from the homogenate by centrifugation for 10 min at 2000 × g to generate a postnuclear supernatant which was subjected to 20,000 × gmax for 30 min at 4 °C in a JS13.1 rotor (Beckman Instr., Inc.) to yield a pellet (20KgP) enriched for peroxisomes and mitochondria and a supernatant (20KgS) enriched for cytosol and high-speed pelletable organelles.
To digest proteins in either WCL or 20KgP fractions, proteins were precipitated by addition of trichloroacetic acid to a final concentration of 10% (v/v) followed by centrifugation at 10,000 × g for 30 min at 4 °C. The pellet was washed twice with ice-cold acetone. Proteins were denatured by incubation with 6 m urea in 50 mm ammonium bicarbonate and reduced for 1 h at 37 °C with 5 mm Tris (2-carboxyethyl) phosphine. Alkylation of cysteine residues was performed by adding iodoacetamide to 30 mm, and incubating for 1 h in the dark, followed by adding dithiothreitol to a final concentration 30 mm and incubating for 1 h. The volume was increased eightfold with 50 mm ammonium bicarbonate to dilute the urea, and samples were incubated overnight at 37 °C with sequencing grade trypsin (at a protein to trypsin mass ratio of 50:1). Samples were desalted using MacroSpin C18 columns (30–300 μg capacity, SMMSS18V, The Nest Group, Southborough, MA) according to the manufacturer's protocol. Eluates were stored at −80 °C.
Mass Spectrometry
All MS experiments were performed on a nanoACQUITY system (Waters, Milford, MA) connected to a hybrid LTQ-Orbitrap XL (Thermo Fisher scientific, San Jose, CA). For each injection, ∼0.5 μg of peptide mixture were loaded, trapped on a 100 μm i.d. × 25 mm long precolumn packed with 200 Å (5 μm) Magic C18 particles (C18AQ; Michrom BioResources Inc., Auburn, CA), and separated in a gravity-pulled 75 μm i.d. × 200 mm analytical column packed with 100 Å (5 μm) Magic C18 particles (C18AQ; Michrom BioResources Inc., Auburn, CA) using a linear gradient of 0–35% acetonitrile in 0.1% formic acid over 60 min. The eluted peptides from the HPLC were directly electrosprayed into the mass spectrometer and analyzed in positive ion mode.
For DIA analysis, all data were acquired in triplicate (using three different technical replicates of each fraction) on the LTQ Orbitrap XL using an accurate PAcIFIC method (8). Each single liquid chromatography (LC)-MS/MS experiment comprised a cycle of 15 data-independent collision-induced dissociation spectra covering 22.5 m/z units with a precursor ion survey scan inserted after every five tandem mass spectra. For each sample (one replicate of one cell fraction), a total of 45 LC-MS/MS analyses were performed in an identical fashion to achieve a 1000 m/z mass range (400–1400 m/z). As 3.5 d are required for 45 injections, the entire analysis required 42 d to complete (3.5 d × 4 fractions × 3 technical replicates).
Preprocessing of Data
For data acquired using accurate PAcIFIC method, data were preprocessed using the workflow described previously (8). Briefly, a feature detection step was first performed on the high resolution survey scans using Hardklor software (16). This information was used to correct the precursor mass of the data-independent spectrum using an in-house Perl program (aPAcIFIC.pl). The output consists of two separate mzXML files containing either modified tandem MS spectra or raw tandem MS spectra for high and low mass accuracy searches, respectively.
Peptide and Protein Identification
Database searches were performed against the yeast ORF database covering 6717 entries (release 2009–05-08) on the Saccharomyces genome database (SGD) website (www.yeastgenome.org) using SEQUEST v. 27 algorithm (17) with the following parameters: one enzyme specific terminus required, one missed cleavage allowed, and cysteine alkylation (+57 Da) as a fixed modification, and methionine oxidation (+16 Da) as variable. For high mass accuracy searches, precursor ion tolerance was set to 10 ppm, and for low mass accuracy searches, fragment ion tolerance was set to 3.75 Da. Sequest results were converted to pepXML files and probability assessments of identified peptides were computed with PeptideProphet and ProteinProphet (18). For all individual searches, peptides with an estimated false discovery rate of less than 0.5% were accepted unless specified. Only peptides mapping to unique protein were considered for protein identification and quantification (supplemental Tables S1A-S1D). Only proteins with multiple unique peptides were considered for further analysis (supplemental Table S2). The raw MS data associated with this manuscript may be downloaded from PeptideAtlas repository (www.peptideatlas.org).
Protein Ratios from Spectral Counts and Statistical Analysis
For spectral counting, the number of peptide tandem mass spectra identified for each peptide from a given protein was used to estimate abundance of that protein relative to all others present in a sample. Only tandem mass spectra that matched to peptide sequences with the above stated criteria were used for quantification. The spectral counts for each protein were summed for all data over the complete m/z range analyzed per sample for each replicate. The raw spectral counts in the three DIA-MS replicates per sample were averaged. In the cases where the peptide was only detected in one sample, 1 was added to the spectral counts to avoid a zero count. Next, the spectral counts for each protein were normalized to generate identical total counts for the two samples. Differences in spectral counts are identified by applying a likelihood ratio test (G-test) for independence (19), which is similar to the χ2 distribution and Fischer's exact tests, and corrects for variations in total counts between data sets. The G statistic is approximately distributed as χ2 with one degree of freedom, allowing p value calculation for each protein to aid in identifying those with differential abundance. Locally weighted scatter plot smoothing (LOWESS) curve fitting (20–22) was performed using R (23) with default parameters.
Gene Ontology Slim Term Enrichment
GO slim terms for each gene in the yeast genome were downloaded from the SGD website (www.yeastgenome.org) on 11/06/2010. For each term, the observed frequencies in each data set were compared with those expected by chance (the frequency of annotation for the 6310 yeast genes). For enriched terms, the probability that the observed distribution would be found by chance was determined by calculating binomial distribution probability using Microsoft Excel and the probability mass function. This algorithm has been used by others to estimate the significance of term enrichments with similar population and sample sizes (24).
Integrative Analysis of Conditional Fraction Enrichment Data with Other Global Data Sets
Data sets that potentially identify proteins responsive to fatty acid exposure were integrated with the data set generated here to identify proteins involved in fatty acid responsive organelle dynamics. They include: (1) A data set of 298 proteins associated with peroxisomal membranes after fatty acid exposure (3). (2) Two data sets of genes necessary for growth on fatty acids, which were taken from our previously published study (25) and included genes necessary for normal growth on fatty acids (either oleic acid or myristic acid) but not glycerol. (3) A data set of genes that are predicted to be controlled by known fatty acid responsive regulators (Oaf1p, Pip2p, and Oaf3p), which includes genes that are bound by any one of these factors in glucose or oleate by previously published chromatin immunoprecipitation analysis (26).
Fluorescence Microscopy and Image Analysis
All images were of live cells harvested during the exponential phase of growth and are representative of data acquired from multiple biological replicates (two or more different cultures). Images were obtained using a DeltaVision PersonalDV deconvolution imaging system (Applied Precision, Issaquah, WA) equipped with an Olympus IX-71 wide field microscope with a 250 watt xenon LED transillumination light source. For each channel, 25 (0.2 micron) z-stack images were generated, deconvolved, and exported as tiff files. Images of cells that were compared across growth conditions were acquired using the same settings and adjusted identically using Adobe Photoshop. To quantify MCC/eisosome density, images of cortical sections of yeast cells expressing Sur7-GFP were used to calculate the number of eisosomes in the section normalized to the cross-sectional area of the cortical slice (eisosomes/kilopixel). Automated enumeration of eisosomes was performed using CellProfiler (27) (http://www.celllprofiler.org/index.shtml) and cross-sectional areas were determined in ImageJ by manually bounding regions of the cortical sections that contained eisosomes. Measurements were imported and analyzed in Excel. In total, ∼15 cells were analyzed for each condition using this semi-automated method. Statistical significance was determined using a two-tailed Student's t test.
RESULTS
Strategy for Measuring Condition-specific Protein Relocalization
The experimental strategy used in the present work is summarized in Fig. 1. To identify proteins that dynamically redistributed between the cytosol and organelles such as mitochondria, peroxisomes, ER, or Golgi in response to the environmental switch, a whole cell lysate (WCL) and a fraction enriched for 20,000 × g pelletable organelles (except nuclei) (20KgP) were prepared from glucose- or oleate-grown cells (Figs. 1A and 1B) (3, 4, 28–30). The tryptic digests of peptides from each sample were analyzed by DIA-MS in triplicate. MS data were processed with an accurate precursor detection workflow as described in the Experimental Procedures Section. Two data analysis methods were combined to measure condition-specific protein relocalization, a differential abundance method (Method I) and a fraction enrichment method (Method II) (Fig. 1C). Method I compares protein abundance in a fraction from oleic acid-grown cells relative to the same fraction from glucose-grown cells. Method II compares the relative abundance of identified proteins in the organelle-enriched fraction versus the crude WCL fraction from the same condition. Relative abundances are then compared across conditions.
Fig. 1.

An overview of the strategy combining DIA-MS with subcellular fractionation. A, Work flow of the experimental and bioinformatics procedures used for analysis of the WCL and 20KgP fractions by DIA MS. B, Equal amounts of protein from WCL and 20KgP fractions from glucose- and oleate-grown cells were separated by SDS-PAGE and visualized by Coomassie blue staining. C, Differential abundance method (Method I) compares protein abundance in a fraction from oleic acid-grown cells relative to the same fraction from glucose-grown cells. Fraction enrichment method (Method II) compares the relative abundance of identified proteins in the organelle-enriched fraction versus the crude WCL fraction within identical conditions.
Three aspects of the MS analysis are important in the context of the goals of the present work: (1) comprehensive identification of as many proteins as possible within each fraction, (2) saturation level in protein identification, and (3) accurate measurement of the relative abundance of all detected proteins across different fractions. To address the first issue, the DIA-MS was used to maximize identification of proteins from each fraction (DIA-MS affords greater dynamic range by at least one order of magnitude compared with traditional DDA-MS approaches (7)). To address the second issue, each fraction was analyzed in triplicate to achieve 95% saturation level in protein identification. This is very important given that a protein can only be meaningfully compared across fractions if each fraction is analyzed systematically and to an equivalent high level of saturation. To address the last issue of relative abundance, spectral counting (31) was used to quantitatively rank identified proteins relative to each other, with respect to two aspects: (a) reliability and (b) reproducibility. First, we compared average spectral counts for proteins in WCL fractions of cells grown in glucose to their abundance determined by quantitative Western blotting against the TAP tag after growth under similar conditions (32). We observed a significant positive correlation between spectral counts and the known protein abundance measured by TAP tagging (Pearson r = 0.58; 2-tailed p value < 0.01; supplemental Fig. S1). This result is consistent with the observation that spectral counting is a good measure for protein quantification (33–37). Second, the correlation of spectral counts for each protein identified from three replicate analyses of WCL from glucose grown cells demonstrates a very consistent linear relationship (supplemental Fig. S2). This was also true for replicates of the 20KgP fraction (supplemental Fig. S2). These data indicate that the spectral count of one protein in a complex sample consisting of many tens of thousands of tryptic peptide ions is a surprisingly stable measurement under these experimental conditions. Taken together, these data indicate that relative abundance of the same protein in different fractions can be measured with high confidence using spectral counting.
Characterization of the Identified Proteome
Table I summarizes the experimental MS data. All MS data for individual peptides are in supplemental Tables S1A–S1D. All MS data for individual proteins are in supplemental Tables S2 and S3. Fig. 2A shows a comparison of the identified proteins between conditions or fractions. Although the comparison shows a high degree of overlap between data sets, each condition or fraction had a unique subset of proteins identified. To investigate the properties of proteins in each fraction, proteins were clustered based on their GO slim term annotations representing their reported cellular component associations and compared across fractions (Fig. 2B). In general, similar numbers of proteins associated with each cellular component were identified from all WCL and 20KgP fractions, suggesting roughly equivalent completeness of coverage in each fraction. However, each fraction contained some protein identifications not found in the other fraction from the same condition, because of either incomplete coverage or bona fide fractional enrichment. In addition, the enriched organelle and/or membrane fraction (20KgP) generally led to the identification of slightly more organelle and membrane proteins compared with WCL, possibly because of the lower relative complexity of the fraction (Fig. 2B).
Table I. Summary of peptide and protein identification.
| WCL |
20KgP |
|||
|---|---|---|---|---|
| Glucose Ave_triplicate | Oleic acid Ave_triplicate | Glucose Ave_triplicate | Oleic acid Ave_triplicate | |
| No. unique peptides | 17,938 | 17,947 | 19,484 | 16,190 |
| No. unique peptides used for protein identificationa | 16,541 | 16,572 | 18,367 | 15,336 |
| No. proteins identified by multiple peptides | 2065 | 2021 | 2369 | 2116 |
a Only peptides which are not shared by multiple proteins are used for protein identification.
Fig. 2.

Comparison of the number of proteins identified between conditions or fractions. A, Venn diagrams comparing proteins identified between different fractions from the same condition (left) or between the same fractions from different conditions (right). In all cases, the overlap was greater than 80%. B, Distribution of cellular component annotations for proteins identified in glucose- (left) and oleic acid-grown cells (right). White regions of each bar represent the number of proteins identified in both fractions. Yellow (or blue) regions in each bar represent the number of proteins identified in each category from only WCL. Green (or pink) regions of each bar represent the number of proteins identified in each category from only 20KgP fraction. Cellular component abbreviations include: SOPG, site of polarized growth; PM, plasma membrane; CMBV, cytoplasmic membrane bound vesicle; MTOC, microtubule organizing centers. The numbers following each cellular component indicate the total number of proteins in each category from SGD. Because certain proteins are classified into multiple localization categories, the total ORF count exceeds the number genomic ORFs.
Differences in the level of representation of individual cellular components between the glucose and oleic acid fractions reflect underlying biological dynamics (Fig. 2B). For example, of the 1124 proteins that localize to, or associate with, mitochondria described in the SGD database, 765 (68%) were identified from the 20KgP of oleic acid-grown cells, whereas only 660 (59%) of these proteins were identified in the same fraction of glucose-grown cells. This likely, reflects the induction of mitochondrial function in the presence of oleic acid compared with glucose (10). Another example is the peroxisome proteome. Of the 62 proteins annotated as localized to peroxisomes or associated with peroxisomes in the SGD database, 50 (81%) were identified from the 20KgP in the presence of oleic acid, whereas only 30 (48%) proteins were identified from the same fraction in the presence of glucose, reflecting the induction of peroxisomal proteins by oleic acid compared with glucose (38).
These data also demonstrate a high level of protein coverage obtained by the approach. In general, most known constituents of each cellular component were identified (Fig 2B). For example, 83% of previously known peroxisomal membrane proteins were identified from the crude organelle fraction 20KgP of the oleate-grown cells by DIA-MS. This is exceptional coverage considering that only 33% of previously known peroxisomal membrane proteins were identified from the highly purified peroxisome fraction by sucrose density gradient centrifugation or immunopurification using DDA-MS (supplemental Fig. S3) (3).
Identification of Proteins with Conditional Fraction Enrichment by Method I or II
To establish a list of proteins with conditional fraction enrichment identified by Method I, the statistical significance of conditional enrichment of each protein was determined for either WCL or 20KgP fractions using a G-test (see Experimental Procedures Section). This analysis generated p values for each protein measuring the significance of conditional fraction enrichment (supplemental Table S3) (19). To support this analysis and to empirically establish G-test p value thresholds, we used LOWESS, a regression modeling method to separate proteins predicted to have true differential expression from those predicted to arise from random quantitative error (21).
To visualize the model, plots of spectral counts for each protein were generated to compare either replicates of the same fraction or fractions from different conditions (Fig. 3). The LOWESS smoothing curves (purple vectors in each panel) were applied to each graph to delineate the calculated upper and lower boundaries of variation because of experimental error.
Fig. 3.

Statistical analysis of differential protein abundance. A and C, Scatter plot of log2 of spectral count ratios versus log2 of summed protein level spectral counts (SpC) comparing replicate samples of either WCL (A) or 20KgP (C) from glucose-grown cells. B and D, The same scatter plots comparing fractions from oleate- and glucose-grown cells. For all scatter plots shown, solid purple curves are the LOWESS smoothing curves defining the upper and lower boundaries of the log2 ratios from control replicates of either WCL (A and B) or 20KgP fractions (C and D). Note that on each plot the proteins with data points that line up in a straight line and bookend the remaining data points are those that were detected in only one of the two fractions.
Plots comparing technical replicates of the same fraction (graphs A and C) showed that most proteins had log2 ratios of spectral counts close to zero, as expected, but in cases of low spectral counts (i.e. low log2 sum of spectral counts), the ratios calculated appeared noisy, consistent with the known limits of spectral counting (39). As expected, the LOWESS smoothing curves bookend almost all of the data, indicating little significant differential expression between the replicates. In contrast, for the comparisons between samples of different conditions (graphs B and D), many proteins were found outside of the LOWESS smoothing curves, suggesting their conditional fraction enrichment.
These curves were used to establish thresholds for p values of significant differential abundance generated using the G-test. Thresholds were chosen as the lowest values that resulted in ∼95% of significantly differentially abundant proteins falling outside of the regions bounded by the LOWESS curves. For analysis of differential abundance between WCLs (panel B), the threshold was set to p value ≤ 0.05. This yielded a total of 293 proteins with significant abundance changes between glucose and oleic acid, of which only 18 proteins (6%) fell into the region bounded by the curve. Of the 293 proteins, 171 and 122 proteins were up- and down-regulated, respectively. For the analysis of differential abundance in 20KgP fractions, the p value cutoff was set to ≤0.1. At this threshold, a total 1526 proteins showed statistically significant abundance changes between glucose and oleic acid, of which 43 (3%) fell into the region bounded by the lines. Of the 1526 genes, 483 and 1043 proteins were up- and down-regulated, respectively. Remarkably, almost fourfold more proteins showed abundance changes in between 20KgP fractions versus WCL fractions. Consistent with this, there are many more scattered spots outside of LOWESS boundaries in the 20KgP plot compared with the WCL plot (Fig. 3 graphs B and 3D). These data suggest that protein redistribution detected by this approach (reflected by comparison of 20KgP fractions), is more evident than differential synthesis (captured by WCL and 20KgP comparisons).
Next, a list of proteins with conditional fractional enrichment identified by Method II was generated. Theoretically, each protein with log2 20KgP/WCL greater than zero is enriched in the 20KgP fraction; however, such a direct assignment could generate a high false positive rate because of experimental noise. Therefore, a threshold ratio of fractional enrichment was established by matching the observed number of 20KgP enriched proteins to an expected number generated from known protein localization data. To calculate expected enrichment, we compared the number of proteins annotated with one or more organelles known to be enriched in the 20KgP (GO Slim terms “endoplasmic reticulum,” “peroxisome,” “mitochondrion,” “vacuole,” “plasma membrane,” and “golgi”) and compared it to the total number of proteins that have been localized to any intracellular compartment (proteins annotated with GO Slim term “cellular component,” which represents proteins of the WCL). By this method, the estimated fraction of 20KgP enriched proteins is 42%. This estimate was used to establish a log2 20KgP/WCL threshold of 0.5 (corresponding to ∼1.4 fold enrichment in 20KgP versus WCL), which resulted in 40% of proteins from glucose-grown cells having 20KgP enrichment.
Subclassification of Proteins with Conditional Fraction Enrichment Profiles
To obtain biological insight from the fractionation analyses, proteins were clustered based on their dynamic differential enrichment patterns identified using both Methods I and II as described below and shown in Fig. 4 and supplemental Fig. S4. This analysis enabled generation of hypotheses about conditional changes in protein abundance and compartmentalization, which may reflect mechanisms of the regulation of biological processes within the cell.
Fig. 4.

Classification of proteins with conditional relocalization using both Method I and Method II. Proteins with significant conditional enrichment identified by Method I were clustered according to their enrichment profiles generated by Method I (Roman numerals in column 1), and then subclustered according to their enrichment profiles generated using Method II (subclasses a, b, and c in column 7). The three largest clusters are shown (all seven clusters are in supplemental Fig. S4). The illustrations show cellular distribution profiles consistent with the fractional enrichment profiles for the proteins of each subcluster. Increased color shading represents increased protein abundance relative to other compartments/conditions in the same row. Equal shading of organelles and cytosol indicates that organelles have equal or lesser protein than cytosol. Clusters highlighted in yellow are those suggested by the data to contain proteins having conditional subcellular redistribution. Note: “no significant change” indicates proteins whose abundance changes were not statistically significant as well as proteins that were not detected under either condition in the given fraction.
First, the 1489 proteins with fractional enrichment identified by Method I were clustered into seven groups based on their significant conditional fractionation changes determined by Method I. The three largest groups are shown in Fig. 4 (columns 1–3) and all seven groups are shown in supplemental Fig. S4 (columns 1–3). Next, the proteins in each of the seven groups were further subcategorized based on conditional organellar enrichment determined using Method II (columns 4–5). This two-step clustering approach provided an objective method for unbiased classification of proteins that showed similar properties of relative abundance and redistribution (illustrated in columns 8–9) and enabled characterization of the proteins of each class by statistical analysis of overrepresented GO annotations in each cluster (columns 10–11). The comprehensive lists of proteins in each cluster, along with their assigned GO annotations, are presented in supplemental Table S4.
Most groups had significant conditional protein abundance changes in the WCL fraction suggesting regulation of cellular protein abundance (supplemental Fig. 4; column 2). However, three groups (II, V, and VII) were differentially enriched between organellar fractions (20KgPs) of the two conditions (column 3) and not WCL fractions, suggesting that these proteins conditionally relocalized. Cellular abundance changes were not detected for these proteins, either because they had little or no abundance changes, or their absolute levels in WCL fractions were too low for accurate quantification. The majority of proteins clustered into one of three groups (groups I, II, and V), suggesting preferred mechanisms to mediate functional changes in response to the stimulus. These groups are shown in Fig. 4 and discussed below.
Group I Proteins Increase in Abundance in Response to Oleate
Group I proteins significantly increased in abundance during fatty acid exposure in both WCL and 20KgP fractions (Fig. 4; columns 2–3), suggesting increased cellular and organellar abundance. The largest subclass of this group (I-a) contained 69 proteins that were enriched in the 20KgP in both conditions (columns 4–5) suggesting localization to organelles. Ninety-seven percent of the proteins in this subclass are known to be localized to mitochondria or peroxisomes, two organelles with well characterized roles in fatty acid metabolism (40, 41). The second largest subclass (I-b) contained 37 proteins that were enriched in the 20KgP fraction only after growth in the presence of oleic acid (Fig. 4; columns 4–5). These data suggest that the proteins have increased abundance and organellar enrichment in response to fatty acid-containing medium. Lack of detectable enrichment in organelles in the presence of glucose might indicate that the proteins are of very low abundance under this condition. In support of this, of the 37 proteins, 23 are known components of either the peroxisomal matrix (including Pox1, Pot1, Mls1, and Faa2) or mitochondria (Cox2, Acs1, and Cta1). The majority of peroxisomal matrix proteins are metabolic enzymes that are known to be exclusively peroxisomal and of very low abundance in the presence of glucose, and highly induced in the presence of oleic acid (3, 4, 42). Group I-c contained proteins that were not enriched in the 20KgP under either condition (columns 4–5), suggesting increased cytoplasmic abundance in the presence of oleic acid, but not redistribution. Even though this set of proteins had no significant enrichment for any of the GO cellular components or functional category, 27 of 31 proteins are known to be cytosolic or cytoplasmic rather than annotated as specific organellar localizations (see supplemental Table S4).
Group II Proteins Associate with Organelles in Response to Oleate
Group II proteins had no significant increase in cellular protein abundance in response to fatty acid-containing medium, but did show significant conditional enrichment in the 20KgP organellar fraction (Fig. 4, columns 2 and 3). These data suggest that the proteins relocalize to organelles in response to fatty acid-containing medium. However, the group may also contain organellar proteins that have conditional cellular abundance changes that were subtle and not detected by this approach.
About half of the proteins in the group were enriched in the 20KgP under both glucose and oleic acid conditions (subclass II-a; columns 4 and 5), but were more abundant in organelles after fatty acid exposure (column 3). Consistent with these data, this subclass contained many peroxisomal proteins (including Pex1, Pex3, Pex6, Pex14, Pex25, and Pex29). The second largest subclass (II-b) consisted of proteins that were enriched in the 20KgP after growth in oleic acid, but not glucose, indicating conditional protein movement from cytosol (or a compartment not enriched in 20KgP) to subcellular compartments including peroxisomes, ER, mitochondria, and plasma membrane. Supporting this idea, this subclass included Gpd1 and Aat2, both of which are known to be redistributed from cytosol to peroxisomes in response to fatty acid exposure (4, 43). In addition, 50 candidates in group II-b are peroxisomal or mitochondrial membrane proteins (including Pex15, Pxa1, Pxa2, Cox1, and Atp18) as annotated on the SGD website (www.yeastgenome.org). The final subclass in group II (II-c) represented proteins that were not enriched in the 20KgP from either condition (columns 4–5), indicating subtle redistribution from cytosol (or other organelles not enriched in 20KgP) to subcellular organelles (enriched in 20KgP).
It should also be noted that although group II is expected to contain proteins that shift their distribution but do not change in abundance in response to fatty acids, the group is also expected to contain some proteins that have conditional abundance changes, which were not detected either because the change was insignificant, or because the overall abundance of the proteins is low leading to sampling error in the MS approach.
Comparison of Groups I and II
Fatty acid metabolism requires two types of proteins, metabolic enzymes, and proteins involved in peroxisome biogenesis. Generally, metabolic enzymes are dramatically transcriptionally responsive to fatty acid exposure, whereas peroxins are not (25). Groups I and II appear to differently represent these two classes of proteins. Groups I and II both contain members that have measured transcriptional induction in oleate versus glucose (as reported in (12)); however, these transcriptionally induced genes are more highly represented in group I than II (55% versus 23% of total, respectively), and have a higher level of induction in group I than II (6.1 versus 4.1 fold, respectively). Additionally, of the members that transcriptionally responded to oleate, group II is more highly enriched for those of low abundance; 44.3% of group II proteins were either not detectable or had extremely low signal in WCL fractions in glucose medium by quantitative Western blotting, whereas 23.7% of group I proteins had this annotation (32). Consistent with these data, groups I and II differently represent peroxisomes; group I is significantly enriched for metabolic enzymes localized to peroxisomes (HGD p value < 1 × 10−30), which tend to be highly induced in the presence of fatty acids; whereas group II is significantly enriched for these proteins (HGD p value of 2.5 × 10−3, but also for peroxins (HGD p value of 9.3 × 10−7), which tend to be of low abundance and not dramatically induced by fatty acids (25).
Group V Proteins are Predicted to Dissociate from Organelles in Response to Oleate
Group V represents proteins that do not have changed levels in response to the stimulus, but are more highly enriched in organelles in the presence of glucose-containing medium (Fig. 4, columns 2–3), suggesting dissociation from organelles in response to oleate. The largest subclass of the group, V-c, contained proteins that were not enriched in the 20KgP under either condition (columns 4–5). The second largest subclass, V-b, included proteins that were conditionally enriched in the 20KgP fraction exclusively in glucose-grown cells. These fractionation patterns suggest that proteins of these subclasses redistribute from organelles to the cytosol (or another subcellular compartment not represented in the 20KgP) in response to fatty acid-containing medium. The final subclass, V-a, included proteins that were enriched in the 20KgP under both conditions, suggesting partial relocalization of these proteins from organelles to the cytosol during the response. The GO annotation enrichments for the three subclasses appear to be consistent with the measured predominant localizations of the proteins; subcategory V-c was enriched for nonpelletable components of cytoplasm and the nucleus (including ribosomes and cytoskeleton and nucleolus), whereas subcategory V-a was enriched for components comprising the 20KgP (including ER and Golgi).
Group IV, the fourth largest group, contained proteins that were significantly depleted during exposure to oleic acid from both WCL and 20KgP fractions, suggestive of a significant decrease in protein abundance (supplemental Fig. S4). The remaining three groups contained relatively small numbers of proteins, suggesting either that they are false positives, or that they represent mechanisms of control that are not widely used for this response.
Investigation of Candidates Using Fluorescent Microscopy
We randomly selected 15 candidates from groups I-b and II for abundance and localization analysis. Strains with genomically integrated gene fusions encoding GFP tagged versions of the proteins were grown in the presence of glucose or oleic acid and analyzed by fluorescence microscopy. Images of 14 candidates are shown in Fig. 5, and the 15th candidate, Fat3, is discussed later. Consistent with the classification analysis (Fig. 4), all three tagged proteins from group I tested (Qcr6, Rdl1, and Ams1) appeared to show increased levels in oleate- versus glucose-grown cells. In addition, some group II proteins also appeared to have similarly increased levels; as discussed earlier, this was expected because some changes in expression are anticipated to be below the level of sensitivity of the analysis. Also consistent with the classification analysis, most of the tagged proteins tested (all except Qcr6, Zta1 and Sds24) showed increased abundance in organelles in response to oleate.
Fig. 5.
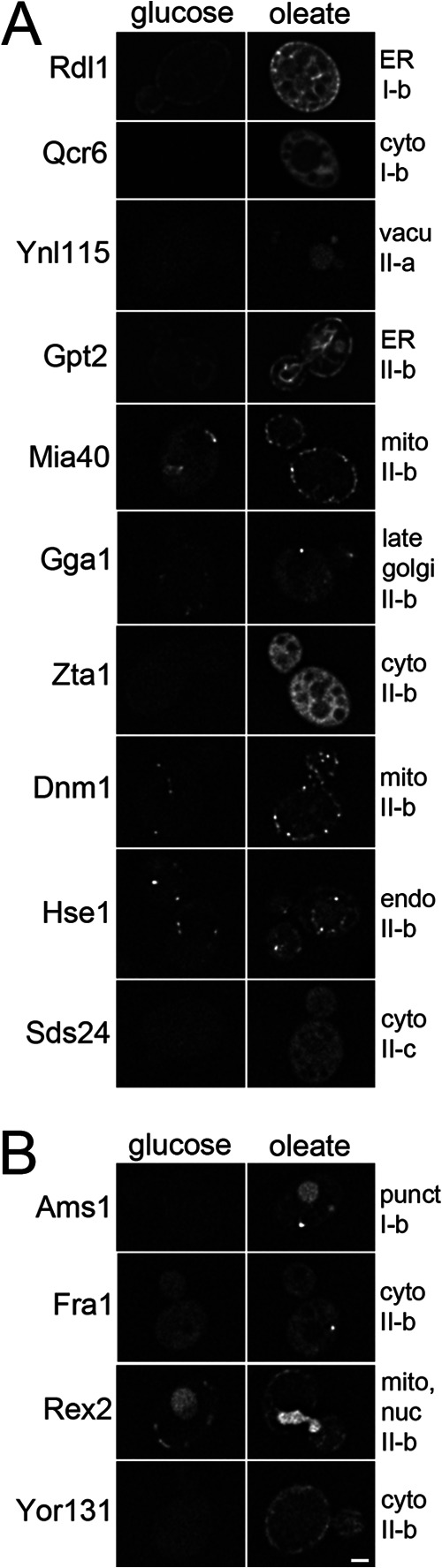
Candidates randomly selected from groups I and II show increased abundance and dynamic redistribution in response to fatty acid exposure. GFP tagged versions of protein candidates were visualized after growth in the presence of glucose or oleate for 24 h. Localization data for GFP fusion proteins in glucose-grown cells from (44) are indicated at the right above the group numbers (mito, mitochondria; cyto, cytoplasm; endo, endosomes; vacu, vacuoles; punct, punctate structures; nuc, nucleus). Panels A and B show candidates that appear to increase in abundance (with our without redistribution) and to redistribute in response to oleate, respectively. Bar, 1 μm.
The data were also analyzed for evidence of conditional redistribution of the proteins. The proteins in Fig. 5A were considered to show no evidence of redistribution in response to oleate because either the pattern of fluorescence in oleate was similar to that in glucose, or, in cases where fluorescence was not detectable in the glucose condition (Qcr6, Sds24, Zta1, Ynl115), because the pattern of fluorescence observed in oleate was similar to that observed in glucose-grown cells by a previously published study (44). The proteins in Fig. 5B showed evidence of conditional redistribution. We did not detect Ams1-GFP in glucose-grown cells, but it has been localized to small punctate structures in a previous study (44). After growth in oleate, we observed Ams1 in small punctate structures and also in large structures resembling vacuoles suggesting redistribution to this organelle in the presence of oleate, consistent with its known function in vacuoles (45). Rex2, a member of group II-b is an RNA exonuclease that has primarily mitochondrial and nuclear localization (44, 46). Interestingly, its pattern of fluorescence was consistent with these localizations under both conditions, but the nuclear signal appeared to be significantly more intense in cells after growth in oleic acid, suggesting condition-specific redistribution. Identification of this candidate by our fractionation analysis is surprising as nuclei are depleted from the 20KgP fraction; however, some contamination is known to be present and may be sufficient for conditional abundance analysis. Fra1p appeared to conditionally redistribute from the cytoplasm to small punctate structures of low abundance (∼1 per cell), whereas Yor131 appeared to redistribute from the cytoplasm to structures at the cell periphery, which appear mitochondrial (based on comparison to the pattern of known mitochondrial proteins, Rex2, Dnm1, and Mia40, in the figure). Our observation of mitochondria at the cell periphery after growth in oleate for 24 h might be because of crowding by lipid droplets in the cell.
For some group II candidates, redistribution was not detected. This could be because they were falsely identified by the fraction enrichment analysis, suggesting that they belong in group I, which is consistent with their overall increased abundance in oleate observed by microscopy. A second possibility is that the GFP tag interfered with normal protein distribution in some cases by masking targeting signals. Alternatively, the first pass single-tag fluorescence microscopy approach may not be sufficiently sensitive to detect redistribution; supporting this idea, the previously characterized redistribution of Dnm1 from mitochondria to peroxisomes in the presence of oleate (47), was not detected.
Integration of Global Fraction Enrichment Data With Other Data Sets Reveals Dynamic Response of Specialized Plasma Membrane Domains to Oleate Exposure
As discussed previously, exposure of cells to fatty acids induces both metabolic and cell structural changes, which appear to be represented primarily by groups I and II, respectively. To understand how peroxisome biogenesis interplays with other subcellular structures during the response, we selected candidates of high priority for further analysis. We did this by integrating the results of other studies including: (1) global data sets of yeast deletion strains specifically necessary for growth on each of two fatty acids (25), (2) a global data set of proteins associated with peroxisomal membranes during fatty acid exposure (3) and, a global data set of genes that physically interact with the known principal transcriptional regulators of the response to fatty acids (26) (see Experimental Methods).
The 347 candidates of group II were filtered to include only those also identified by at least two of the four assays listed above, generating a short list of 23 proteins including 12 known peroxisomal proteins (supplemental Table S4). Remarkably, this list also contained Nce102 and Ynl194, two integral membrane components of specialized plasma membrane domains made up of integral membrane proteins (MCC portion) as well as a complex of peripherally associated proteins (eisosome portion) (48). As they have only recently been identified, the biological functions of these structures are not well characterized. Generally, they are known to be involved in membrane organization and function. Specifically, a role in endocytosis has been proposed, but it is unclear whether they are active sites of an endocytic pathway (49), or if they are protective regions that shield PM proteins from internalization (50). Various MCC components have been implicated in coping with nutrient availability and environmental stresses, including osmotic and nitrogen stress (48).
As only 28 proteins have been localized to MCC/eisosomes (and only nine of which are in the MCC domain) (supplemental Table S4) (48), the presence of two MCC components in the group II short list of 23 proteins is a significant over-representation (HGD p value < 0.008). These two proteins, Nce102 and Ynl194, are tetraspanners of the Nce102 and Sur7 families of MCCs, respectively. Interestingly, an uncharacterized candidate in the short list, Ykl187, which we have renamed Fat3 (FATty acid transporter 3), is also predicted to be a member of the Sur7 protein family (pfam 00687) by PSI-BLAST (blast.ncbi.nlm.nih.gov) (E-value = 7.56e−3) and is predicted to be an integral membrane protein, suggesting that it may also be an MCC component. In addition to this, the unfiltered list of 346 candidates in group II includes Lsp1 and Pil1, the two primary components of eisosomes (48).
To investigate the involvement of MCC/eisosomes in the response to fatty acid exposure, we tracked the abundance and subcellular distributions of the three MCC-related proteins in the short list (Nce102, Ynl194 and Fat3) under different growth conditions. Nce102-GFP distribution was analyzed in the presence of glucose, oleate, or glycerol (nonfermentative carbon source conferring a yeast growth rate similar to that of oleate) (Fig. 6). In the presence of each carbon source, Nce102-GFP localized to punctate cortical domains as expected; however, the patches appeared more numerous and diffuse after growth in glycerol, and even more so after growth in oleate. The same dynamics were observed using a different MCC marker, Sur7-mCherry, and quantitative analysis of the images demonstrated a significant increase in MCC abundance after growth in glycerol versus glucose and after growth in oleate versus glycerol (t test p values of 3.9 × 10−12 and 4.0 × 10−3, respectively). These data suggest MCCs (and associated eisosomes) change in abundance and compactness in response to environmental cues such as available carbon sources, and that there is a specific response to fatty acids above a general response to nonfermentative growth.
Fig. 6.
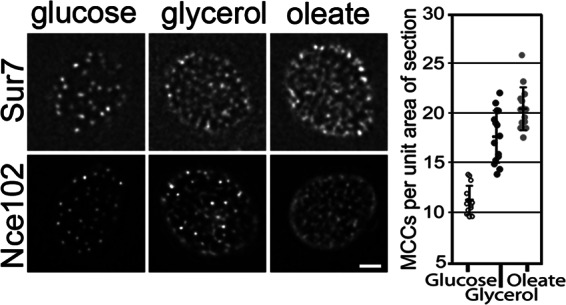
MCCs significantly increase in abundance in response to fatty acid exposure. MCCs, membrane domains associated with eisosomes, were visualized with Sur7-mCherry or Nce102-GFP in cells under three growth conditions. In comparison to the glucose condition, MCCs appear more abundant and diffuse in the presence of the nonfermentative carbon source, glycerol, and even more so in the presence of oleic acid. For each condition, both cortical and mid-sections of the cell are shown. Bar, 1 μm. The graph at the left shows the results of quantitative analysis of MCCs visualized with Sur7-mCherry in cortical sections. These data are in agreement with the qualitative analysis. The differences in the number of MCCs between the samples are statistically significant with Student's t test p values of 3.9 × 10−12 (glucose versus glycerol) and 4.0 × 10−3 (oleate versus glycerol).
Ynl194p Partially Colocalizes with MCCs in the Presence of Fatty Acids
It has been shown that Ynl194p is not readily detectable in glucose-grown cells and partially colocalizes with Sur7-labeled MCC domains associated with eisosomes after growth in high salt (51). To determine the effects of fatty acids on Ynl194-GFP distribution, it was localized in Sur7-mCherry labeled cells after growth under three different conditions (Fig. 7). As expected, it was not detectable in glucose grown cells. Interestingly, it was increasingly more abundant after growth in glycerol and oleate, respectively, and partially colocalized with Sur7-mCherry under both conditions. Faint signals for both markers were also observed in structures resembling vacuoles. These data suggest that the dynamic association of Ynl194p to MCCs is not only responsive to salt stress, but also fatty acid exposure.
Fig. 7.

Ynl194 (group II) increases in abundance in response to fatty acid exposure and partially colocalizes with MCCs under this condition. Localization of Ynl194-GFP was compared with that of Sur7-mCherry under three growth conditions. Ynl194-GFP is not detectable in glucose-grown cells, and is increasingly more abundant in glycerol- and oleate-grown cells. It localizes primarily to cortical patches, which partially colocalize with the MCC marker, Sur7, and also weakly to subcellular structures resembling vacuoles. For each condition, both cortical and mid-sections of the cell are shown. Bar, 1 μm. Inset images are enlarged three times.
Fat3 Localizes to Membrane Domains that Appear Distinct From Eisosome-associated Plasma Membrane Domains
Fat3-GFP was localized in Sur7-mCherry labeled cells after growth in medium containing glucose, glycerol or oleate (Fig. 8). As with Ynl194-GFP, the fusion was not detectable in glucose grown cells, and became increasingly more abundant in glycerol- and oleate-grown cells. Although a portion of the fusion protein was localized to cortical patches, the domains appeared more dissipated than MCC domains, and colocalization with Sur7-mCherry was not apparent, suggesting that it is localized to plasma membrane domains distinct from eisosomes. As with Ynl194-GFP, Fat3-GFP was also observed in structures resembling vacuoles.
Fig. 8.

Levels of Fat3 (group II) increase in response to fatty acid exposure and localize to diffuse cortical patches distinct from MCC/eisosomes. Localization of Fat3-GFP was compared with that of Sur7-mCherry under three growth conditions. Fat3-GFP is not detectable in glucose grown cells, but is increasingly more abundant in glycerol- and oleate-grown cells, respectively. It localizes to subcellular structures resembling vacuoles and to cortical patches; however, the cortical staining is more diffuse than the sur7-mCherry signal and does not appear to colocalize with Sur7. For each condition, both cortical and mid-sections of the cell are shown. Bar, 1 μm.
We have previously identified FAT3 as a gene necessary for growth in the presence of fatty acids (25). These data along with the cortical localization of the protein suggest a role for Fat3p in fatty acid uptake. This role was investigated using a previously developed fatty acid import assay (52). The assay is based on the fact that in the presence of cerulenin, a fatty acid synthesis inhibitor, yeast are auxotrophic for fatty acids. Therefore, wild-type cells are able to grow in the presence of cerulenin and fatty acids; however, cells that are deficient in fatty acid uptake have a growth defect under this condition. A FAT3 gene deletion strain was analyzed with various other strains for growth in the presence of cerulenin, or both cerulenin and myristic acid (a C14:0 fatty acid) (Fig. 9). As expected, ΔFAT1, a strain with a known deficiency of fatty acid uptake (53), had a growth defect compared with wild-type cells in the presence of cerulenin and myristic acid. Interestingly, ΔFAT3 but not ΔYNL194C also had a growth defect under this condition suggesting a role for Fat3 in fatty acid uptake into the cell. Because our previous global chromatin immunoprecipitation analysis predicted that the fatty acid responsive transcription factor Oaf1 regulates FAT3, both in the presence of glucose and in the presence of fatty acids (26), ΔOAF1 was also tested in the assay. It appeared to grow even better than the wild type strain in the presence of cerulenin and fatty acid, suggesting it has augmented fatty acid availability and/or uptake. One possible explanation for this observation is that Oaf1p acts as a negative regulator of FAT3 under the condition tested here. Supporting this idea, we have previously shown that Oaf1p can be an activator or repressor depending on the binding context, and that Oaf1p binds upstream of FAT3 in a repressive context (with Adr1 and Oaf3, but not Pip2) in the presence of glucose (26), which is present in the assay medium. Another possibility is that the phenotype is due to the known role of Oaf1 as a positive regulator of fatty acid β-oxidation; deficient β-oxidation in ΔOAF1 might result in increased cellular fatty acid levels, which improves cell growth or viability during inhibition of fatty acid synthesis in the assay.
Fig. 9.
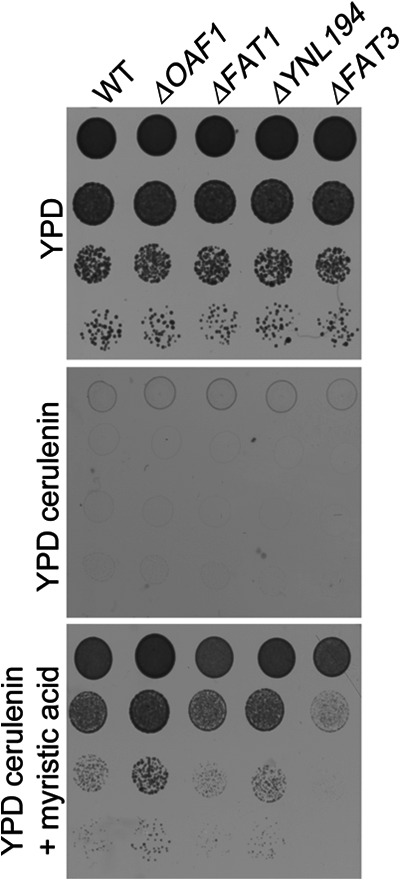
Cerulenin toxicity assay suggests role for Fat3 in fatty acid uptake. Approximately 1 × 105 yeast cells of each strain were spotted along with 10-fold serial dilutions onto the plates indicated and grown at 30 °C for 24 h. Cerulenin is an inhibitor of fatty acid synthesis and is toxic to yeast in the absence of exogenously added fatty acids. In the presence of both cerulenin and the fatty acid, myristate, growth is restored to wild type cells that can import fatty acids. Like ΔFAT1, a strain with a known fatty acid import deficiency, ΔFAT3 has a growth defects under this condition suggesting that it has a role in fatty acid uptake. A growth defect was not detected for ΔYNL194, but interestingly, ΔOAF1, a strain with a deletion of a transcriptional regulator of fatty acid metabolism, showed augmented growth.
DISCUSSION
Dynamic changes in protein levels and localization are important responses to a variety of physiological stimuli. The subcellular redistribution of proteins plays an essential role in a variety of subcellular processes by controlling access of proteins to all types of molecular interaction partners. Here, we introduce and validate an approach for quantifying global conditional changes in protein abundance and compartmentalization. First, we performed quantitative proteomic analysis of subcellular fractions from cells grown under different conditions. Next, various fraction enrichment ratios were calculated and used to cluster proteins with similar dynamics. This method identified seven clusters representing two classes of responsive proteins: (1) those with measured cellular abundance changes (i.e. proteins with differential enrichment between conditions at the level of whole cell lysate) and (2) those with measured redistribution (i.e. those without cellular abundance changes, but with differential enrichment between the same cell fractions of different conditions).
Of particular interest to us were clusters I and II, containing proteins that appeared to have increased levels and redistribution to organelles, respectively, in response to the stimulus. GO annotation enrichment analysis revealed general cellular dynamics of proteins in these clusters. Cellular component terms mitochondria and peroxisomes were significantly enriched in both groups I and II (Fig. 4, column 10). However, biological process term enrichments were quite different between the groups; mitochondrion organization and peroxisome organization were specifically enriched in group II, but not group I, whereas generation of precursor metabolites and energy and other metabolic processes were specifically enriched in group I, but not group II. These data suggest that although proteins involved in biogenesis/organization of organelles responded to the stimulus (by relocalization), their abundance changes were generally more subtle than the metabolic enzymes involved in the response. These data are consistent with our previous analysis showing a bias of transcriptional regulation toward metabolic enzymes in response to fatty acid exposure (25) and with recent data showing that this bias is generally applicable to many cellular responses (54).
As mentioned above, cluster II was enriched for organelle organization and proteins of this cluster are predicted to redistribute into organelles (including mitochondria, ER, peroxisomes, and plasma membranes) (Fig. 4) in response to the stimulus. What specific aspects of organization does this cluster represent? Of all proteins in cluster II, those in subcluster II-b, which are only enriched in the 20KgP fraction in fatty acid-containing medium, appear to have the most dramatic redistribution into organelles (Fig. 4). Importantly, this cluster contains Gpd1 and Aat2, which have previously been shown to move from cytosol to peroxisomes during this response (4, 43), thus validating our approach. These proteins are involved in coping with stress and nitrogen metabolism, respectively, but the biology underlying their fatty acid-induced redistribution is not known. For proteins in subcluster II-a (those enriched in organelles in both conditions, with higher abundance in fatty acid-induced organelles), redistribution could reflect dynamics of organelle biogenesis. This cluster is enriched for peroxins involved in peroxisome organization (Fig. 4). During de novo peroxisome biogenesis, most peroxins traffic through the ER into preperoxisomal vesicles, which are not functional until they undergo heterotypic fusion with other preperoxisomal vesicles (55). As these vesicles are not pelletable at 20,000 g (and not in the 20KgP fraction), the measured relocalization to the 20KgP fraction could reflect a shift in distribution of peroxins from preperoxisomal vesicles to mature peroxiosmes in the presence of fatty acids. This is consistent with the fact that peroxisomes are dispensable in the presence of glucose, but are necessary for growth in the induction medium where fatty acids are the only carbon source. To further characterize these other hypotheses, studies involving higher resolution localization will be necessary.
Remarkably, cluster II contained a total of 346 proteins (Fig. 4). These, along with the ∼900 proteins in clusters V and VII that also have redistribution potential, represent a large portion of the proteome. Do all of these proteins redistribute in response to the stimulus? We suspect that some of the candidates for relocalization are falsely categorized and do in fact change in abundance in response to the stimulus. This is because the quantitative accuracy of shotgun proteomics is dependent on sampling depth (56). Therefore, we expected greater power to detect significant protein abundance changes in 20KgP fractions, which are less complex than WCLs, especially for proteins with low copy numbers. This error is expected to be low as 76% of proteins detected in WCLs (762 proteins) had log2 spectral count sums > 3, the threshold above which noise rapidly diminishes (see Fig. 3, graph A). Nonetheless, some proteins are likely to have changes in abundance at the WCL level that were undetected, which explains why some proteins of group II appeared to show redistribution (Fig. 5B), whereas others appeared to have increased protein levels (Fig. 5A) after oleate exposure when analyzed by fluorescence microscopy. It should be noted, however, that the fluorescence microscopy approach used to validate the candidates can also have inaccuracies. C-terminal tagging of proteins can potentially affect protein turnover, or mask motifs such as the HDEL and SKL signals used for retention and targeting to the ER and peroxisomes, respectively (57, 58, 59); thus, failure to detect redistribution by this fluorescence microscopy method is not necessarily conclusive. For these reasons, further experimentation is required to conclusively categorize each candidate. For example, as this error is related to protein abundance, supporting evidence can be obtained by integrating global gene expression and protein abundance data from the literature.
To validate our approach, a group of candidates were investigated further. Integrative analysis of group II candidates with other data sets in the literature led to the characterization of a role for Fat3 in fatty acid transport into yeast cells. Fat3 was shown here to increase in abundance in patches in the cell cortex in response to oleate exposure (Fig. 8) and to be necessary for uptake of myristic acid in a fatty acid import assay (Fig. 9). These data are supported by the fact that Fat3 is necessary for robust growth on fatty acids as a sole carbon source (25), which we have reconfirmed (data not shown), and that regulatory network analysis implicates fatty acid specific up-regulation of the FAT3 gene by fatty acid responsive factors Adr1p, Oaf1, Oaf3, and Pip2 (26). In addition, an association of Fat3 with peroxisomal membranes of fatty acid-grown cells was suggested by our global quantitative proteomic analysis (3). It will be interesting to investigate multiple dynamic subcellular localizations of Fat3 and/or possible interactions between peroxisomes and plasma membrane domains to follow this up.
The analysis also implicates MCCs, specialized plasma membrane domains associated with eisosomes, in the response to fatty acid exposure. The 23 high-priority candidates of group II include two of nine known MCC proteins (Nce102 and Ynl194), and Fat3, a predicted member of the Sur7 family of MCC components. In addition, the unfiltered list of candidates in group II includes Lsp1 and Pil1, the two primary components of eisosomes (48). Our data suggest that these structures are dynamic and can change in appearance, abundance, and content in response to environmental cues including fatty acid exposure. One MCC component, Nce102, which showed oleate-specific dynamics in our screen, has been implicated in sphingolipid signaling (60), but we do not yet know the roles of MCCs or eisosomes in the response to fatty acid exposure. Our previous global analysis did not detect abnormal peroxisome morphology in any of the corresponding deletion strains (ΔFAT3, ΔYNL194C, ΔNCE102), suggesting that they are not involved in peroxisome biogenesis per se (61). As discussed above, one Sur7-like protein, Fat3, has a role in fatty acid uptake, but it localizes to cortical domains that appear to be distinct from Sur7-containing MCCs. This lack of colocalization of Fat3, along with the conditional and partial colocalization of Ynl194p-containing domains with Sur7, suggests heterogeneity and plasticity of MCC/eisosome structure and function. It will be interesting to investigate pair-wise conditional colocalization of all MCC/eisosome-related proteins to test this hypothesis.
In conclusion, we have combined the established principles of subcellular fractionation with a DIA-MS proteomic method referred to as PAcIFIC that, unlike DDA-MS methods, systematically profiles all available m/z channels to provide large-scale and quantitative assessment of condition-specific protein relocalization. Although here, we analyzed WCL versus an enriched fraction of organelles (including mitochondria, peroxisomes, and ER), this approach can be also extended to measure the redistribution of proteins between other subcellular compartments, as long as an established fractionation procedure is established. This global approach to identify proteins that change distribution or abundance in response to a stimulus is complementary to large-scale assays, based on screening of fluorescently tagged proteins, which have their faults. This method would be particularly useful for comprehensively measuring conditional trafficking of transcriptional regulators to and from the nucleus, which could be integrated with other large-scale data sets to improve our understanding of transcriptional regulatory networks, and for uncovering new relationships between subcellular compartments.
Supplementary Material
Acknowledgments
We thank the Luxembourg Centre for Systems Biomedicine and the University of Luxembourg for support.
Footnotes
* This work was funded by NIH/NIGMS (R01-GM075152, P50-GM076547, U54-RR022220, 5R33CA099139-04, 3R33CA099139-04S1, and 1S10RR023044-01) and supported in part by the University of Washington's Proteomics Resource (UWPR95794).
 This article contains supplemental Figs. S1 to S4 and Tables S1 to S4
This article contains supplemental Figs. S1 to S4 and Tables S1 to S4
1 The abbreviations used are:
- DDA
- data-dependent acquisition
- DIA
- data-independent acquisition
- MCC
- membrane compartment occupied by Can1
- PAcIFIC
- Precursor Acquisition Independent From Ion Count
- PNS
- post nuclear supernatant
- SGD
- Saccharomyces genome database
- SpC
- spectral counts
- WCL
- whole cell lysate.
REFERENCES
- 1. Yates J. R., 3rd, Gilchrist A., Howell K. E., Bergeron J. J. (2005) Proteomics of organelles and large cellular structures. Nat. Rev. Mol. Cell Biol. 6, 702–714 [DOI] [PubMed] [Google Scholar]
- 2. Dudley A. M., Janse D. M., Tanay A., Shamir R., Church G. M. (2005) A global view of pleiotropy and phenotypically derived gene function in yeast. Mol. Syst. Biology 1, 2005.0001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marelli M., Smith J. J., Jung S., Yi E., Nesvizhskii A. I., Christmas R. H., Saleem R. A., Tam Y. Y., Fagarasanu A., Goodlett D. R., Aebersold R., Rachubinski R. A., Aitchison J. D. (2004) Quantitative mass spectrometry reveals a role for the GTPase Rho1p in actin organization on the peroxisome membrane. J. Cell Biol. 167, 1099–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jung S., Marelli M., Rachubinski R. A., Goodlett D. R., Aitchison J. D. (2010) Dynamic changes in the subcellular distribution of Gpd1p in response to cell stress. J. Biol. Chem. 285, 6739–6749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 6. Liu H., Sadygov R. G., Yates J. R., 3rd (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201 [DOI] [PubMed] [Google Scholar]
- 7. Panchaud A., Scherl A., Shaffer S. A., von Haller P. D., Kulasekara H. D., Miller S. I., Goodlett D. R. (2009) Precursor acquisition independent from ion count: how to dive deeper into the proteomics ocean. Anal. Chem. 81, 6481–6488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Panchaud A., Jung S., Shaffer S. A., Aitchison J. D., Goodlett D. R. (2011) Faster, quantitative, and accurate precursor acquisition independent from ion count. Anal. Chem. 83, 2250–2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Entian K. D., Barnett J. A. (1992) Regulation of sugar utilization by Saccharomyces cerevisiae. Trends Biochem. Sci. 17, 506–510 [DOI] [PubMed] [Google Scholar]
- 10. Schüller H. J. (2003) Transcriptional control of nonfermentative metabolism in the yeast Saccharomyces cerevisiae. Current Gen. 43, 139–160 [DOI] [PubMed] [Google Scholar]
- 11. Trotter P. J. (2001) The genetics of fatty acid metabolism in Saccharomyces cerevisiae. Annu. Rev. Nutr. 21, 97–119 [DOI] [PubMed] [Google Scholar]
- 12. Smith J. J., Marelli M., Christmas R. H., Vizeacoumar F. J., Dilworth D. J., Ideker T., Galitski T., Dimitrov K., Rachubinski R. A., Aitchison J. D. (2002) Transcriptome profiling to identify genes involved in peroxisome assembly and function. J. Cell Biology 158, 259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shaner N. C., Campbell R. E., Steinbach P. A., Giepmans B. N., Palmer A. E., Tsien R. Y. (2004) Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567–1572 [DOI] [PubMed] [Google Scholar]
- 14. Titorenko V. I., Smith J. J., Szilard R. K., Rachubinski R. A. (1998) Pex20p of the yeast Yarrowia lipolytica is required for the oligomerization of thiolase in the cytosol and for its targeting to the peroxisome. J. Cell Biol. 142, 403–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Titorenko V. I., Chan H., Rachubinski R. A. (2000) Fusion of small peroxisomal vesicles in vitro reconstructs an early step in the in vivo multistep peroxisome assembly pathway of Yarrowia lipolytica. J. Cell Biol. 148, 29–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoopmann M. R., Finney G. L., MacCoss M. J. (2007) High-speed data reduction, feature detection, and MS/MS spectrum quality assessment of shotgun proteomics data sets using high-resolution mass spectrometry. Anal. Chem. 79, 5620–5632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eng J. K., McCormack A. L., Yates J. R., 3rd (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 18. Deutsch E. W., Shteynberg D., Lam H., Sun Z., Eng J. K., Carapito C., von Haller P. D., Tasman N., Mendoza L., Farrah T., Aebersold R. (2010) Trans-Proteomic Pipeline supports and improves analysis of electron transfer dissociation data sets. Proteomics 10, 1190–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sokal R. R., Rohlf F. J. (1995) The principles and practice of statistics in biological research Biometry, W. H. Freeman, New York: 1995 [Google Scholar]
- 20. Becker R. A., Chambers J. M., Wilks A. R. (1988) The New S Language, Wadsworth & Brooks/Cole [Google Scholar]
- 21. Cleveland W. (1981) LOWESS: A program for smoothing scatterplots by robust locally weighted regression. Am. Stat. 35, 54 [Google Scholar]
- 22. Cleveland W. S. (1981) LOWESS: A program for smoothing scatterplots by robust locally weighted regression. Am. Stat. 35, 54 [Google Scholar]
- 23. Ihaka R., Gentleman R. (1996) R: A lanugage for data analysis and graphics. J. Computational Graph. Stati. 5, 299–314 [Google Scholar]
- 24. Begley T. J., Rosenbach A. S., Ideker T., Samson L. D. (2004) Hot spots for modulating toxicity identified by genomic phenotyping and localization mapping. Mol. Cell 16, 117–125 [DOI] [PubMed] [Google Scholar]
- 25. Smith J. J., Sydorskyy Y., Marelli M., Hwang D., Bolouri H., Rachubinski R. A., Aitchison J. D. (2006) Expression and functional profiling reveal distinct gene classes involved in fatty acid metabolism. Mol. Syst. Biol. 2, 2006.0009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith J. J., Ramsey S. A., Marelli M., Marzolf B., Hwang D., Saleem R. A., Rachubinski R. A., Aitchison J. D. (2007) Transcriptional responses to fatty acid are coordinated by combinatorial control. Mol. Syst. Biol. 3, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carpenter A. E., Jones T. R., Lamprecht M. R., Clarke C., Kang I. H., Friman O., Guertin D. A., Chang J. H., Lindquist R. A., Moffat J., Golland P., Sabatini D. M. (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aitchison J. D., Rachubinski R. A. (1990) In vivo import of Candida tropicalis hydratase-dehydrogenase-epimerase into peroxisomes of Candida albicans. Current Gen. 17, 481–486 [DOI] [PubMed] [Google Scholar]
- 29. Eitzen G. A., Aitchison J. D., Szilard R. K., Veenhuis M., Nuttley W. M., Rachubinski R. A. (1995) The Yarrowia lipolytica gene PAY2 encodes a 42-kDa peroxisomal integral membrane protein essential for matrix protein import and peroxisome enlargement but not for peroxisome membrane proliferation. J. Biol. Chem. 270, 1429–1436 [DOI] [PubMed] [Google Scholar]
- 30. Yi E. C., Marelli M., Lee H., Purvine S. O., Aebersold R., Aitchison J. D., Goodlett D. R. (2002) Approaching complete peroxisome characterization by gas-phase fractionation. Electrophoresis 23, 3205–3216 [DOI] [PubMed] [Google Scholar]
- 31. Old W. M., Meyer-Arendt K., Aveline-Wolf L., Pierce K. G., Mendoza A., Sevinsky J. R., Resing K. A., Ahn N. G. (2005) Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 4, 1487–1502 [DOI] [PubMed] [Google Scholar]
- 32. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 33. Au C. E., Bell A. W., Gilchrist A., Hiding J., Nilsson T., Bergeron J. J. (2007) Organellar proteomics to create the cell map. Curr. Opin. Cell Biol. 19, 376–385 [DOI] [PubMed] [Google Scholar]
- 34. Delahunty C. M., Yates J. R., 3rd (2007) MudPIT: multidimensional protein identification technology. BioTechniques 43, 563, 565,, 567 passim [PubMed] [Google Scholar]
- 35. Dilworth D. J., Saleem R. A., Rogers R. S., Mirzaei H., Boyle J., Aitchison J. D. (2010) QTIPS: a novel method of unsupervised determination of isotopic amino acid distribution in SILAC experiments. J. Am. Soc. Mass Spectrom. 21, 1417–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hengel S., Murray E., Langdon S., Hayward L., O'Donoghue J., Panchaud A., Hupp T., Goodlett D. R. (2011) Data-independent Proteomic Screen Identifies Novel Tamoxifen Agonist that Mediates Drug Resistance. J. Proteome Res. in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parker S. J., Halligan B. D., Greene A. S. (2010) Quantitative analysis of SILAC data sets using spectral counting. Proteomics 10, 1408–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Titorenko V. I., Rachubinski R. A. (2001) The life cycle of the peroxisome. Nat. Rev. Mol. Cell Biol. 2, 357–368 [DOI] [PubMed] [Google Scholar]
- 39. Zhang B., VerBerkmoes N. C., Langston M. A., Uberbacher E., Hettich R. L., Samatova N. F. (2006) Detecting differential and correlated protein expression in label-free shotgun proteomics. J. Proteome Res. 5, 2909–2918 [DOI] [PubMed] [Google Scholar]
- 40. Epstein C. B., Waddle J. A., Hale W. T., Dave V., Thornton J., Macatee T. L., Garner H. R., Butow R. A. (2001) Genome-wide responses to mitochondrial dysfunction. Mol. Biol. Cell 12, 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hiltunen J. K., Mursula A. M., Rottensteiner H., Wierenga R. K., Kastaniotis A. J., Gurvitz A. (2003) The biochemistry of peroxisomal beta-oxidation in the yeast Saccharomyces cerevisiae. FEMS Microbiol. Rev. 27, 35–64 [DOI] [PubMed] [Google Scholar]
- 42. Henke B., Girzalsky W., Berteaux-Lecellier V., Erdmann R. (1998) IDP3 encodes a peroxisomal NADP-dependent isocitrate dehydrogenase required for the beta-oxidation of unsaturated fatty acids. J. Biol. Chem. 273, 3702–3711 [DOI] [PubMed] [Google Scholar]
- 43. Verleur N., Elgersma Y., Van Roermund C. W., Tabak H. F., Wanders R. J. (1997) Cytosolic aspartate aminotransferase encoded by the AAT2 gene is targeted to the peroxisomes in oleate-grown Saccharomyces cerevisiae. Eur. J. Biochem. 247, 972–980 [DOI] [PubMed] [Google Scholar]
- 44. Huh W. K., Falvo J. V., Gerke L. C., Carroll A. S., Howson R. W., Weissman J. S., O'Shea E. K. (2003) Global analysis of protein localization in budding yeast. Nature 425, 686–691 [DOI] [PubMed] [Google Scholar]
- 45. Yoshihisa T., Anraku Y. (1990) A novel pathway of import of alpha-mannosidase, a marker enzyme of vacuolar membrane, in Saccharomyces cerevisiae. J. Biol. Chem. 265, 22418–22425 [PubMed] [Google Scholar]
- 46. Hanekamp T., Thorsness P. E. (1999) YNT20, a bypass suppressor of yme1 yme2, encodes a putative 3′-5′ exonuclease localized in mitochondria of Saccharomyces cerevisiae. Current Gen. 34, 438–448 [DOI] [PubMed] [Google Scholar]
- 47. Kuravi K., Nagotu S., Krikken A. M., Sjollema K., Deckers M., Erdmann R., Veenhuis M., van der Klei I. J. (2006) Dynamin-related proteins Vps1p and Dnm1p control peroxisome abundance in Saccharomyces cerevisiae. J. Cell Sci. 119, 3994–4001 [DOI] [PubMed] [Google Scholar]
- 48. Douglas L. M., Wang H. X., Li L., Konopka J. B. (2011) Membrane Compartment Occupied by Can1 (MCC) and Eisosome Subdomains of the Fungal Plasma Membrane. Membranes 1, 394–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Walther T. C., Brickner J. H., Aguilar P. S., Bernales S., Pantoja C., Walter P. (2006) Eisosomes mark static sites of endocytosis. Nature 439, 998–1003 [DOI] [PubMed] [Google Scholar]
- 50. Grossmann G., Malinsky J., Stahlschmidt W., Loibl M., Weig-Meckl I., Frommer W. B., Opekarová M., Tanner W. (2008) Plasma membrane microdomains regulate turnover of transport proteins in yeast. J. Cell Biol. 183, 1075–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Young M. E., Karpova T. S., Brügger B., Moschenross D. M., Wang G. K., Schneiter R., Wieland F. T., Cooper J. A. (2002) The Sur7p family defines novel cortical domains in Saccharomyces cerevisiae, affects sphingolipid metabolism, and is involved in sporulation. Mol. Cell. Biol. 22, 927–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Johnson D. R., Knoll L. J., Levin D. E., Gordon J. I. (1994) Saccharomyces cerevisiae contains four fatty acid activation (FAA) genes: an assessment of their role in regulating protein N-myristoylation and cellular lipid metabolism. J. Cell Biol. 127, 751–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Faergeman N. J., DiRusso C. C., Elberger A., Knudsen J., Black P. N. (1997) Disruption of the Saccharomyces cerevisiae homologue to the murine fatty acid transport protein impairs uptake and growth on long-chain fatty acids. J. Biol. Chem. 272, 8531–8538 [DOI] [PubMed] [Google Scholar]
- 54. Yeger-Lotem E., Riva L., Julie Su L., Gitler A. D., Cashikar A. G., King O. D., Auluck P. K., Geddie M. L., Valastyan J. S., Karger D. R., Lindquist S., Fraenkel E. (2009) Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity. Nat. Genet. 41, 316–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. van der Zand A., Gent J., Braakman I., Tabak H. F. (2012) Biochemically distinct vesicles from the endoplasmic reticulum fuse to form peroxisomes. Cell 149, 397–409 [DOI] [PubMed] [Google Scholar]
- 56. Hackett M. (2008) Science, marketing and wishful thinking in quantitative proteomics. Proteomics 8, 4618–4623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pelham H. R. (1989) Control of protein exit from the endoplasmic reticulum. Annu. Rev. Cell Biol. 5, 1–23 [DOI] [PubMed] [Google Scholar]
- 58. Platta H. W., Erdmann R. (2007) Peroxisomal dynamics. Trends Cell Biol. 17, 474–484 [DOI] [PubMed] [Google Scholar]
- 59. Thoms S., Erdmann R. (2006) Peroxisomal matrix protein receptor ubiquitination and recycling. Biochim. Biophys. Acta 1763, 1620–1628 [DOI] [PubMed] [Google Scholar]
- 60. Frohlich F., Moreira K., Aguilar P. S., Hubner N. C., Mann M., Walter P., Walther T. C. (2009) A genome-wide screen for genes affecting eisosomes reveals Nce102 function in sphingolipid signaling. J. Cell Biol. 185, 1227–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Saleem R. A., Long-O'Donnell R., Dilworth D. J., Armstrong A. M., Jamakhandi A. P., Wan Y., Knijnenburg T. A., Niemisto A., Boyle J., Rachubinski R. A., Shmulevich I., Aitchison J. D. (2010) Genome-wide analysis of effectors of peroxisome biogenesis. PloS One 5, e11953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.