

Background: A novel chaperone, cpSRP43, disassembles a family of membrane protein aggregates.
Results: cpSRP43-mediated disaggregation requires two steps, recognition and remodeling, each with distinct molecular requirements.
Conclusion: cpSRP43 uses distinct substrate binding interactions to recognize and then remodel and disrupt the protein aggregate.
Significance: Mechanism of this novel ATP-independent disaggregase guides the understanding of analogous systems and design efforts to target protein aggregates of interest.
Keywords: Enzyme Mechanisms, Kinetics, Molecular Chaperone, Mutagenesis Mechanisms, Protein Aggregation, ATP-independent Disaggregase, Membrane Proteins, Protein Biogenesis, Signal Recognition Particle
Abstract
The ability of molecular chaperones to overcome the misfolding and aggregation of proteins is essential for the maintenance of proper protein homeostasis in all cells. Thus far, the best studied disaggregase systems are the Clp/Hsp100 family of “ATPases associated with various cellular activities” (AAA+) ATPases, which use mechanical forces powered by ATP hydrolysis to remodel protein aggregates. An alternative system to disassemble large protein aggregates is provided by the 38-kDa subunit of the chloroplast signal recognition particle (cpSRP43), which uses binding energy with its substrate proteins to drive disaggregation. The mechanism of this novel chaperone remains unclear. Here, molecular genetics and structure-activity analyses show that the action of cpSRP43 can be dissected into two steps with distinct molecular requirements: (i) initial recognition, during which cpSRP43 binds specifically to a recognition motif displayed on the surface of the aggregate; and (ii) aggregate remodeling, during which highly adaptable binding interactions of cpSRP43 with hydrophobic transmembrane domains of the substrate protein compete with the packing interactions within the aggregate. This establishes a useful framework to understand the molecular mechanism by which binding interactions from a molecular chaperone can be used to overcome protein aggregates in the absence of external energy input from ATP.
Introduction
Protein homeostasis is vital to all cells and requires the proper production, folding, localization, assembly, and degradation of all cellular proteins (1). Crucial to the maintenance of protein homeostasis is an elaborate network of “molecular chaperones” (2–4), which prevents the misfolding and aggregation of proteins by protecting exposed hydrophobic residues in non-native states or unstructured regions and, in some cases, actively promotes protein folding (2). However under stress conditions, the folding capacity of the chaperone network could be exceeded or impaired, leading to protein aggregation. A special set of chaperone machineries, the “disaggregases,” plays a crucial role in rescuing these detrimental processes. The best studied disaggregases belong to the Hsp100 family: Hsp104 in yeast and ClpB in bacteria (5). Both are members of the “ATPases associated with various cellular activities” (AAA+) superfamily that assemble into hexameric ring structures (5). These disaggregases use repetitive ATPase cycles and, in collaboration with their co-chaperones, remodel large protein aggregates via translocation of the substrate protein through their central pores (6–8).
Despite the fascinating activity displayed by ClpB/Hsp104, their homologues have not been found beyond bacteria and yeast cells. Nevertheless, multiple lines of evidence indicate that maintenance of protein homeostasis in mammalian cells is critically dependent on cellular programs to overcome the deleterious effects of protein aggregation (9). Recently, it was demonstrated that ATP-independent actions of the mammalian Hsp110 and small heat shock proteins can engage and facilitate the remodeling of protein aggregates in collaboration with Hsp70 and Hsp40 homologues (10, 11). These observations suggest that cells, especially higher eukaryotic cells, have evolved alternative strategies and mechanisms to rescue protein aggregates.
Recently, we described a novel disaggregase system that operates independently of ATP: the 38-kDa subunit of the chloroplast signal recognition particle (cpSRP43)4 (12). The substrates of this chaperone belong to the light-harvesting chlorophyll a/b-binding (LHC) family of proteins, which are delivered by the cpSRP from the chloroplast stroma to the thylakoid membrane (13). The most abundant member of this family, LHCP, comprises up to 50% of the protein content in the thylakoid membrane and is likely the most abundant membrane protein on earth (13, 14). LHC proteins contain three hydrophobic transmembrane (TM) helices, making them highly prone to aggregation as they traverse aqueous compartments in the cell (14, 15). Recently, we and others showed that the cpSRP43 subunit of cpSRP acts as an effective molecular chaperone for the LHC proteins (12, 16). Intriguingly, cpSRP43 also efficiently reverses the aggregation of LHC proteins without the requirements for ATP hydrolysis or co-chaperones (12, 16).
cpSRP43 provides a valuable example of a novel category of ATP-independent chaperones/disaggregases that operates with energy derived solely from binding interactions with its substrate proteins. Understanding its mechanism of action will provide valuable insights into alternative principles and approaches that can be used to overcome protein aggregation problems. An increasing number of examples speak to the generality of this phenomenon. Mitochondria import stimulation factor (MSF) reverses the aggregation of mitochondrial precursor proteins and restores their import in an ATP-independent mode (17, 18). Small heat shock proteins play crucial roles in remodeling protein aggregates and facilitate their resolubilization by Hsp70/100 (10, 11). Cyclophilins reactivate the aggregates of adenosine kinase (19). ATP-independent disaggregase activities have also been reported in nematode and mammalian tissue (9, 20, 21). However, the mechanism by which protein aggregates can be disassembled based solely on the substrate binding energy of a chaperone remains elusive.
Many questions arise in addressing these mechanisms. First, what are the precise binding interactions between cpSRP43 and its substrate proteins? Previous work has demonstrated a specific interaction of cpSRP43 with a highly conserved 18-amino acid loop, L18, between TM2 and TM3 of LHC proteins (22, 23). However, the ability of cpSRP43 to prevent LHC proteins from aggregation implies that it must also protect the hydrophobic TMs of the substrate protein. Consistent with this notion, the binding affinity between cpSRP43 and full-length LHCP is at least an order of magnitude higher than that for the L18 peptide (12, 24). Thus additional interactions most likely exist between LHCP and cpSRP43, but the nature of these interactions remains to be determined. Second, how does cpSRP43 use these binding interactions to effect the reversal of protein aggregation? Previous kinetic analyses revealed that disaggregation is a cooperative process during which multiple cpSRP43 molecules recognize and actively remodel the LHCP aggregate (12). However, how the recognition and remodeling of the protein aggregate were accomplished by cpSRP43 has been elusive.
By combining molecular genetics with kinetic and thermodynamic analysis, here we present evidence that the interaction of cpSRP43 with its substrate proteins is composed of two components: sequence-specific recognition of the L18 motif and highly promiscuous interactions with hydrophobic TMs. These interactions enable distinct steps in the cpSRP43-mediated disaggregation of LHC proteins: initial recognition and subsequent remodeling and disruption of the aggregate. The balance of these binding interactions with the energetics of packing interactions within the aggregate dictates the efficiency of the disaggregation reaction.
EXPERIMENTAL PROCEDURES
Materials
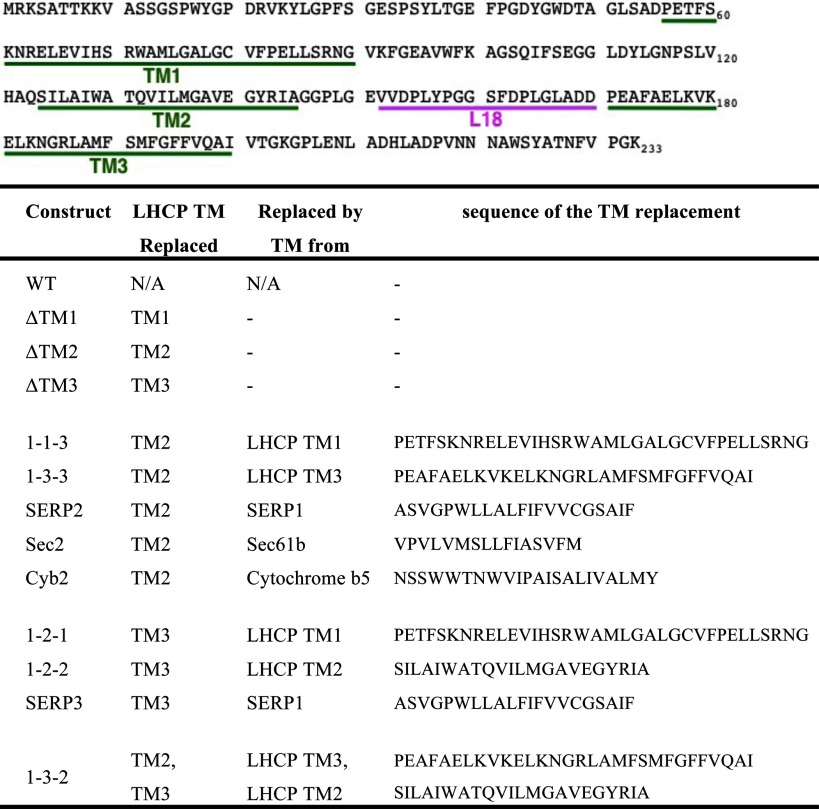
To construct the LHCP TM mutants (see Table 1), a pair of unique restriction sites was introduced into the expression plasmid encoding LHCP before and after the sequences encoding TM1, TM2, or TM3. The sequences coding for the TMs were replaced with PCR fragments encoding alternative TMs using the corresponding restriction sites. TM deletion mutants and Lhcb5 cysteine mutants were constructed using the QuikChange mutagenesis procedure (Stratagene). cpSRP43, LHCP, and its variants were purified as described (12).
TABLE 1.
Description of the LHCP TM mutants
Determination of Binding Affinity between cpSRP43 and Soluble Protein Substrates
Two independent methods were used to determine the apparent dissociation constant Kdapp for cpSRP43-substrate binding. (i) The first method was prevention of LHCP aggregation by cpSRP43, monitored by light scattering at 360 nm after a 10-min incubation of the substrate protein with varying concentrations of cpSRP43 (12). The light scattering is linearly proportional to the concentration of LHCP except at very low concentrations (Ref. 12 and the accompanying manuscript (34)). The percentage of soluble substrates (% soluble) was analyzed as a function of cpSRP43 concentration. The data were fit to Equation 1,
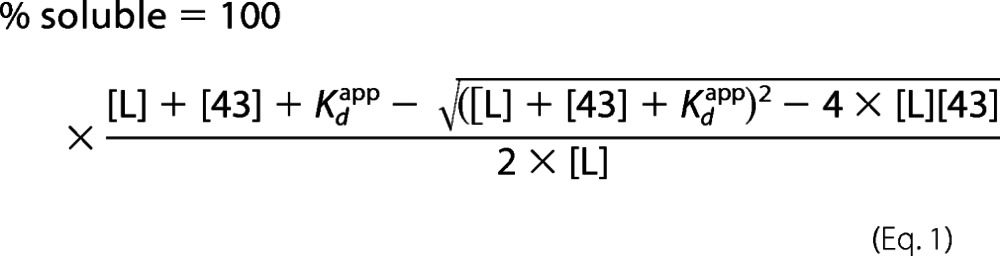 |
in which [L] is the LHC protein concentration and [43] is the cpSRP43 concentration. (ii) The second method was fluorescence anisotropy, as described previously (12). Briefly, all anisotropy measurements were conducted at room temperature using a Fluorolog 3-22 spectrofluorometer (HORIBA Jobin Yvon). Fluorescein-labeled LHCP or its variants (100 nm) were diluted into buffer containing different concentrations of cpSRP43. The samples were excited at 450 nm, and the fluorescence anisotropy was recorded at 524 nm. The data were fit to Equation 2,
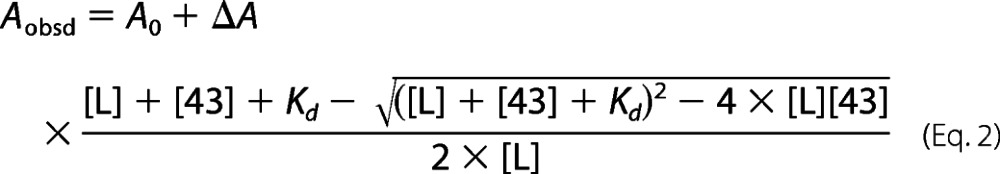 |
in which Aobsd is the observed anisotropy value, A0 is the anisotropy value without cpSRP43, ΔA is the total change in anisotropy, and Kd is the equilibrium dissociation constant. The Kd values measured by these two methods produced consistent results for the substrates tested (see Fig. 2A).
FIGURE 2.

cpSRP43 can interact with a variety of LHCP TM mutants. A, binding of cpSRP43 to TM mutants as measured by changes in anisotropy. Fits of data gave Kd values of 22 nm for ΔTM3 and 713 nm for 1-2-2. For comparison, the Kdapp values measured by light scattering were 26 and 489 nm, respectively (Table 4). B–E, binding of cpSRP43 to LHCP and its TM mutants as measured by the ability of cpSRP43 to prevent the aggregation of substrate proteins (see “Experimental Procedures”). The data were fit to Equation 1 (see “Experimental Procedures”) and gave Kdapp values that are summarized in Table 4. F, summary of the Kdapp values of all the LHCP TM mutants characterized in this study. Values of Kdapp were determined by a combination of light scattering and fluorescence anisotropy assays.
Thermodynamic and Kinetic Analyses of cpSRP43-mediated Disaggregation
Disaggregation reactions were performed as described previously (12), with the exception that aggregate formation was allowed to proceed for 1 min before the addition of cpSRP43. The observed light scattering intensity was normalized to that prior to the addition of cpSRP43. The disaggregation time courses were fit to an exponential function, Equation 3,
in which A is the observed light scattering, Af is the amount of light scattering at t → ∞, ΔA is the extent of light scattering change, and kobsd is the observed rate constant. The fractions disaggregated (K) were calculated as (ΔA/(ΔA + Af)). The cpSRP43 concentration dependences of the value of K were fit to Equation 4,
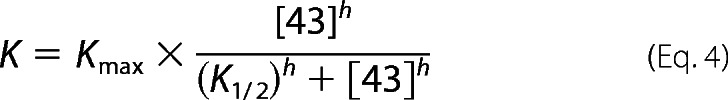 |
in which Kmax is the extent of disaggregation at saturating cpSRP43 concentration, K½ is the concentration of cpSRP43 that enables 50% solubilization of the aggregates, and h is the Hill coefficient.
Kinetic analysis was performed and analyzed as described previously (12) to obtain the forward rate disaggregation rate constant, kf, from the observed rate constants (kobsd, Equation 3) and the extent of disaggregation (K). The cpSRP43 concentration dependence of the kf values was fit to Equation 5,
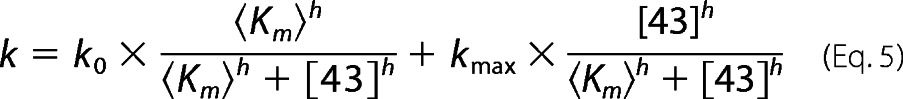 |
in which k0 is the rate of spontaneous LHCP disaggregation in the absence of the chaperone, 〈Km〉 is the concentration of cpSRP43 required to achieve half-maximal disaggregation rate, h is the Hill coefficient, and kmax is the disaggregation rate constant at saturating cpSRP43 concentration.
For some of the irreversible mutants (see Table 4, red), both the reaction equilibrium and the kinetics did not show detectable cooperative concentration dependences. Therefore, the data were fit to Michaelis-Menten equations (Equations 6 and 7).
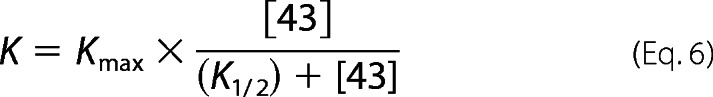 |
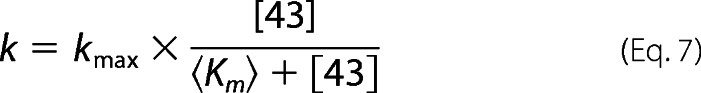 |
TABLE 4.
Summary of the thermodynamic and kinetic parameters of the LHCP TM mutants
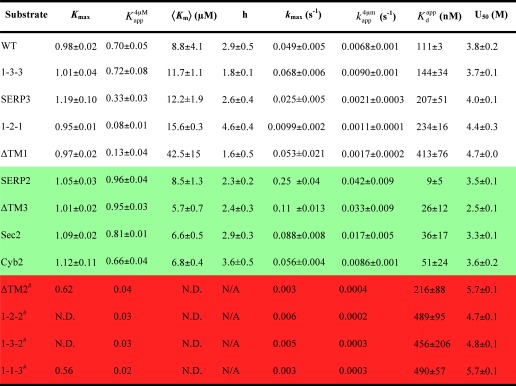
N.D. = not determined. Values reported are averages from two or more independent experiments ± S.D. # denotes mutants that are fit with Equations 6 and 7 under “Experimental Procedures.” kmax values were estimated for these mutants from the observed disaggregation rate constants at the highest cpSRP43 concentrations, where saturation was reached. Accurate 〈Km〉 values could not be determined due to the extremely slow reaction of these mutants at low cpSRP43 concentrations and are hence not reported. N/A = not applicable.
Although direct evidence remains to be obtained, the following strongly suggests that the formation of an initial recognition complex between cpSRP43 and LHCP aggregates is fast when compared with subsequent remodeling and disassembly of the aggregate. First, in all the binding experiments, the cpSRP43-LHCP interaction is complete within the timescale of manual mixing (≤15 s), much faster than the overall disaggregation rates. Second, given an affinity of ∼2 μm for the cpSRP43-L18 motif interaction (12, 24) and the typical range of macromolecular association rate constants (106-108 m−1 s−1), the dissociation rate constant of the recognition complex would be in the range of 2–200 s−1, much faster than the overall disaggregation reaction. Together, these observations suggest that the remodeling and disassembly of the aggregate is the rate-limiting step in the disaggregation reaction. Therefore, the cpSRP43 concentration required to achieve half of the maximal rate of disaggregation, 〈Km〉, provides an empirical measure for the average binding affinity of cpSRP43 to the aggregate.
Determination of the Energetics of Aggregate Formation
The energetics of packing interactions that drive aggregate formation were probed with a sedimentation assay. Briefly, preformed LHCP aggregates (10 μm) were resolubilized by various concentrations of guanidinium hydrochloride or urea for 30 min at 25 °C. The mixtures were centrifuged at 18,000 × g for 30 min, and soluble (S) and pellet (P) fractions were visualized by SDS-PAGE. The intensities of the Coomassie Blue-stained bands for the pellet and soluble fractions were quantified using ImageJ (25). The data were fit to a two-state model (Equation 8) analogous to that for protein folding (26),
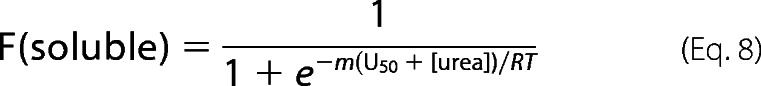 |
in which fraction soluble is calculated as (S/(S+P)), R is the gas constant, T is temperature, U50 is the urea concentration to achieve 50% solubilization, and m is a constant of proportionality.
Mathematical Analyses
Linear regression analysis was performed using Mathematica to identify a weighted linear combination of U50 and lnKd values that best reproduces the lnkmax values in Table 4. This was carried out by identifying the global minimum for the scoring function (Equation 9)
LHCP Scanning Mutagenesis
All molecular biology manipulations, including site-directed mutagenesis, transformation, and plating, were performed on a Tecan Freedom EVO liquid-handling robot.5 Constructs were sequence-verified and rearrayed into master plates. These master plates served to inoculate 10-ml volumes of Instant TB autoinduction media in 24-well plates. After overnight expression at 37 °C, the hexahistidine-tagged proteins were purified under denaturing conditions. Cysteine scanning of the L18 motif was carried out using QuikChange mutagenesis (Stratagene). Single cysteine mutants of LHCP were analyzed using the light scattering assay as described under “Determination of Binding Affinity between cpSRP43 and Soluble Protein Substrates.”
Plate-based Aggregation Prevention and Disaggregase Activity Assay
All LHCP variants were normalized to 45 μm and then diluted to 5 μm in 384-well plates by the liquid-handling robot. In the aggregation prevention assay, either no cpSRP43 or an equimolar amount (5 μm) or a 1:3 molar ratio (15 μm) of traditionally purified cpSRP43 was already present in each reaction well. The reaction was followed by absorbance at 360 nm and allowed to proceed for at least 20 min. When measuring disaggregase activity, the cpSRP43 concentration was raised to a 1:6 molar ratio (30 μm) and added ∼1 min after diluting the LHCP protein into aqueous buffer. Again, the reaction was followed for 20 min by measuring the absorbance at 360 nm. For both types of assays, the first time point was measured ∼5 min after mixing, and the final time point was used for data analysis. The percentage of chaperone activity is defined as
 |
where the equimolar chaperoneA360 value can be substituted for the A360 values of other chaperone conditions. Relative chaperone activity is then calculated by normalizing the percentage of chaperone activity to the WT LHCP value for each assay plate.
RESULTS
Bipartite Interactions of cpSRP43 with Soluble LHCP
To identify binding interactions of cpSRP43 with LHCP that are crucial to its chaperone and disaggregase activities, we performed exhaustive alanine-scanning mutagenesis in LHCP and assayed the chaperone activity using automated protocols on a Tecan Freedom EVO liquid-handling robot.5 Residues in the conserved L18 sequence between TM2 and TM3 of LHCP were further mutated to glycine and lysine. We tested the mutational effects on the interaction of cpSRP43 with LHCP by measuring the ability of cpSRP43 to (i) bind and thus prevent the aggregation of LHCP (Fig. 1, A, B, D, and E); and (ii) reverse existing LHCP aggregates (Fig. 1, C and F). Single mutations of every residue in an FDPLGL motif in L18 had large deleterious effects on both activities (Fig. 1, D–F), indicating that this motif plays a crucial role in the ability of cpSRP43 to bind and chaperone LHCP. In contrast, mutations in the remainder of the LHCP had modest to marginal effects (Fig. 1, A–F). An independent cysteine mutagenesis scan of the L18 sequence yielded the same results (Fig. 1, G and H). These results extend previous studies (12, 24), and together, they show that cpSRP43 makes highly sequence-specific interactions with the XDPLGX motif in the L18 sequence.
FIGURE 1.

cpSRP43 makes highly sequence-specific interactions with the FDPLGL motif in the L18 sequence. A–F, alanine-scanning mutagenesis of the entire LHCP (A-C), and alanine-, glycine-, and lysine-scanning mutagenesis within the L18 sequence of LHCP (D–F). The aggregation prevention activity of cpSRP43 was assayed at 1:1 (A and D) and 1:3 (B and E) molar ratios of LHCP to cpSRP43. aa, amino acids; mut, mutant. In C and F, the disaggregase activity was measured at 1:6 molar ratio of LHCP to cpSRP43. All assays were performed in 384-well plates using a Tecan Freedom EVO liquid-handling robot, as described under “Experimental Procedures.” G and H, single-cysteine substitutions at individual residues in L18 were tested for their ability to prevent the aggregation of LHCP (G) and to resolubilize existing LHCP aggregates (H). In G, a 1:1 ratio of cpSRP43 and LHCP was used. In H, a 5:1 ratio of cpSRP43 relative to LHCP (in aggregates) was used.
The absence of significant defects from point mutations of the remainder of LHCP (Fig. 1, A–C) suggests that the interactions of cpSRP43 with the TMs of LHCP are likely promiscuous. To provide independent evidence for this notion and to further probe the nature of the interaction of cpSRP43 with the TMs of the substrate protein, we constructed LHCP variants in which the individual TMs are deleted or swapped. In addition, the TMs in LHCP were replaced with those from unrelated membrane proteins, including the tail-anchored proteins SERP1, Sec61β, and cytochrome b5 (see Table 1 for nomenclature and composition of LHCP TM mutants used in this study). If the interaction of cpSRP43 with the TMs is sequence-specific, these mutations should significantly reduce the ability of cpSRP43 to bind and chaperone LHCP (12). On the other hand, if these interactions arise from generic hydrophobic interactions or backbone contacts, these TM replacements should not substantially disrupt the chaperone activity. We quantitatively measured the binding interactions of the TM mutants with cpSRP43 using two independent approaches: (i) the ability of cpSRP43 to bind and thus prevent the aggregation of substrate proteins, which provides a convenient measure for the apparent binding affinity (Kdapp) between this chaperone and the soluble LHCP; and (ii) equilibrium titrations based on changes in the fluorescence anisotropy of fluorescein-labeled LHCP upon its binding to cpSRP43 (12). The values of Kdapp obtained from the two assays were comparable with one another(12) (Fig. 2A).
All the LHCP TM mutants tested could be efficiently bound and protected from aggregation by cpSRP43 (Fig. 2), with efficiencies that differ no more than 5-fold from wild-type LHCP. Some mutants, such as ΔTM3, SERP2, and Sec2, bound cpSRP43 with even higher affinity than wild-type LHCP and are hence more readily protected by this chaperone (Fig. 2, A–E, green). Collectively, all the TM replacement mutants exhibit moderate to high affinities for cpSRP43, which are 10–100-fold higher than that of the isolated L18 peptide (24) (Fig. 2F). This strongly suggests that the hydrophobic TMs contribute additional binding interactions with cpSRP43. Further, these interactions are fairly generic and highly adaptable, in contrast to the strictly sequence-specific interactions of the L18 motif. Finally, these results show that cpSRP43 can protect a variety of aggregation-prone proteins, as long as the L18 motif is present to provide specific recognition.
A Quantitative Framework to Analyze cpSRP43-mediated Disassembly of LHC Aggregates
To understand how the substrate binding interactions of cpSRP43 are used to drive the disassembly of LHC aggregates, it is crucial to establish a quantitative framework that describes the energetics of the individual steps of this reaction. The disaggregation reaction mediated by cpSRP43 can be studied under single turnover conditions (26), minimizing complications from multiple turnover and facilitating interpretation of data. Both the kinetics and the equilibrium of this reaction exhibit saturable cooperative concentration dependences (Fig. 3, A and B) (12), strongly suggesting that the reaction involves at least two steps: (i) a higher-order step dependent on cpSRP43 concentration, presumably the assembly of a recognition complex between cpSRP43 and the aggregate (Fig. 3C, step 1) followed by (ii) a unimolecular step independent of chaperone concentration, presumably involving the remodeling and disruption of the aggregate to generate resolubilized cpSRP43·LHC complexes (Fig. 3C, step 2). Important parameters can be extracted from these data to empirically report on the energetics of these steps (Fig. 3 and Table 2). Assuming that the initial recognition step is fast when compared with the subsequent remodeling (see “Experimental Procedures”), the cpSRP43 concentration required to achieve half of the maximal disaggregation rate provides an empirical measure for the average binding affinity of cpSRP43 to the LHC aggregate (Fig. 3, A and C, and Table 2, 〈Km〉). The Hill co-efficient, h, denotes the minimum number of cpSRP43 molecules that cooperatively act together to disrupt the aggregate (Fig. 3C and Table 2). The maximal rate of disaggregation at saturating chaperone concentration, kmax, measures the energetic barrier for remodeling and disrupting the aggregate once the initial recognition complex is formed (Fig. 3, A and C, and Table 2). In equilibrium measurements, the fraction of LHC proteins resolubilized at saturating cpSRP43 concentrations, Kmax, reports on the extent to which the interactions between LHC and cpSRP43 overcome the forces that stabilize the aggregate. Finally, at a subsaturating cpSRP43 concentration, the observed kinetics and equilibrium of LHC resolubilization (kapp and Kapp, respectively) measure the overall barrier to reach the transition state and the final cpSRP43·LHC complex, respectively.
FIGURE 3.
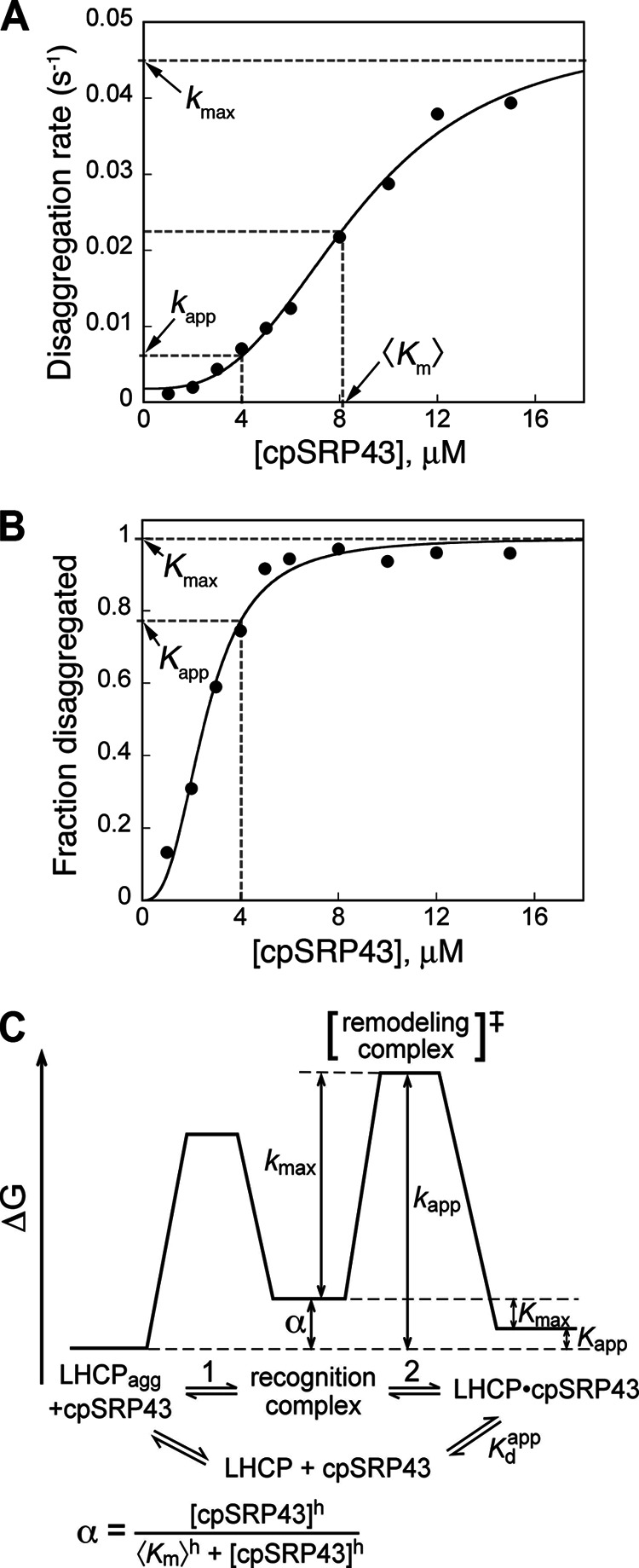
Schematics depicting quantitative analysis of the cpSRP43-mediated disaggregation reaction. Concentration dependences of the kinetics (A) and equilibrium (B) of the cpSRP43-mediated reversal of wild-type LHCP aggregate yield important parameters that report on the energetics of different steps of the disaggregation pathway (C).
TABLE 2.
Thermodynamic and kinetic parameters in the disaggregation reaction
| Parameter | Definition | Assay | Equationa |
|---|---|---|---|
| 〈Km〉 | cpSRP43 concentration that achieves half-maximal rate of disaggregation | Disaggregation | 5 |
| h | Hill coefficient | Disaggregation | 5 |
| kmax | Maximal disaggregation rate constant at saturating cpSRP43 concentration | Disaggregation | 5 |
| Kmax | Maximal fraction disaggregated at saturating cpSRP43 concentration | Disaggregation | 4 |
| kapp | Rate constant of disaggregation at a subsaturating cpSRP43 concentration | Disaggregation | 5 |
| Kapp | Fraction disaggregated at a subsaturating cpSRP43 concentration | Disaggregation | 4 |
| Kdapp | Apparent dissociation constant of the soluble cpSRP43·LHCP complex | 1) Prevention of aggregation and 2) fluorescence anisotropy | 1, 2 |
| U50 | Urea concentration required for 50% resolubilization of the aggregate | Sedimentation | 8 |
a Numbers refer to equations under “Experimental Procedures.”
To provide independent evidence that the disaggregation reaction can be experimentally dissected into distinct steps and to probe the molecular determinants that underlie each step, we characterized mutant cpSRP43 or LHC proteins that exhibit different defects in the disaggregation reaction. Below, we present evidence for two distinct classes of mutants that uncouple the initial recognition of the aggregate from its subsequent remodeling and solubilization and for the distinct molecular determinants that underlie these steps.
Interaction with the L18 Motif Is Essential for Initial Recognition of the Aggregate
In the accompanying manuscript (Nguyen et al. (34)), the results of both electron paramagnetic resonance (EPR) and chemical modification experiments showed that in LHC aggregates, the hydrophobic TMs are buried in the interior, whereas the L18 motif is displayed on the exterior. These results suggest an attractive model in which cpSRP43 could recognize the L18 motif presented on the surface of the aggregate, initiating its action as a disaggregase. If this were the case, mutant LHC or cpSRP43s that specifically disrupt the L18-cpSRP43 interaction would impair the initial recognition of the aggregate, exhibiting defects in disaggregation at low chaperone concentrations. As binding is a higher-order process, the defects of these mutants could be overcome when a sufficiently high chaperone concentration is used to drive the initial binding. To test this hypothesis, we examined how mutations in the L18 motif of LHC or in the L18-binding sites of cpSRP43 affect the efficiency of disaggregation.
We identified two mutations in the L18 motif of Lhcb5 (a close homologue of LHCP), H160C and L170C, that weaken substrate binding with cpSRP43. Equilibrium binding assays showed that wild-type Lhcb5 binds tightly to cpSRP43, with a Kdapp value of 10 nm, whereas mutants H160C and L170C exhibited weakened binding, with Kdapp values of 30 nm and 1.1 μm, respectively (Fig. 4A and Table 3). Reciprocally, mutation of Arg-161 in cpSRP43 (R161A), which provides an important hydrogen bond partner with L18 (24), significantly reduces the binding affinity of cpSRP43 to LHCP (Kdapp = 1.2 μm, when compared with 138 nm with wild-type cpSRP43 (12)).
FIGURE 4.

L18-binding mutants uncouple initial recognition of the aggregate from its subsequent solubilization. A, binding of cpSRP43 to wild-type Lhcb5 and L18 mutants H160C and H170C. The data were fit to Equation 1 (see “Experimental Procedures”) and gave Kdapp values of 10 nm for wild-type Lhcb5 (black), 30 nm for Lhcb5-H160C (blue), and 1.1 μm for Lhcb5-L160C (red). B, concentration dependences for the equilibrium of disaggregation of LHCP by wild-type cpSRP43 (black) or mutant cpSRP43(R161A) (magenta). C and D, chaperone concentration dependences for the equilibrium (C) and kinetics (D) of disaggregation of Lhcb5 (black), Lhcb5-H160C (blue), and Lhcb5-L170C (red) by wild-type cpSRP43.
TABLE 3.
Summary of the thermodynamic and kinetic parameters of the L18-binding mutants
Values reported are from Figure 1. N.D. = not determined.
| Construct | Kdapp | 〈Km〉 | Kmax | Kapp4μm | kmax | Kapp4μm |
|---|---|---|---|---|---|---|
| μm | μm | s−1 | s−1 | |||
| cpSRP43 R161A | 1200a | >502 | >0.40b | 0.02 | ND | ND |
| Lhcb5 | 10 | 8.8 | 1.06 | 0.51 | 0.029 | 0.0042 |
| Lhcb5 H160C | 30 | 64 | 0.85 | 0.12 | 0.025 | 0.0021 |
| Lhcb5 L170C | 1100 | >902 | >0.352 | 0.03 | >0.014 | N.D. |
a Previously determined by fluorescence anisotropy (12).
b Denotes the values at the highest cpSRP43 concentration used.
Consistent with defects in recognition of the LHCP aggregate, mutant cpSRP43-R161A exhibited severe defects in the reversal of LHCP aggregates at low chaperone concentrations (Fig. 4B, magenta versus black; Table 3, Kapp and kapp). However, when the concentration of the mutant chaperone was raised to compensate for the binding defect, cpSRP43-R161A could reverse LHCP aggregation. At saturating chaperone concentrations, close to 50% solubilization of the aggregate could be attained (Fig. 4B, magenta and Table 3). Analogously, the aggregates formed by the Lhcb5 mutants, H160C and L170C, exhibited defects in the disaggregation reaction that can be rescued by higher cpSRP43 concentrations (Fig. 4, C and D, and Table 3). At saturating chaperone concentrations, the equilibrium and kinetics of disaggregation with the mutant aggregates are within 2-fold of those of wild-type Lhcb5 (Fig. 4, C and D, and Table 3). Finally, all three mutants exhibited much higher values of 〈Km〉 in the disaggregation reaction when compared with the wild-type protein (Fig. 4 and Table 3), which correlated with their reductions in substrate binding affinity (12) (Fig. 4A). Together, these results showed that L18 binding is a key requirement for the initial recognition of the aggregate by cpSRP43; further, this recognition event can be uncoupled from the subsequent concentration-independent step(s) in the disaggregation reaction.
A Class of LHCP TM Mutants Specifically Blocks the Disaggregation Process
Because the LHCP TM mutants contain intact L18 motifs, they provide a collection of substrates to probe for additional molecular requirements that underlie the disaggregase activity of cpSRP43. Surprisingly, although all the TM mutants can be efficiently bound and prevented from aggregation by cpSRP43 (Fig. 2), they exhibit striking differences in the thermodynamics and kinetics of the disaggregation reaction (Fig. 5 and Table 4).
FIGURE 5.

LHCP TM mutants exhibit a wide range of disaggregation efficiencies. A–H, representative concentration dependences of the equilibrium (A, C, E, and G) and kinetics (B, D, F, and H) for disassembly of the aggregates formed by the LHCP TM mutants. The data for wild-type LHCP (black) were shown as a reference of comparison in all four sets. The data in A, C, E, and G were fit to Equation 4 (black, blue, and green) or Equation 6 (red) to obtain Kmax values and extract Kapp values at 4 μm cpSRP43. The data in B, D, F, and H were fit to Equation 5 (black, blue, and green) or Equation 7 (red) to obtain kmax, 〈Km〉, and h values and to extract kapp values at 4 μm cpSRP43. All the thermodynamic and kinetic parameters were reported in Table 4.
The aggregates formed by some of the TM mutants, notably those of ΔTM3, SERP2, Sec2, and Cyb2, showed disaggregation kinetics and efficiencies that are comparable with or even higher than that of wild-type LHCP (Fig. 5 and Table 4, green). Notably, the aggregates formed by a group of mutants, especially 1-1-3, ΔTM2, 1-2-2, and 1-3-2, were virtually irreversible even when saturation in disaggregation rate constants had been reached at high cpSRP43 concentrations (Fig. 5 and Table 4, red). To a lesser extent, mutants 1-2-1 and ΔTM1 also exhibited significant reductions in the disaggregation rates even when saturation was reached at high cpSRP43 concentrations (Fig. 5, E–H, blue, and Table 4). In the aggregate formed by all these mutants, the L18 motif is highly accessible and solvent-exposed (Fig. 6, A–C); this and the observation that saturation in disaggregation kinetics can be reached with these mutants indicate that their defects could not be accounted for by the inability of cpSRP43 to recognize the aggregate. In addition to substantial reductions in maximal disaggregation rates, the disaggregation reaction of these mutants lost cooperative dependence on cpSRP43, further supporting a specific defect in the ability of cpSRP43 to remodel and resolubilize the aggregate. Together, these results provide strong evidence for the presence of an additional remodeling step in the disaggregase mechanism of cpSRP43, whose molecular requirements are distinct from the initial recognition step.
FIGURE 6.

Time courses for the alkylation reactions of cysteine residues in the L18 shows accessibility of WT (A) and mutant (B and C) LHC proteins. A–C, Lhcb5 L170C (A), ΔTM2 G158C (B), and ΔTM3 G158C (C) were labeled with 30-fold excess N-ethyl-maleimide in denaturant guanidinium hydrochloride (GdmHCl), pH 7.5 (black traces) and in aqueous buffer, pH 7.5 (red traces), and the reactions were quenched at different time points with DTT and subjected to intact protein mass spectrometry as in the accompanying manuscript (34).
The Irreversible LHCP TM Mutants Form Ultrastable Aggregates
Unlike the L18-binding mutants, the irreversible LHCP TM mutants bind reasonably well to cpSRP43. What caused their defects in disaggregation? The results of chemical modification and EPR experiments showed that the TM segments are buried inside the aggregate and engage in strong interactions (see accompanying manuscript (34)). We hypothesized that the internal packing interactions within the aggregates are altered in these TM mutants, which could present higher barriers for cpSRP43 to remodel and disrupt the aggregate. To test this hypothesis, we probed the energetics of the packing interactions that stabilize the aggregate by quantitatively analyzing its solubility in chemical denaturants. Using the sedimentation assay, we showed that both guanidinium hydrochloride and urea could effectively solubilize the LHCP aggregate in a concentration-dependent manner (Fig. 7A). Quantification of the amount of solubilized LHCP as a function of urea concentration gave an aggregate solubilization curve analogous to protein unfolding curves (26) (Fig. 7, B–E). Based on a two-state model, quantitative analyses of these data yielded information about the energetics of transfer of LHCP from urea to water (ΔG°) and the urea concentration required to achieve 50% solubilization (Table 4, U50; see “Experimental Procedures”). These parameters provide quantitative empirical measures of the energetics of the internal packing interactions that drive aggregate formation.
FIGURE 7.

The irreversible TM mutants form ultrastable aggregates. A, sedimentation analysis of the ability of guanidinium chloride (GdmHCl) and urea to resolubilize LHCP aggregates. M denotes the protein marker lane. B–E, urea solubilization curves of LHCP and its TM mutants. The data were fit to Equation 8 (see “Experimental Procedures”) and gave U50 values (Table 4).
The aggregates formed by the LHCP TM mutants exhibited a wide range of stabilities, with U50 values ranging from 2.5 to 5.7 m (Fig. 7, B–E, and Table 4). Notably, the four irreversible mutants whose aggregates could not be efficiently resolubilized exhibited the highest U50 values (4.7–5.7 m; Fig. 7 and Table 4, red). In contrast, some of the mutant aggregates that are more readily resolubilized by cpSRP43, such as ΔTM3, displayed the lowest U50 values (2.5–3.3 m; Fig. 7 and Table 4, green). These results strongly suggest that the internal packing interactions within the aggregate provide a crucial barrier to the efficiency with which cpSRP43 can disrupt protein aggregates.
Linear Free Energy Analysis to Probe the Energetic Determinants of Disaggregation Efficiency
The collection of LHCP TM mutants, which display a wide range of disaggregation efficiencies and kinetics (Table 4), further allowed us to systematically probe the contributions of different molecular features and the nature of the rate-limiting remodeling complex (Fig. 3C, ‡) during the disaggregation reaction. To this end, we evaluated how the maximal rate of disaggregation (kmax) correlates with the two energetic parameters that were varied in this set of mutants: (i) the binding affinity between cpSRP43 and the solubilized LHCP (Kdapp), which ultimately drives the disaggregation reaction; and (ii) the energetics of packing interactions that drive aggregate formation (U50), which must be overcome by cpSRP43 during disaggregation. A strong correlation was found between the maximal disaggregation rate constant and a weighted combination of the U50 and Kdapp values (Fig. 8A, R2 = 0.96), but not with either of the parameters alone. This correlation strongly suggests that once a recognition complex is formed, the competition between the packing interactions that stabilize the aggregate and additional binding interactions that cpSRP43 establishes with the TMs of LHCP dictates the resolubilization of the aggregate. Finally, two of the mutants, ΔTM1 and ΔTM3, exhibit significant deviations from this correlation (Fig. 8A, blue), suggesting preferences in the disaggregation pathway of cpSRP43 that are not accounted for by these two parameters (see “Discussion”).
FIGURE 8.

Linear free energy analysis of the cpSRP43-mediated disaggregation reaction. A, the maximal disaggregation rate constant strongly correlates with a weighted combination of the U50 and Kdapp values. Values for the analysis are from Table 4. The black line represents a linear fit to the data (R2 = 0.96). ΔTM1 and ΔTM3 (blue) were marked as outliers and were not included in the correlation. B, ϕ value analysis of LHCP disaggregation. The values of Kapp and kapp were calculated from fits of disaggregation equilibrium and kinetic data to Equations 4 and 5 (“Experimental Procedures”), respectively, and the concentration of cpSRP43 was chosen at 4 mm. Linear fit of the data (black line, R2 = 0.98) gave a slope (ϕ value) of 0.73.
Analysis of the relationship between the equilibrium and rate constants of disaggregation provides further insights into the nature of the rate-limiting remodeling complex (Fig. 3C, ‡). At a subsaturating cpSRP43 concentration (chosen at 4 μm), the free energy barrier (ΔΔG ∼ ln Kapp) and the activation barrier (ΔΔG‡ ∼ ln kapp) of the disaggregation reaction showed an excellent linear correlation for the entire set of LHCP mutants (Fig. 8B), giving a slope of Φ = 0.73. Analogous to the Φ-value analysis of protein folding (26), this analysis could be used to infer the nature and structure of the transition state relative to the reaction substrate (the LHCP aggregate) and product (the solubilized cpSRP43·LHCP complex). The observation of a Φ-value that approaches unity implies a fairly late transition state in which a substantial fraction of packing interactions within the LHCP aggregate is disrupted and those interactions with cpSRP43 are formed, albeit not to the same extent as those in the resolubilized cpSRP43·LHCP complex.
DISCUSSION
cpSRP43 provides an example of a novel class of chaperones that can effect the reversal of insoluble protein aggregates based solely on ATP-independent binding interactions with its substrate protein. The simplicity of this system makes it an accessible model system to delineate the molecular mechanisms as well as the capability and limitations of ATP-independent disaggregases. Here, mutational analyses revealed distinct sets of binding interactions that this chaperone establishes with its substrate proteins. Further, molecular genetics combined with thermodynamic and kinetic analyses allowed us to dissect the molecular steps during the cpSRP43-mediated disaggregation reaction and revealed distinct molecular requirements and interactions that underlie each step. These results, combined with previous work (Ref. 12 and the accompanying manuscript (34)), led us to propose a two-step working model for the action of cpSRP43 as a protein disaggregase (Fig. 9A).
FIGURE 9.

A, Working model for cpSRP43-mediated disaggregation reaction. Step 1 depicts initial binding of cpSRP43 (magenta) to the LHCP aggregate (green), which occurs via recognition of the solvent-exposed L18 motif (red). Step 2 depicts the cooperative action of cpSRP43 molecules to compete with and disrupt the packing interactions between the LHCP TM segments within the aggregate, leading to its resolubilization. B and C, qualitative free energy diagrams summarizing the effects of the L18-binding mutants that disrupt the initial binding step (B) and the irreversible TM mutants that disrupt the remodeling step (C), as described under “Results.” The figures are not drawn to scale.
Bipartite Interactions of cpSRP43 with Substrate Protein
Previous work has established a specific interaction of cpSRP43 with L18, a conserved and relatively hydrophilic segment between TM2 and TM3 of the LHC family of proteins (22–24). The mutagenesis results here further demonstrated that this interaction is localized to the most conserved FDPLGL motif within L18, emphasizing the highly specific nature of this recognition. This is consistent with crystallographic observations in which DPLG was found to form a “turn” that wraps around Tyr-204 of cpSRP43, whereas the side chains in the remainder of the L18 peptide were not well resolved (24). Nevertheless, the ability of cpSRP43 to protect LHC proteins from aggregation implies that additional interactions must exist between this chaperone and the hydrophobic TMs on its substrate proteins. Although the precise motif(s) that mediate these additional interactions remain to be determined, the results here demonstrate that the interactions of cpSRP43 with the TMs are highly promiscuous, enabling it to bind and chaperone a variety of substrate variants in which the TMs were removed or replaced. Some of the substrate variants could be bound and chaperoned by cpSRP43 even more effectively than wild-type LHCP. Together, these results establish two important components of the binding interaction of cpSRP43 with substrate protein: highly specific recognition of the FDPLGL motif in the L18 segment and generic hydrophobic interactions with the TMs of the substrate protein that are highly adaptable. As discussed below, these two sets of binding interactions contribute to distinct stages in the action of cpSRP43 as a disaggregase.
Different Binding Interactions Drive Distinct Stages of the Disaggregase Activity of cpSRP43
Recognition of the Aggregate
To initiate the disaggregation reaction, cpSRP43 must first recognize and engage the LHC aggregates (Fig. 9A, step 1). Although additional interactions cannot be excluded, an attractive mechanism to drive this initial recognition is the binding of cpSRP43 to the L18 motif, which is displayed on the solvent-accessible exterior of the LHC aggregate (Fig. 9A, step 1). In support of this model, mutant proteins that disrupt the interaction of cpSRP43 with the L18 motif exhibit defects in disaggregation at low cpSRP43 concentrations and require much higher chaperone concentrations to reach saturation (Fig. 9B, ΔΔG1). Consistent with a specific defect of these mutants in a binding step, their defects could be overcome by increasing the chaperone concentration such that the maximal rate and efficiency of the disaggregation reaction with these mutants are within 2-fold of that of the wild-type protein (Fig. 9B).
Intriguingly, the values of 〈Km〉, which provide a proxy for the binding of cpSRP43 to the LHCP aggregate, are considerably weaker than the binding of cpSRP43 to the L18 peptide (24) and, with the exception of ΔTM1, vary from ∼6 to 16 μm across different TM mutants (Table 4, white and green). On the one hand, this variation is much smaller than the up to 50-fold changes in the binding affinity between cpSRP43 and the soluble substrate protein (Table 4, Kdapp), supporting the notion that interaction with the L18 motif is a major driving force for the initial recognition step and is less sensitive to variations in the TMs of LHCP. On the other hand, such variation, although modest, could not be explained by the simplest model in which the recognition step is equivalent to interaction of cpSRP43 with an isolated L18 peptide. It is possible that the L18 motif is presented in different configurations on the aggregates formed by the different TM mutants, which could cause the observed variations. Consistent with this possibility, EPR experiments showed that in the aggregate, spin probes in the DPLG motif exhibit much lower mobility than the remainder of the L18 sequence (see accompanying manuscript (34)), suggesting that this motif might contact the remainder of the aggregate and needs to undergo a rearrangement to interact with cpSRP43. Additional structural or sequence elements presented on the aggregate surface could also be recognized by cpSRP43. Consistent with this possibility, cpSRP43 cross-links to residues at the N terminus of TM3 (27); this segment is also exposed on the surface of the LHC aggregate (see accompanying manuscript (34)) and available for recognition by cpSRP43.
Remodeling and Disruption of the Aggregate
The class of irreversible TM mutants (Table 4, red), which exhibits severe defects in maximal disaggregation rate constants (kmax), provides strong evidence for a distinct remodeling step in the disaggregation reaction (Fig. 9A, step 2) that has different molecular and energetic requirements than the initial recognition step. As cpSRP43 effectively prevents the aggregation of these mutant LHCPs, the defects of these mutants in disaggregation are most likely kinetic, rather than thermodynamic in origin. Further, the observation that all the irreversible mutants form significantly more stable aggregates than wild-type LHCP (Fig. 9C, ΔΔGagg) strongly suggests that the packing interactions within the aggregate present a major barrier for disaggregation (Fig. 9C, kmax′ ≫ kmax) and that these packing interactions need to be substantially disrupted in the rate-limiting remodeling complex (Fig. 9A, species in brackets).
Additional insights into the remodeling step are provided by analyses of the entire series of TM mutants, which display a wide range of binding interactions with cpSRP43, packing interactions within the aggregate, and kinetics of disaggregation. In this series of mutants, the best predictor for disaggregation kinetics is provided by a combination of two energetic parameters: the packing interactions within the aggregate (U50) compensated by the available binding interactions between cpSRP43 and soluble LHCP (Kdapp; Fig. 8A). This correlation is striking and implies that the transition state (or rate-limiting remodeling complex) for the disaggregation reaction involves substantial global disruption of the aggregate. Further, these disruptions must be compensated by the establishment of additional binding interactions between cpSRP43 and the TMs of the dislodged LHCP molecules. A notable example of the latter is SERP2, which forms an aggregate with a U50 value comparable with wild-type LHCP, 1-3-3 or Cyb2, but displays the fastest disaggregation kinetics as cpSRP43 establishes the strongest binding interactions with this mutant. Together, these results strongly support the model that, once cpSRP43 recognizes and “latches” onto the LHCP aggregate, the competition between its binding interactions with the TMs of LHCP and the packing interactions that stabilize the aggregate dictates the efficiency of the disaggregation process.
Φ value analysis, which compares the extent to which mutations affect the barrier to reach the transition state versus that to the resolubilized cpSRP43·LHCP complex, provide additional insights into the nature of the rate-limiting remodeling complex. The Φ value of 0.73 observed here rules out early (Φ ∼ 0) transition states and suggests a fairly late structure for the rate-limiting remodeling intermediate (26), in which a substantial portion of the packing interactions within the LHCP aggregate is disrupted and significant binding interactions with cpSRP43 have been established. A slightly alternative model, which takes into account potential heterogeneity in the action of cpSRP43, is that cpSRP43 disrupts the packing interactions at certain parts of the aggregate more extensively than at others, giving rise to a Φ value less than unity. As formation of the aggregate is a highly cooperative process (see accompanying manuscript (34)), it is conceivable that extensive disruption at multiple parts of the LHC aggregate could lead to the collapse of the network of packing interactions that drive aggregate formation (Fig. 9A), thus leading to its solubilization.
Perspective
The analyses here established two key requirements for how a chaperone can use binding interactions to reverse a protein aggregate. First, the chaperone must efficiently recognize and latch onto the target aggregate, through interactions with structural or sequence motifs displayed on the exterior of the aggregate. Second, the chaperone must effectively compete with and replace the internal packing interactions of the aggregate, by interacting with and protecting the segments of the substrate protein buried in the aggregate interior. Although the specifics of each system differ, these principles may be general to other ATP-independent chaperones that participate in or facilitate protein disaggregation processes.
Nevertheless, these do not represent all the features in the cpSRP43-mediated disaggregation reaction. Most notably, mutants ΔTM1 and ΔTM3 are clear outliers in the correlation analysis (Fig. 8A, blue squares). These deviations imply that factors in addition to the two parameters explored here (U50 and Kd) contribute to the disaggregation reaction, and suggest preferred pathways in the action of cpSRP43. For example, ΔTM3 forms the loosest aggregate and can bind cpSRP43 tightly, but its rate of disaggregation is significantly slower than that expected from these considerations. Coupled with the observation that N-terminal residues of TM3 are also highly accessible on the aggregate (see accompanying manuscript (34)) and can contact cpSRP43 (27), this raises the intriguing possibility that cpSRP43 preferentially exerts its action on TM3 during the remodeling process to most effectively disrupt the aggregate. In contrast, ΔTM1 forms one of the tightest aggregates and has weaker binding interactions with cpSRP43, yet its maximal rate of disaggregation by cpSRP43 far exceeded what would be expected based on these parameters. This led us to speculate that TM1 is not a preferred site of action of cpSRP43 during the disaggregation process. Finally, despite these deviations, the aggregates formed both by mutants and by “hybrid” substrates containing TMs from unrelated membrane proteins are efficiently reversed by cpSRP43, demonstrating the remarkable adaptability of this chaperone.
It is noteworthy to compare the mechanism of aggregate remodeling by cpSRP43 to that of the ClpB/Hsp104 disaggregases. Many of the insights into the action of ClpB/Hsp104 are based on analogy with the ClpAP/ClpXP proteases (8), which use cycles of ATP binding and hydrolysis to drive repetitive movements of the substrate-binding loops, forcing the polypeptides through a constricted pore in the hexameric assembly and thus unfolding the substrate protein. By analogy, ClpB/Hsp104 could use ATPase cycles to drive translocation of a polypeptide, extracting it out of protein aggregates (28, 29). In this mechanism, each disaggregase machine can locally sever an aggregate without disrupting the remainder of the aggregate. This is consistent with the observation that local, rather than global, structure and stability near the recognition sites dictate the efficiency of ClpA/ClpX (30, 31) and with the ability of Hsp104 to generate more amyloid fragments and thus promote amyloid propagation (32, 33). Although the precise molecular details remain to be elucidated, our results suggest that cpSRP43 acts globally, rather than locally, on the protein aggregate. The rate-limiting step in the reaction pathway of cpSRP43 involves the generation of a late intermediate in which the packing interactions within the entire aggregate are extensively disrupted and which requires the cooperative action of multiple cpSRP43 molecules. Conceivably, in the absence of external energy input, individual cpSRP43 molecules cannot compete with the packing interactions inside the aggregate and extract a soluble LHC molecule from it. Instead, multiple chaperones collectively disrupt and collapse the entire aggregate (Fig. 9A). The results here provide a valuable framework to probe the capability, effectiveness, and limitations of this alternative ATP-independent chaperone mechanism and to understand the design principles by which binding energy can be used to overcome the problems of protein aggregation.
Acknowledgments
We thank Drs. W. M. Clemons, J. Chartron, and C. Suloway for the plasmids of SERP1, Sec61β, and cytochrome b5 and the Shan laboratory for helpful comments on the manuscript.
A. Nisthal and S. L. Mayo, manuscript in preparation.
- cpSRP
- chloroplast signal recognition particle
- LHC
- light-harvesting chlorophyll a/b-binding
- LHCP
- LHCb1 gene product
- TM
- transmembrane domain
- SERP
- transmembrane of the stress-associated endoplasmic reticulum protein 1
- Sec
- transmembrane of Sec61β
- Cyb
- transmembrane of cytochrome b5.
REFERENCES
- 1. Balch W. E., Morimoto R. I., Dillin A., Kelly J. W. (2008) Adapting proteostasis for disease intervention. Science 319, 916–919 [DOI] [PubMed] [Google Scholar]
- 2. Hartl F. U., Hayer-Hartl M. (2002) Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295, 1852–1858 [DOI] [PubMed] [Google Scholar]
- 3. Chang H. C., Tang Y. C., Hayer-Hartl M., Hartl F. U. (2007) Snapshot: molecular chaperones, Part I. Cell 128, 212–213 [DOI] [PubMed] [Google Scholar]
- 4. Tang Y. C., Chang H. C., Hayer-Hartl M., Hartl F. U. (2007) Snapshot: molecular chaperones, Part II. Cell 128, 412–413 [DOI] [PubMed] [Google Scholar]
- 5. Doyle S. M., Wickner S. (2009) Hsp104 and ClpB: protein disaggregating machines. Trends Biochem. Sci. 34, 40–48 [DOI] [PubMed] [Google Scholar]
- 6. Glover J. R., Lindquist S. (1998) Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94, 73–82 [DOI] [PubMed] [Google Scholar]
- 7. Goloubinoff P., Mogk A., Zvi A. P. B., Tomoyasu T., Bukau B. (1999) Sequential mechanism of solubilization and refolding of stable protein aggregates by a bichaperone network. Proc. Natl. Acad. Sci. U.S.A. 96, 13732–13737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haslberger T., Bukau B., Mogk A. (2010) Towards a unifying mechanism for ClpB/Hsp104-mediated protein disaggregation and prion propagation. Biochem. Cell Biol. 88, 63–75 [DOI] [PubMed] [Google Scholar]
- 9. Cohen E., Bieschke J., Perciavalle R. M., Kelly J. W., Dillin A. (2006) Opposing activities protect against age-onset proteotoxicity. Science 313, 1604–1610 [DOI] [PubMed] [Google Scholar]
- 10. Shorter J. (2011) The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS One 6, e26319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rampelt H., Kirstein-Miles J., Nillegoda N. B., Chi K., Scholz S. R., Morimoto R. I., Bukau B. (2012) Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 31, 4221–4235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jaru-Ampornpan P., Shen K., Lam V. Q., Ali M., Doniach S., Jia T. Z., Shan S. (2010) ATP-independent reversal of a membrane protein aggregate by a chloroplast SRP subunit. Nat. Struct. Mol. Biol. 17, 696–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schuenemann D., Gupta S., Persello-Cartieaux F., Klimyuk V. I., Jones J. D., Nussaume L., Hoffman N. E. (1998) A novel signal recognition particle targets light-harvesting proteins to the thylakoid membranes. Proc. Natl. Acad. Sci. U.S.A. 95, 10312–10316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jansson S. (1999) A guide to the Lhc genes and their relatives in Arabidopsis. Trends Plant Sci. 4, 236–240 [DOI] [PubMed] [Google Scholar]
- 15. Liu Z., Yan H., Wang K., Kuang T., Zhang J., Gui L., An X., Chang W. (2004) Crystal structure of spinach major light-harvesting complex at 2.72 Å resolution. Nature 428, 287–292 [DOI] [PubMed] [Google Scholar]
- 16. Falk S., Ravaud S., Koch J., Sinning I. (2010) The C terminus of the Alb3 membrane insertase recruits cpSRP43 to the thylakoid membrane. J. Biol. Chem. 285, 5954–5962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hachiya N., Alam R., Sakasegawa Y., Sakaguchi M., Mihara K., Omura T. (1993) A mitochondrial import factor purified from rat liver cytosol is an ATP-dependent conformational modulator for precursor proteins. EMBO J. 12, 1579–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hachiya N., Komiya T., Alam R., Iwahashi J., Sakaguchi M., Omura T., Mihara K. (1994) MSF, a novel cytoplasmic chaperone which functions in precursor targeting to mitochondria. EMBO J. 13, 5146–5154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chakraborty A., Das I., Datta R., Sen B., Bhattacharyya D., Mandal C., Datta A. K. (2002) A single-domain cyclophilin from Leishmania donovani reactivates soluble aggregates of adenosine kinase by isomerization-independent chaperone function. J. Biol. Chem. 277, 47451–47460 [DOI] [PubMed] [Google Scholar]
- 20. Bieschke J., Cohen E., Murray A., Dillin A., Kelly J. W. (2009) A kinetic assessment of the C. elegans amyloid disaggregase activity enables uncoupling of disassembly and proteolysis. Protein Science 18, 2231–2241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Murray A. N., Solomon J. P., Wang Y.-J., Balch W. E., Kelly J. W. (2010) Discovery and characterization of a mammalian amyloid disaggregation activity. Protein Science 19, 836–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tu C. J., Peterson E. C., Henry R., Hoffman N. E. (2000) The L18 domain of light-harvesting chlorophyll proteins binds to chloroplast signal recognition particle 43. J. Biol. Chem. 275, 13187–13190 [DOI] [PubMed] [Google Scholar]
- 23. DeLille J., Peterson E. C., Johnson T., Moore M., Kight A., Henry R. (2000) A novel precursor recognition element facilitates posttranslational binding to the signal recognition particle in chloroplasts. Proc. Natl. Acad. Sci. U.S.A. 97, 1926–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stengel K. F., Holdermann I., Cain P., Robinson C., Wild K., Sinning I. (2008) Structural basis for specific substrate recognition by the chloroplast signal recognition particle protein cpSRP43. Science 321, 253–256 [DOI] [PubMed] [Google Scholar]
- 25. Abramoff M. D., Magelhaes P. J., Ram S. J. (2004) Image processing with Image J. Biophotonics Int. 11, 36–42 [Google Scholar]
- 26. Fersht A. (1998) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, pp. 508–614, W. H. Freeman and Company, New York [Google Scholar]
- 27. Cain P., Holdermann I., Sinning I., Johnson A. E., Robinson C. (2011) Binding of chloroplast signal recognition particle to a thylakoid membrane protein substrate in aqueous solution and delineation of the cpSRP43-substrate interaction domain. Biochem. J. 437, 149–155 [DOI] [PubMed] [Google Scholar]
- 28. Weibezahn J., Tessarz P., Schlieker C., Zahn R., Maglica Z., Lee S., Zentgraf H., Weber-Ban E. U., Dougan D. A., Tsai F. T., Mogk A., Bukau B. (2004) Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell 119, 653–665 [DOI] [PubMed] [Google Scholar]
- 29. Schlieker C., Weibezahn J., Patzelt H., Tessarz P., Strub C., Zeth K., Erbse A., Schneider-Mergener J., Chin J. W., Schultz P. G., Bukau B., Mogk A. (2004) Substrate recognition by the AAA+ chaperone ClpB. Nat. Struct. Mol. Biol. 11, 607–615 [DOI] [PubMed] [Google Scholar]
- 30. Lee C., Schwartz M. P., Prakash S., Iwakura M., Matouschek A. (2001) ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol. Cell 7, 627–637 [DOI] [PubMed] [Google Scholar]
- 31. Kenniston J. A., Burton R. E., Siddiqui S. M., Baker T. A., Sauer R. T. (2004) Effects of local protein stability and the geometric position of the substrate degradation tag on the efficiency of ClpXP denaturation and degradation. J. Struct. Biol. 146, 130–140 [DOI] [PubMed] [Google Scholar]
- 32. Shorter J., Lindquist S. (2004) Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 304, 1793–1797 [DOI] [PubMed] [Google Scholar]
- 33. Shorter J., Lindquist S. (2006) Destruction or potentiation of different prions catalyzed by similar Hsp104 remodeling activities. Mol. Cell 23, 425–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nguyen T. X., Jaru-Ampornpan P., Lam V. Q., Cao P., Piszkiewicz S., Hess S., Shan S. (2013) Mechanism of an ATP-independent protein disaggregase. I. Structure of a membrane protein aggregate reveals a mechanism of recognition by its chaperone. J. Biol. Chem. 288, 1340–13430 [DOI] [PMC free article] [PubMed] [Google Scholar]