

Abstract
Introduction
Cancer stem cells are a high profile drug target for cancer therapeutics due to their indispensable role in cancer progression, maintenance, and therapeutic resistance. Restoring wild-type p53 function is an attractive new therapeutic approach for the treatment of cancer due to the well-described powerful tumor suppressor function of p53. As emerging evidence intimately links p53 and stem cell biology, this approach also provides an opportunity to target cancer stem cells.
Areas covered
Therapeutic approaches to restore the function of wild-type p53, cancer and normal stem cell biology in relation to p53, and the downstream effects of p53 on cancer stem cells.
Expert opinion
The restoration of wild-type p53 function by targeting p53 directly, its interacting proteins, or its family members holds promise as a new class of cancer therapies. This review examines the impact that such therapies may have on normal and cancer stem cells based on the current evidence linking p53 signaling with these populations.
Keywords: cancer, cancer stem cells, cancer therapy, p53, p53 restoration, stem cells, p53 pathway, p53 pathway restoration
1. Introduction
p53 is a tumor suppressor gene commonly referred to as the guardian of the genome. Cellular stresses such as hypoxia, DNA damage and oncogenic stress cause accumulation of p53 protein within normal cells. p53 induces cell cycle arrest, senescence or apoptosis in response to cellular stresses, preventing the build-up of genetic mutations within cells. p53 is a transcription factor that regulates the expression of several target genes such as p21/WAF1, Bid and DR5 to elicit its functions. Mice with engineered loss of function in p53 develop spontaneous tumors that confirm its tumor suppressor function [1–12]. p53 mRNA is expressed constitutively but the level of expression varies across tissue types [2, 3]. Cellular p53 protein is short-lived and its levels are tightly regulated under physiological conditions [3, 4, 7]. In the absence of stress signals, p53 protein is rapidly degraded in normal cells by ubiquitination and proteasomal degradation via E3 ligases murine/human double minute 2 (MDM2/HDM2). MDM2 protein is released from p53 binding when both proteins are phosphorylated in the DNA damage response, which leads to stabilization of the p53 protein. p53 regulation involves a negative feedback loop since MDM2 is a transcriptional target of p53 and so the negative regulator is positively regulated by p53 leading to dampening of the p53 response [1, 2, 4, 7]. MDM4/MDMX, a homolog of MDM2, can enhance MDM2-mediated p53 degradation by forming heterodimers with MDM2. However MDMX is not a major part of the negative feedback loop since its expression is not regulated by p53 and it lacks intrinsic ubiquitin ligase activity [4]. A second feedback loop regulating p53 involves the tumor suppressor ARF (p14ARF in humans, p19ARF in mice) and the transcription factor E2F-1. ARF stabilizes p53 by inhibiting MDM2-mediated degradation of p53. E2F-1 activates ARF, which promotes degradation of E2F-1 and is repressed by p53[13]. The Rb tumor suppressor also regulates the p53 pathway by repressing the E2F family of transcription factors [14]. Thus, p53 is regulated by a complex Rb-E2F-ARF-MDM2 signaling network. Post-translational modifications of p53 such as phosphorylation, acetylation and sumoylation are also important for its activation and for determining p53-dependent cellular outcomes [1, 13, 15]. p53 is phosphorylated by a broad range of stress-activated kinases such as ATM, ATR, Chk1 and Chk2 while acetylation of p53 is carried out by transcriptional co-activators such as p300/CREB-binding protein (CBP) and p300/CBP-associated factor [16, 17].
p53 is part of a protein family that includes two other transcription factors, p63 and p73. They are similar in structure and biological function to p53. A high degree of homology in the DNA-binding domain has been reported among p53 family members. When overexpressed, both p63 and p73 have been shown to cause cell cycle arrest and apoptosis. p63 and p73 activate a large number of p53 target genes, however each member is known to activate additional target genes independently. p63 and p73 have several isoforms with diverse functions. Gene knock-out mouse models have helped to identify the functional specificity of each family member, including a more restricted role for p63 as a guardian in the female germline [6, 18, 19].
The p53 gene is mutated in more than half of all human cancers [1–5, 7, 12, 20]. Loss of WT p53 function is critical for the progression of human cancers [1–4, 6, 7, 19]. Mutations may lead to conformational changes in p53 that disrupt binding to DNA response elements and therefore target gene transcription [3]. Many tumors overexpress mutant (mt) p53, which contributes to increased resistance to chemotherapy and radiation as compared to tumors that do not over-express mt p53. Alternatively, some p53 mutations lead to a gain-of-function phenotype [4]. In human cancers with wild-type (WT) p53, post-translational modification of p53 as well as p53 signaling pathways are disrupted. Very few mutations in p63 and p73 have been reported in human tumors. p63 and p73 are sometimes inactivated in tumors by complexes formed with mt p53 or by antagonistic effects of ΔNp63 or ΔNp73 (N-terminal truncated isoforms), as well as epigenetic events. Certain isoforms of p63 and p73 are overexpressed in tumors, suggesting their oncogenic potential [1–4, 6, 7, 19]. p53 status of tumors correlates with prognosis and therapeutic response in the clinic [3, 4]. The effectiveness of chemotherapy and radiation treatments depends on functional WT p53 [2, 4]. Aberrant regulation of p53 due to overexpression of MDM2/MDM4 and deletion of ARF has also been related to tumorigenicity. Clearly, the p53 family is an important molecular target in human cancer and restoring WT p53 function, specifically in cancer cells, could provide novel and effective alternatives for cancer therapy [3, 4, 7].
2. p53 restoration for cancer therapy
Restoration of WT p53 function alone has been shown to cause tumor regression in mice through induction of apoptosis and senescence without affecting normal tissues [12, 21–24]. Cancer cells are more sensitive to effects of p53 restoration than normal cells. p53 restoration specifically in cancer cells maybe a useful therapeutic strategy that may also help to avoid the side effects associated with current chemotherapy [2]. WT p53 activity is essential for the efficacy of chemotherapy and radiation, thus p53 restoration compounds may be used in combination to enhance chemo- and radio-sensitivity [25]. p53 remains an attractive but challenging target for cancer therapy development considering its structure, physiological function and diverse modes of inactivation in cancer [2]. Due to the multifaceted regulation of p53 activity, therapeutic restoration of p53 can be achieved through multiple approaches that involve WT or mt p53, proteins that interact with p53, or other p53 family members (Figure 1).
Figure 1. Therapeutic strategies to restore wild-type p53 function.
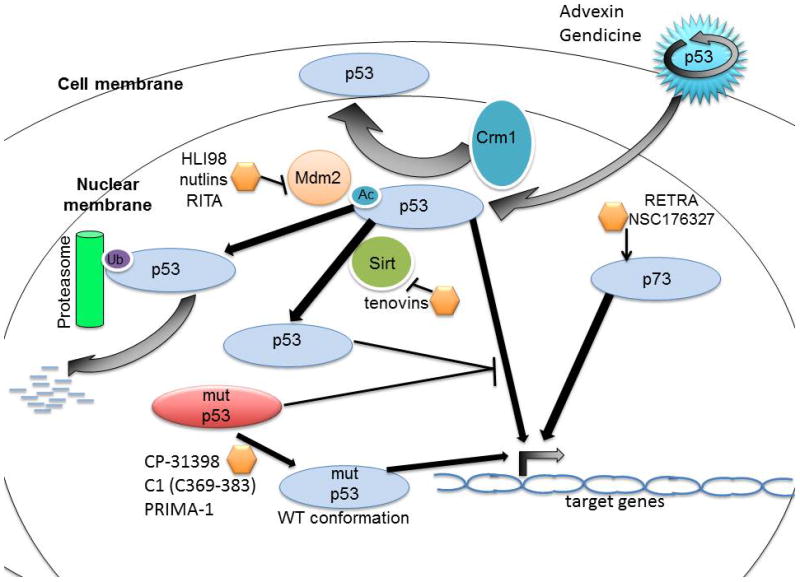
p53 is degraded under homeostatic conditions by its negative regulator MDM2, which is an E3 ubiquitin ligase. MDM2 monoubiquitinates p53 and consequently induces the proteasomal degradation of p53. Several small molecules that disrupt the interaction between p53 and MDM2 are being investigated including HLI98, the nutlins, RITA and others. The activity of wild-type p53 may also be restored by inhibiting Crm1-mediated nuclear export or by inhibiting Sirt-1 and Sirt-2, which deacetylate p53 to decreases its activity. The tenovins are a class of small molecules that inhibit Sirt. Mutant p53 can be therapeutically targeted using small molecules such as CP-31398 or PRIMA-1 or the peptide C1(C369–383) to a cause a conformational shift in the mutant p53 to restore WT conformation and therefore activity. Adenovirus-based therapies such as Advexin and Gendicine can also be used to deliver wild-type p53. NSC176327 and RETRA restore WT p53 function by increasing p73 levels.
2.1 Activation of wild-type p53
The first approach to p53 restoration is targeting the WT p53 pathway. Adenovirus-based gene therapies such as Advexin and Gendicine deliver the WT p53 gene into cancer cells. This approach is currently being tested in clinical trials [2]. Advexin has shown promising results in phase III clinical trials with respect to patient survival, tumor response and safety profile in the USA for head and neck cancer. Gendicine was approved for the treatment of head and neck cancer in China and is being further evaluated in clinical trials [26]. ONYX-015, an oncolytic adenoviral therapy, was developed to selectively replicate in cancer cells with dysfunctional p53 and cause tumor cell lysis [2]. However, studies later showed that the tumor selectivity of ONYX-015 is independent of p53 status of cells. Instead its effects related to mRNA export and protein synthesis have been shown to contribute to its selective replication in tumor cells [27, 28]. Several clinical studies have shown efficacy of ONYX-015 in the treatment of various tumor types. Clinical trials with ONYX-015 were discontinued in USA and randomized clinical trials were not completed. A similar oncolytic adenovirus H101 was approved for treatment of head and neck cancer in China [26, 28, 29]. The gene therapy and oncolytic adenoviral therapy may be used to treat tumors expressing WT p53 and mt p53. However, mt p53 can oligomerize with the expressed WT p53 to effectively cause p53 inactivation. Hence, the expression level of WT p53 must be higher than that of the endogenous mt p53 [7].
Other compounds used to target the WT p53 pathway include tenovins, nuclear export inhibitors and HLI98 [2]. Tenovin-1 and its water soluble analog, Tenovin-6, prevents deacetylation of p53 by inhibiting Sirt-1 and Sirt-2. p53 acetylation enhances its DNA binding and interferes with MDM2-mediated degradation of p53 [2, 4]. Nuclear export inhibitors target the export protein CRM1 to increase p53 protein levels. HLI98 inhibits the E3 ubiquitin ligase activity of HDM2 preventing p53 degradation [2]. Small molecules such as nutlins, MI-219, JNJ-26854165 and RITA have been used to target the protein-protein interaction of WT p53 with its negative regulator MDM2. Nutlins disrupt this interaction by occupying the binding pocket for p53 in MDM2. Nutlin RG7112 is being tested in clinical trials for advanced solid tumors and hematologic malignancies. Preliminary results have shown acceptable safety profiles. MI-219 binds with high affinity and selectivity to MDM2 while RITA interacts with WT p53. JNJ-26854165 binds the RING domain of MDM2 and prevents interaction of the MDM2-p53 complex with the proteasome. The initial results of phase I clinical trials for refractory solid tumors are encouraging as JNJ-26854165 was well tolerated at clinically effective doses. NSC279287, NSC66811 and terphenyl compounds are also small molecules targeting the MDM2-p53 interaction. TDP521252, TDP665759, PXN727, PXN822 and isoindolinones are other compounds currently under pre-clinical development that target MDM2 to increase p53 levels by inhibiting the MDM2-p53 interaction [2, 4, 7, 30].
Targeting the MDM2-p53 interaction may be problematic. MDM2 is a p53 target gene and targeting the MDM2-p53 interaction may eventually lead to MDM2 induction, thereby limiting efficacy. Considering the pro-oncogenic effects of p53 mutations, interfering with MDM2-mediated mt p53 degradation would be problematic for such therapies [4]. Recently, the compounds SJ172550 and XI-006 have been shown to restore WT p53 by modulating MDMX. SJ172550 interferes with MDMX-p53 interaction while XI-006 decreases MDMX transcription [4, 7].
2.2 Targeting p53 family member p73
Activating p73 can also restore WT p53-like function. p73 plays a crucial and specific role in tumor suppression along with p53. Unlike p53, p73 is not frequently mutated in cancer cells and therefore this therapeutic approach could be useful for the treatment of tumors expressing WT p53 as well as mt p53. [2, 6, 18]. This has been exploited for p53-like restoration with compounds such as NSC176327, RETRA, NSC143491 and NSC254681. NSC176327 exerts anti-tumor effects by increasing p73 protein expression. NSC176327 also increased target genes of p53 such as DR5 and p21 without inducing DNA-damage signaling [31]. Similarly, NSC143491 and NSC254681 were shown to restore p53 pathway transcription in a p73-dependent manner. However, DNA-damage signaling may be involved in the anti-tumor effects of these compounds [32]. The small molecule RETRA increases p73 expression and blocks the inhibitory effect of mt p53 on p73 by preventing protein-protein interactions [33]. Mammalian target of rapamycin (mTOR) is also known to regulate p73 levels and the mTOR inhibitor rapamycin has been shown to increases p73 levels in cancer cells [34].
2.3 Reactivation of mt p53
The second approach for restoration of p53 in tumors is reactivation of mt p53, which is a tumor-specific target. Due to its prevalence in cancer, restoring WT p53 function in cancer cells harboring mt p53 could have therapeutic impact on 50% of all human cancers. Although a large number of p53 mutations have been identified in various cancers, the majority of the mutations in the p53 gene in human cancer occur in the core DNA-binding domain. Small-molecules that specifically bind to the DNA-binding domain in mt p53 and stabilize the WT conformation would be useful for p53 restoration [2, 4]. Such compounds would be effective in a broad spectrum of tumors as the antitumor activity would be irrespective of the particular mutation present. This approach is not expected to affect p53 in normal cells, owing to its proper conformation and continuous MDM2-mediated degradation [4].
Various peptides and compounds have been identified for reactivation of mt p53. The peptide C1 (C369–383) activates p53 by allosteric stabilization of mt p53 WT conformation. 9-hydroxy-ellipticine induces apoptosis in tumor cells in a mt p53-dependent manner [2]. The small molecule CP-31398 restores mt p53 WT conformation and represses tumor growth in mice [2, 4]. The compound PRIMA-1 forms adducts with thiol groups in the core DNA-binding domain of mt p53 to reactivate wt p53 function and inhibit tumor growth in vivo. PRIMA-1 is now in phase I clinical trials [2, 4, 7]. The compounds MIRA-1, WR2721 (amifostine) and peptide CDB3 are also known to re-activate mt p53 but the exact molecular mechanisms are unclear. PhiKan083, a small molecule, binds to a specific mt p53 commonly found in tumors and stabilizes its WT conformation [2, 4]. Another small molecule, SCH529074, enhances the DNA-binding activity of mt p53 and reduces growth of xenograft tumors [4]. Recently, NSC319726 was shown to selectively kill cancer cells expressing the p53R175 mt in vitro and in vivo. The small molecule specifically restores WT p53 function for the p53R175 mutation based on reactive oxygen species changes and chelation of zinc ions [35]. Problems with delivering peptides to tumors have hindered their progress to the clinic. Continued identification of compounds for mt p53 reactivation is ongoing with multiple approaches that include cell-based assays, protein-binding assays, computational techniques and in silico screens [2, 4].
3. Cancer stem cell (CSC) hypothesis
The CSC hypothesis is based on the concept that, similar to normal proliferative tissues, tumors are composed of a heterogeneous population of cells with varying capacity for self-renewal. Tumors mainly consist of cells that are incapable of self-renewal, such as rapidly proliferating cells and post-mitotic differentiated cells. Emerging evidence suggests that tumors also contain a smaller population of stem/progenitor cells (CSCs) capable of self-renewal that are essential for long-term sustenance of the tumor [36]. It is believed that CSCs can not only self-renew, but are also quiescent, resistant to chemotherapy and radiation, and capable of migration that drives metastasis. The CSC hypothesis indicates that selective targeting of CSCs in combination with chemotherapy and radiation treatment could lead to more effective treatment regimens for cancer [36–39].
Recently, the CSC hypothesis has been extensively investigated and has generated considerable interest amongst oncologists and cancer biologists with regard to its therapeutic relevance. However, the biological origins of CSCs are unclear and a number of challenges and controversies have surfaced [36, 38, 40]. CSCs could arise from accumulation of genetic damage in normal tissue stem cells/progenitor cells or by dedifferentiation of existing cancer cells that may arise from differentiated cells or progenitor cells [38, 39]. A CSC may not necessarily be the cell of origin for a tumor. Some evidence even indicates that the CSC phenotype may not be a stable trait and that selective targeting of CSCs for therapy should be reconsidered [36].
The validity of preclinical models for studying CSCs remains an area of contention. The tumor-sphere assay fails to account for the importance of extracellular matrix interactions [38]. Xenograft assays used to define CSCs have been criticized due to the associated cellular stress and the fact that stem cell behavior may change following transplantation and therefore, the assay may not reveal the true fate of CSCs in the original tissue [36]. Lack of site-specific extracellular matrix interactions and immune cells may also affect tumorigenicity of transplanted cells [38]. There is a need to develop novel assays for studying CSCs avoiding the use of cell sorting and transplantation, especially to clarify the existence of CSCs in primary tumors [36].
The differences between CSCs and normal stem cells need to be further investigated [38]. The CSC markers used for solid tumors have generated conflicting results pointing towards a need for understanding stem cell biology of these tissues under physiological conditions. Whether there is a group of markers that define CSCs in enriched or divergent populations needs further clarification. CSC frequency calculations have generated some debate about the therapeutic relevance of the CSC hypothesis [36]. There is also an unmet need to understand the relationship between the CSC hypothesis and the clonal evolution model in most malignancies [36, 38]. On the positive side, significant progress in the field has emerged. Recent studies have clarified the existence of a CSC phenotype in squamous skin tumors [41], intestinal adenomas [42] and glioblastoma [43] using mouse models. These studies do not rely on transplantation assays in immunocompromised mouse models for detection of CSCs. Instead, genetic lineage tracing was employed to validate the CSC hypothesis. Clinical studies have uncovered the existence of chemotherapy-resistant CSCs and the importance of CSCs in metastasis [36, 38]. Other studies have identified approaches to selectively target CSCs [37, 38]. Further work in understanding the biology of CSCs and their clinical relevance will enable development of CSC-targeting therapeutics for cancer patients [38, 40].
4. p53 and stem cells
Recent evidence suggests that the p53 protein plays a key role in stem cell biology. p53 regulates stem cell differentiation and self-renewal, signaling pathways known to influence stem cell self-renewal, hematopoietic stem cell quiescence and multidrug resistance (Figure 2).
Figure 2. p53 inactivation enriches for stem cells.
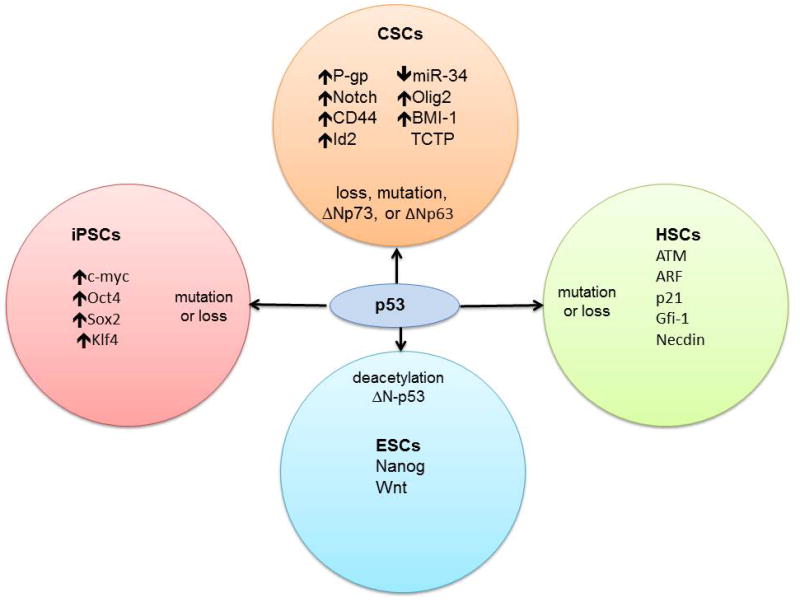
Inactivation of p53 by deletion, mutation, or dominant-negative isoforms of p53 family members enriches stem cell populations that include cancer stem cells (CSCs), induced pluripotent stem cells (iPSCs) and hematopoietic stem cells (HSCs). p53 is known to regulate self-renewal and differentiation of embryonic stem cells (ESCs). Some of these genes such as P-gp and miR-34 are direct targets of p53 activity whereas others such as Notch and c-myc are transcription factors that crosstalk with the p53 pathway and drive stem-like characteristics. Described mechanisms of p53 inactivation are noted for each stem cell type along with genes that are important for the specific population and that intersect with the p53 pathway.
4.1 p53 and embryonic stem cells (ESCs)
ESCs are pluripotent stem cells that are derived from blastocysts and are of therapeutic value as they are capable of differentiating into all cell lineages in the body. During self-renewal ESCs maintain genetic stability by stringent mechanisms and have a low rate of spontaneous mutations. However, compared to somatic cells mouse ESCs usually have attenuated G1 and G2 phases of the cell cycle, have higher level of p53 deacetylation and truncated ΔN-p53. As a result, DNA damage response pathways such as classical p53-mediated cell cycle arrest and apoptosis seem to be inefficient. Some studies report non-classical p53-dependent cell death in ESCs while other studies indicate p53-independent cell death and differentiation upon DNA damage. p53 suppresses self-renewal and pluripotency in response to certain types of DNA damage in mouse and human ESCs.
p53 also directly suppresses the expression of Nanog, a transcription factor that suppresses differentiation and maintains self-renewal of ESCs. Other p53 family members, p63 and p73, may also be involved in suppressing Nanog expression. Thus, in DNA-damaged ESCs p53 induces differentiation of ESCs into other cell types that are responsive to p53-dependent apoptosis and cell cycle arrest. In addition, during ESC differentiation, p53 is known to suppress progenitor cell dedifferentiation into pluripotent stem cells. These findings suggest that p53 could play a vital role in cellular differentiation and may be involved in the differentiation of tissue stem cells following DNA damage. During cancer progression, loss of WT p53 function may hinder tissue stem cell differentiation following DNA damage and give rise to CSCs. Restoration of WT p53 function in cancer cells may restore tissue stem cell differentiation and suppress CSC genesis [1, 44–46].
Few studies have evaluated the effects of p53 restoration compounds in ESCs. Lee et al demonstrated that nutlin treatment resulted in p53-dependent Nanog-independent activation of Wnt signaling in mouse ESCs. However, p53-mediated Wnt activation was attenuated in neural progenitor cells derived from mouse ESCs [47]. Maimets et al reported that treatment with nutlin resulted in rapid differentiation of human ESCs along with p53 and p21 activation [48]. Thus, p53-mediated effects on differentiation seem to be context and cell-type dependent.
4.2 p53 and induced pluripotent stem cells (iPSCs)
Somatic cells can be reprogrammed into pluripotent stem cells with similar capabilities and therapeutic potential as ESCs. iPSCs are obtained from somatic cells with enforced expression of genes that act as reprogramming factors. Specific combinations of activated transcription factors such as Oct4, Sox2, Klf4, c-Myc, Nanog and other proteins such as Lin28 have been used to reprogram both mouse and human somatic cells into iPSCs. Oct4 and Sox2 seem to be indispensible for reprogramming [1, 49, 50]. Five recent papers suggest that p53 acts as a barrier to successful reprogramming of somatic cells and increased reprogramming frequency has been observed in p53-deficient cells [49–56]. ARF, an activator of p53 during oncogenic stress also suppresses induced pluripotency. p53-mediated cell cycle arrest and senescence have been shown to be involved in suppressing reprogramming while the role p53-mediated apoptosis is unclear [1, 50].
Reprogramming factors such as c-Myc and Klf4 are oncoproteins whereas Nanog, Oct4, Lin28 and Sox2 have oncogenic potential [1, 49, 50]. p53 inactivation seems to be a major function of these reprogramming factors [1, 56, 57]. Considering p53 plays an important role in maintaining genetic stability, the tumorigenicity of iPSCs lacking p53 is a major concern [1, 49, 50, 56]. Interestingly, mouse embryonic fibroblasts with mt p53 exhibited higher reprogramming efficiency and were more tumorigenic in vivo than p53 knock-out mouse embryonic fibroblasts [56, 58]. The similarities between cancer progression and cellular reprogramming may lead to new knowledge in cancer biology [49]. p53 blocks both cancer progression and pluripotency and serves as a link between cancer and stem cells [1, 50, 56]. Loss of WT p53 function is critical for cancer progression and the reprogramming factors such as c-Myc, Oct4, Sox2 and Klf4 are also overexpressed in human cancers [1, 49]. Thus, it has been hypothesized that p53 inactivation along with overexpression of reprogramming factors could lead to cancer cell dedifferentiation to generate CSCs [1, 49]. Preventing cellular dedifferentiation may be an important component of p53-mediated tumor suppression. p53 restoration compounds could be used to prevent cancer cell dedifferentiation and therefore to prevent the creation of CSCs [1, 50].
4.3 p53 and adult tissue stem cells
p53 plays a critical role in regulating the adult prostate, breast, neural, hematopoietic and epidermal stem cell/progenitor cell compartment. p53 loss increased the proliferation of epidermal stem cells with dysfunctional telomeres [59]. Simultaneous loss of p53 and Rb resulted in formation of prostate neoplasms in a region enriched for stem/progenitor cells [60]. p53 is expressed in neural stem cells (NSCs) and negatively regulates their proliferation, since p53 loss resulted in increased growth of neurosphere cultures [61, 62]. Loss of p53 in neurosphere cultures resulted in a differentiation pattern that was different from that of WT cultures [63]. These studies clearly indicate that p53 regulates NSC proliferation and differentiation [62, 64]. p53 and various members of the p53 pathway such as ATM, ARF and p21 are known to regulate hematopoietic stem cell (HSC) abundance and function [46, 65, 66].
4.4 p53 and stem cell signaling pathways
Wnt, Notch and Hedgehog signaling pathways are known to regulate the self-renewal of normal stem cells [67]. Dysregulation of these pathways is associated with cancer [68–72] and these pathways have been considered as therapeutic targets for CSCs [73–75]. Cross-talk of p53 with Wnt, Notch and Hedgehog signaling pathways may be one of the links between p53 and CSCs. The Wnt signaling pathway has been identified as a major target of p53 in mouse ESCs. Activated Wnt signaling is known to inhibit mouse ESC differentiation. Lee et al have shown that in mouse ESCs with UV-mediated DNA damage, anti-differentiation mechanisms are activated that depend on p53 and Wnt signaling [47]. As described above, p53 is also known to induce differentiation of mouse ESCs by repression of Nanog expression [1]. p53 seems to be the key regulator of mouse ESC differentiation that maintains a balance between stem cell population size and genomic stability [47]. Recently, Tao et al reported that p53 loss results in an increased number of mammospheres in vitro and an increase in stem cell frequency in vivo. The increased number of mammospheres upon p53 loss was reduced by treatment with a γ-secretase inhibitor commonly used to inhibit Notch activation. Interestingly, mammosphere cultures were resistant to radiation-induced apoptosis. The data indicates that p53 regulates Notch signaling to maintain a constant mammary stem cell pool independent of p53-mediated apoptosis. An increase in mammary stem cells upon p53 loss could due to elevated Notch signaling [76].
4.5 p53 and HSC quiescence
Liu et al have shown that p53 regulates HSC quiescence during steady-state hematopoiesis [77]. HSC quiescence is required to maintain a population of HSCs that are resistant to cytotoxic agents, oxidative stress and radiation to ensure the essential production of blood cells. In p53-deficient mice, the proportion of quiescent HSCs is decreased but the overall HSC pool size is increased. These p53-mediated effects are independent of p21 but dependent on other direct targets such as Gfi-1 and Necdin. However, p21 has been reported to regulate HSC quiescence in other studies [78]. Thus, p53 negatively regulates mouse HSC self-renewal and promotes quiescence of mouse HSCs [77, 79, 80]. It is possible that during leukemia initiation, hematopoietic progenitor cells may acquire the ability to self-renew with loss of WT p53 function. Since quiescence can contribute to resistance to chemotherapy and radiation treatment it is possible that p53 mutations in leukemia do not affect the ability of p53 to promote quiescence of stem/progenitor cells [80].
4.6 p53, MDR1 and stem cells
Human multidrug resistance-1 (MDR1) is expressed in multidrug-resistant cancer cells [19]. P-glycoprotein (P-gp), encoded by MDR1, belongs to a larger family of transport proteins called the ATP-binding cassette (ABC) transporters. Many small molecule chemotherapeutic and targeted agents are substrates of ABC transporters, resulting in their efflux from cells and conferring multidrug resistance. ABC transporters also efflux many other small molecules including fluorescent dyes such as Hoechst 33342, which can be detected by flow cytometry to identify the rare dye-effluxing sub-population known as the side-population (SP). SP analysis is commonly used to identify stem cells in normal and tumor tissues of the bone marrow, brain and mammary gland. Overexpression of P-gp in murine bone marrow cells caused expansion of SP cells [81–83].
WT p53 acts as a transcriptional repressor of the MDR1 gene. Interestingly, p63 and p73 have been reported to activate MDR1. The differential effects of p53 family members may be due to binding to different regions of the MDR1 promoter [19]. Also, ΔNp73, a truncated isoform of p73 has been shown to activate MDR1 in human gastric carcinoma by inhibiting WT p53 function [84]. Restoration of wt p53 function independent of p63/p73 in CSCs could reduce P-gp activity and therefore chemotherapy efflux. This would support greater efficacy and reduced toxicity from chemotherapy or targeted therapy used in combination with p53 restoration compounds.
4.7 p53 family members, stem cells and differentiation
p63 regulates epithelial stem/progenitor cell proliferation and differentiation while p73 is important for NSC renewal and neural precursor cell differentiation. Epidermal progenitor cells predominantly express ΔNp63, which is critical for their maintenance. p63 is known to regulate pathways involved in stem cell self-renewal and differentiation such as Wnt, Notch and Hedgehog signaling. p63 is also known to induce the expression of CD44, a commonly used marker for breast CSCs. ΔNp63 overexpression results in Notch pathway activation and expansion of the CSC pool.
Regulation of differentiation by the p53 family is thought to be a part of the tumor suppressor activity [46, 85–88]. p53 regulates the differentiation of neurons, macrophages and other cell types. Accumulation of p53 mutation in cancer cells is often associated with an undifferentiated state. Also, ΔNp73 expression in tumors could disrupt p53 and p73 regulation of differentiation. Thus, the p53 family contains proteins with various isoforms that have antagonistic effects on differentiation. Consequently, a homeostatic balance between proliferation of stem/precursor cells and differentiation can be maintained. Disruption of this balance by p53 mutation or overexpression of ΔNp73 could lead to tumor formation. p53 restoration compounds could be useful in repairing defective differentiation in tumor cells [6]. Nutlin 3 has been shown to induce neuronal differentiation in neuroblastoma cell lines in a p53-dependent manner [89]. Cellular differentiation is promoted by p53 activation in various other cancers [46].
5. p53 and CSCs
Recent studies on the direct and indirect links between the CSC population and p53 have further solidified the importance of therapeutic targeting of p53 in CSCs.
5.1 p53 loss and CSCs
Several studies have delineated the effects of p53 loss and p53 mutation on the CSC pool. Committed myeloid progenitors expressing oncogenic KRAS were transformed into leukemic stem cells (LSCs) capable of initiating acute myeloid leukemia upon p53 inactivation [90]. Breast cancers with mt p53 or functionally inactive wt p53 were highly associated with transcriptional patterns representing inhibited differentiation and stem-like characteristics [91]. p53 loss was correlated with poorly differentiated tumors in thyroid, lung, skin and colorectal cancers [46]. p53 loss promotes the expression of CD44 [87] and elevates levels of the target gene, Inhibitor of DNA binding 2 (Id2), resulting in increased self-renewal of neural progenitor cells. Id2 levels are elevated in glioma cell lines with mt p53 and overexpression of Id2 results in enhanced proliferation of glioma stem cells [92]. Simultaneous inactivation of p53 and PTEN stimulates c-Myc expression and enhances the self-renewal and differentiation of NSCs. c-Myc inactivation in tumor neurospheres lacking p53 and PTEN restored differentiation and suppressed tumorigenic potential [93].
5.2 p53, BMI-1 and CSCs
ARF is known to regulate the p53 pathway and, similarly, the tumor suppressor p16Ink4a (Ink4a) regulates the Rb pathway. Crosstalk of the Rb and p53 pathway occurs via E2F-1 as described above. Ink4a/Rb and ARF/p53 signaling are frequently disrupted in tumors due to their role in tumor suppression, cell growth and proliferation. Ink4a and ARF are known to regulate self-renewal of murine HSCs while Rb is essential for the maintenance of murine placenta trophoblast stem cells [94]. BMI-1, an oncogene, represses Ink4a and ARF expression and is essential for the maintenance of murine HSCs and NSCs. Ink4a and ARF are important for mediating the effects of BMI-1 on the stem cell population. BMI-1 is essential for the proliferation of LSCs and is highly expressed in CSCs obtained from brain tumors [94–97]. It has been demonstrated that BMI-1 mediates the effects of Hedgehog signaling pathway on mammary stem cell self-renewal. Both Hedgehog signaling and BMI-1 are activated in breast CSCs [98]. BMI-1 could serve as one of the links between Hedgehog and p53 signaling in CSCs. Thus, the complex regulatory network of p53 may play a critical role in regulating the CSC pool and may provide alternative therapeutic opportunities.
5.3 p53, miR34 and CSCs
The miR34 family of microRNAs are transcriptionally regulated by p53 and have been linked to CSCs. The tumor suppressor miR34 family regulates target proteins that include Notch pathway proteins and Bcl-2 [99]. miR34a negatively regulates CD44 expression in prostate cancer cells and inhibits prostate CSC properties [100]. Nalls et al recently reported that miR34a expression was reduced in pancreatic CSCs. Epigenetic restoration of miR34a with chromatin-modifying agents reduced growth, induced apoptosis and inhibited self-renewal, epithelial-mesenchymal transition, invasion, migration, colony and spheroid formation in pancreatic CSCs [101]. Similarly, upon lentiviral restoration of miR34, Ji et al reported reduction in pancreatic and gastric CSCs with mt p53, high Bcl-2 expression and low miR34 [99, 102]. Thus, these studies demonstrate a potential for therapeutic targeting of CSCs by activation of the miR34 family that includes important downstream targets of p53.
5.4 p53, Translationally Controlled Tumor Protein (TCTP) and CSCs
TCTP is a critical regulator of apoptosis and is highly expressed in cancer cells [103]. TCTP has been previously described in the regulation of Oct4 and Nanog transcription during nuclear reprogramming [104]. Recently, Amson et al described a negative feedback regulatory loop between TCTP and p53 whereby TCTP promotes MDM2-mediated degradation of p53 while p53 represses TCTP gene transcription [103]. PKH26 dye retention has been described previously as a useful method to isolate normal and tumor mammary stem cells [105]. The authors demonstrate that TCTP is highly expressed in PKH26 positive cells as compared to the PKH26 negative fraction. Knockdown of TCTP in breast tumor mammospheres resulted in increased p53 levels and reduced tumor mammosphere formation efficiency. Thus, TCTP knockdown represents another mechanism for p53 restoration and targeting CSCs. However, p53 restoration by TCTP knockdown is mediated by MDM2 [103] and it remains to be seen whether targeting the TCTP pathway provides an advantage over currently available compounds disrupting MDM2-mediated p53 degradation.
5.5 p21, Olig2 and glioma stem cells
Olig2, a transcription factor expressed in the central nervous system regulates the proliferation of NSCs. Olig2 is highly expressed in glioma progenitor/stem cells and is essential for the growth of gliomas obtained from tumor neurospheres. In NSCs and glioma cells, Olig2 directly represses the p53 target gene p21 [106], which negatively regulates the proliferation of NSCs [107]. Thus, p53 restoration could help target glioma stem cells by preventing Olig2-mediated p21 repression.
5.6 Mt p53, mevalonate pathway and breast CSCs
The mevalonate metabolic pathway is involved in important cellular processes such as sterol biosynthesis, hormone production and maintenance of cell membrane integrity. Mevalonate pathway intermediates such as geranylgeranyl pyrophosphate are involved in post-translational modification of Ras and Rho proteins. The mevalonate pathway plays a key role in various stages of tumorigenesis. Several inhibitors of the mevalonate pathway are being evaluated as anti-cancer therapies in preclinical and clinical settings [108, 109]. Recently, mt p53 was shown to be associated with upregulation of genes involved in the mevalonate pathway. The mevalonate pathway mediates the disruption of acinar morphology in breast tissues by mt p53. Protein geranylgeranylation was shown to play an important role in these mt p53-mediated effects [110]. Another study identified mevalonate metabolism as a key regulator of breast CSCs. Inhibition of mevalonate pathway with simvastatin and inhibition of protein geranylgeranylation depleted basal breast CSCs expressing mt p53 [111]. Thus, mt p53 potentially regulates breast CSCs, at least in part, through mevalonate metabolism [112]. These studies provide further support for the gain-of-function phenotype of mt p53 and emphasize the importance of targeting mt p53 for cancer therapy.
5.7 p53, epithelial-mesenchymal transition and CSCs
The epithelial-mesenchymal transition (EMT) enables cancer cells to acquire the abilities of invasion, migration and resistance to apoptosis such that they can orchestrate the metastatic cascade. EMT is regulated by a well-defined set of transcription factors such as Snail, Slug, Twist and Zeb1 that repress the epithelial adhesion molecule E-cadherin. The loss of an epithelial trait is accompanied by acquisition of a mesenchymal phenotype, involving up-regulation of N-cadherin and Vimentin expression. This mesenchymal phenotype enables cancer cells to migrate and invade surrounding tissue and resist apoptosis [39, 113]. During later stages of metastasis cancer cells must adapt to a foreign tissue and self-renew to give rise to micro- and macro-metastasis. These properties are associated with a stem cell phenotype [114]. Recent evidence suggests that the EMT program induces a stem cell phenotype in normal epithelial cells as well as epithelial cancer cells [115]. Several studies have confirmed that the EMT process can give rise to CSCs [116–119]. Thus, EMT potentially causes dedifferentiation of more differentiated cancer cells giving rise to CSCs [114].
p53 has been shown to negatively regulate EMT and stem cell properties [120] and furthermore, p53 mutation has been demonstrated to promote EMT [121, 122]. p53 directly regulates mir-200c expression, which suppresses the EMT and CSC phenotypes by inhibition of Zeb1 and BMI-1 [120]. Recently, it has also been shown that p73 negatively regulates EMT [123]. Thus, the p53 family is potentially involved in the regulation of an EMT-associated CSC phenotype.
5.8 p53 restoration and CSCs
Few studies have directly evaluated the effects of p53 restoration on the CSC population. Li et al recently identified the deacetylase SIRT1 as an important therapeutic target in chronic myelogenous leukemia stem cells (CMLSC). CMLSC depend on p53 deacetylation mediated by SIRT1 for survival and resistance to imatinib. Inhibition of SIRT1 by tenovin-6 resulted in enhanced p53 acetylation in CMLSC and inhibited imatinib-resistant CMLSC patient cells in vivo. The combination of imatinib and tenovin-6 impaired CMLSC growth and in vivo engraftment without affecting normal cells [124, 125].
Cicalese et al demonstrated that p53 loss results in increased stem cell frequency, increased frequency of symmetric self-renewing divisions and increased replicative potential in mammary stem cells. p53 loss also enhances Nanog expression in mammary stem cells. p53 restoration with Nutlin 3 treatment reduced the frequency of mammary CSCs by limiting their replicative potential and symmetric division without affecting wt mammary stem cells [126].
Allen et al developed a non-toxic fluorescent assay utilizing Calcein AM for the detection of colon cancer stem cells. This assay is similar to the Hoechst SP assay in principle but provides the advantage of being non-toxic to the side population, potentially improving the in vitro and in vivo evaluation of CSCs by enabling longitudinal studies using a functional assay. Using this assay system along with SP analysis, Allen et al demonstrated that loss of wt p53 function enriches putative colon CSCs. Restoration of wt p53 in p53-null HCT116 cells using an adenoviral vector eliminated the dye-effluxing CSC population [83].
Using a single cell p53-regulated green fluorescent protein-reporter system in DLD-1 human colon tumor cells that express mt p53 protein, Huang et al have demonstrated p53 restoration by ellipticine in putative colon CSCs. The chemotherapeutic drug 5-Fluorouracil (5-FU) caused enrichment of dye-effluxing colon CSCs. Huang et al have also reported depletion of p53-null and mt-p53 expressing putative colon CSCs by ellipticine in combination with 5-FU [127].
6. Conclusion
It is clear that regulation of stem cell self-renewal and differentiation forms an important part of the tumor suppressive function of p53. The CSC population is known to contribute to therapeutic resistance, tumor recurrence and metastatic relapse. The regulation of p53 is multimodal, complex and often disrupted in human cancer and is intimately linked to CSCs. Restoration of WT p53 activity represents a promising tumor-specific regimen capable of targeting the therapy-resistant CSC population.
7. Expert Opinion
The emerging field of p53 restoration for targeting CSCs hasprovided a new dimension to our vast understanding of the role of p53 in cancer. The field illustrates the complexities involved in the relation between stem cells and cancer [46]. Targeting the CSC population should be approached with caution as this cellular pool is susceptible to phenotypic changes and plasticity and effects on normal stem cells may impart toxicity and impact the therapeutic index. The simplistic model of the existence of a uniform cancer stem cell pool is now obsolete. It is possible that the transient pool of CSCs can shuttle between differentiated and stem like states as evidenced by studies involving cellular reprogramming. It has been difficult to uniformly quantify the CSC population and characterize it with definitive markers [39, 46]. Understanding the molecular and cellular pathways that define this transient population could lead to novel targets for cancer therapy [46]. More insight into molecular markers and groups of markers that define enriched CSC populations is needed.
From the various approaches for p53 restoration it is clear that personalized medicine would be critical for the success of p53 restoration in the clinic. Genetic diagnosis may aid in identification of compounds to be used for each patient [2]. p53 is capable of activating pro-survival pathways by expression of anti-apoptotic genes, protection of cells from oxidative stress and by facilitating DNA repair pathways. Concerns over any possible pro-survival effects in tumor cells following p53 restoration need to be addressed. The effect of p53 restoration compounds on normal cells and tissues needs to be further evaluated [4, 5, 12, 95, 128, 129]. In vitro and in vivo experiments indicate that p53 activation may contribute to organismal aging due to reduction in adult tissue stem cell function [12, 130, 131]. Combinations of p53 restoration compounds with chemotherapeutic agents need to be further evaluated. p53 restoration compounds may sensitize cancer cells to chemotherapy and radiation treatments to yield clinically relevant combinations with reduced toxicity. Also, in order to overcome new p53 mutations it is essential to constantly identify novel small molecules capable of targeting them. As cell-based and biochemical screens are implemented to develop novel approaches to target p53 signaling, small-molecules for p53 restoration will eventually progress to the clinic [2, 4].
Several studies have uncovered the role of p53 in regulating adult tissue stem cells. Few studies have determined the effects of restoration of p53 in cancer stem cells. Methods used to make these conclusions such as transplantation involve cellular stresses that can induce p53. Thus the effects of p53 loss on the stem cell pool need to be carefully interpreted as they may represent a lack of p53-dependent cell cycle arrest and apoptosis rather than effects on self-renewal and differentiation. Currently, most methods such as mammosphere cultures rely on detection of an enriched population of CSCs rather than a pure population due to limitations in the specificity of markers. This may affect the calculation and interpretation of stem cell frequency and number [46]. Considering these experimental challenges, there is a need to identify robust markers for in vivo non-invasive imaging and quantification of tissue and cancer stem cells at the single cell level. This would enable unbiased interpretation of regulation of tissue and CSCs by p53.
Article Highlights.
The prevalence of p53 mutations in human cancer and the ability of p53 to induce apoptosis and senescence selectively in tumors make p53 restoration an attractive therapeutic strategy. Advexin and ONYX-105, which are adenovirus-based therapies that deliver wild-type p53, are in late stage clinical trials. Wild-type p53 can also be stabilized to upregulate its activity by inhibiting the E3 ubiquitin ligase MDM2 as exemplified by the small molecule nutlins.
Reactivation of mutant p53 by causing a shift to the wild-type conformation is being explored with small molecules such as PRIMA-1, CP31398 and the peptide CDB3.
p63 and p73 are members of the p53 family that also induce several p53 target genes while possessing other exclusive target genes. p73 is not typically mutated in tumors and therefore is a feasible target for restoring p53 function. This modality of p53 restoration is utilized by the small molecules NSC176327 and RETRA.
p53 generally suppresses stem cells as exemplified by p53-mediated suppression of Nanog, a transcription factor that inhibits differentiation. p53 is also a critical barrier in creating induced pluripotent stem cells while loss of p53 increases the number of neurospheres and mammospheres in vitro.
Restoration of p53 is expected to eradicate the cancer stem cell population as mutations or loss of p53 has been directly linked to induction or enrichment of cancer stem cells. Furthermore, targets of p53 such as MDR1, miR34, and TCTP intersect with signaling pathways that are essential for cancer and normal stem cells.
Footnotes
Declaration of interest
The authors state no conflict of interest and have received no payment in preparation of this manuscript.
References
(Papers of particular interest have been highlighted as: • = of interest, •• = of considerable interest)
- 1.Zhao T, Xu Y. p53 and stem cells: new developments and new concerns. Trends Cell Biol. 2010;20:170–5. doi: 10.1016/j.tcb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Chen F, Wang W, El-Deiry WS. Current strategies to target p53 in cancer. Biochem Pharmacol. 2010;80:724–30. doi: 10.1016/j.bcp.2010.04.031. [DOI] [PubMed] [Google Scholar]
- 3.Wang W, El-Deiry WS. Restoration of p53 to limit tumor growth. Curr Opin Oncol. 2008;20:90–6. doi: 10.1097/CCO.0b013e3282f31d6f. [DOI] [PubMed] [Google Scholar]
- 4.Mandinova A, Lee SW. The p53 pathway as a target in cancer therapeutics:obstacles and promise. Sci Transl Med. 2011;3:64rv1. doi: 10.1126/scitranslmed.3001366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–30. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stiewe T. The p53 family in differentiation and tumorigenesis. Nat Rev Cancer. 2007;7:165–8. doi: 10.1038/nrc2072. [DOI] [PubMed] [Google Scholar]
- 7•.Essmann F, Schulze-Osthoff K. Translational approaches targeting the p53 pathway for anti-cancer therapy. Br J Pharmacol. 2012;165:328–44. doi: 10.1111/j.1476-5381.2011.01570.x. Recent description of various approaches for p53 restoration in cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sax JK, Fei P, Murphy ME, et al. BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol. 2002;4:842–9. doi: 10.1038/ncb866. [DOI] [PubMed] [Google Scholar]
- 9.el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 10.Wu GS, Burns TF, McDonald ER, 3rd, et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet. 1997;17:141–3. doi: 10.1038/ng1097-141. [DOI] [PubMed] [Google Scholar]
- 11.Sax JK, El-Deiry WS. p53 downstream targets and chemosensitivity. Cell Death Differ. 2003;10:413–7. doi: 10.1038/sj.cdd.4401227. [DOI] [PubMed] [Google Scholar]
- 12.Stegh AH. Targeting the p53 signaling pathway in cancer therapy - the promises, challenges and perils. Expert Opin Ther Targets. 2012;16:67–83. doi: 10.1517/14728222.2011.643299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin S, Levine AJ. The p53 functional circuit. J Cell Sci. 2001;114:4139–40. doi: 10.1242/jcs.114.23.4139. [DOI] [PubMed] [Google Scholar]
- 14.Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10:699–703. doi: 10.1093/hmg/10.7.699. [DOI] [PubMed] [Google Scholar]
- 15.Tang Y, Zhao W, Chen Y, et al. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–26. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y, Lu S, Wu L, et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1) Mol Cell Biol. 2006;26:2782–90. doi: 10.1128/MCB.26.7.2782-2790.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–22. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collavin L, Lunardi A, Del Sal G. p53-family proteins and their regulators: hubs and spokes in tumor suppression. Cell Death Differ. 2010;17:901–11. doi: 10.1038/cdd.2010.35. [DOI] [PubMed] [Google Scholar]
- 19.Johnson RA, Shepard EM, Scotto KW. Differential regulation of MDR1 transcription by the p53 family members. Role of the DNA binding domain. J Biol Chem. 2005;280:13213–9. doi: 10.1074/jbc.M414646200. [DOI] [PubMed] [Google Scholar]
- 20.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 21.Ventura A, Kirsch DG, McLaughlin ME, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–5. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 22.Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–34. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 23.Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kastan MB. Wild-type p53: tumors can’t stand it. Cell. 2007;128:837–40. doi: 10.1016/j.cell.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 25.Lu C, El-Deiry WS. Targeting p53 for enhanced radio- and chemo-sensitivity. Apoptosis. 2009;14:597–606. doi: 10.1007/s10495-009-0330-1. [DOI] [PubMed] [Google Scholar]
- 26.Lee J, Moon C. Current status of experimental therapeutics for head and neck cancer. Exp Biol Med (Maywood) 2011;236:375–89. doi: 10.1258/ebm.2010.010354. [DOI] [PubMed] [Google Scholar]
- 27.O’Shea CC, Johnson L, Bagus B, et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell. 2004;6:611–23. doi: 10.1016/j.ccr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 28.O’Shea CC, Soria C, Bagus B, McCormick F. Heat shock phenocopies E1B-55K late functions and selectively sensitizes refractory tumor cells to ONYX-015 oncolytic viral therapy. Cancer Cell. 2005;8:61–74. doi: 10.1016/j.ccr.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 29.Garber K. China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst. 2006;98:298–300. doi: 10.1093/jnci/djj111. [DOI] [PubMed] [Google Scholar]
- 30.Yuan Y, Liao YM, Hsueh CT, Mirshahidi HR. Novel targeted therapeutics: inhibitors of MDM2, ALK and PARP. J Hematol Oncol. 2011;4:16. doi: 10.1186/1756-8722-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu C, Wang W, El-Deiry WS. Non-genotoxic anti-neoplastic effects of ellipticine derivative NSC176327 in p53-deficient human colon carcinoma cells involve stimulation of p73. Cancer Biol Ther. 2008;7:2039–46. doi: 10.4161/cbt.7.12.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang W, Kim SH, El-Deiry WS. Small-molecule modulators of p53 family signaling and antitumor effects in p53-deficient human colon tumor xenografts. Proc Natl Acad Sci U S A. 2006;103:11003–8. doi: 10.1073/pnas.0604507103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kravchenko JE, Ilyinskaya GV, Komarov PG, et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc Natl Acad Sci U S A. 2008;105:6302–7. doi: 10.1073/pnas.0802091105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosenbluth JM, Mays DJ, Pino MF, et al. A gene signature-based approach identifies mTOR as a regulator of p73. Mol Cell Biol. 2008;28:5951–64. doi: 10.1128/MCB.00305-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu X, Vazquez A, Levine AJ, Carpizo DR. Allele-Specific p53 Mutant Reactivation. Cancer Cell. 2012;21:614–25. doi: 10.1016/j.ccr.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36•.Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313–9. doi: 10.1038/nm.2304. Includes elaborate discussions on various aspects related to the cancer stem cell hypothesis. [DOI] [PubMed] [Google Scholar]
- 37.Gupta PB, Onder TT, Jiang G, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–59. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shipitsin M, Polyak K. The cancer stem cell hypothesis: in search of definitions, markers, and relevance. Lab Invest. 2008;88:459–63. doi: 10.1038/labinvest.2008.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 40.O’Brien CA, Kreso A, Dick JE. Cancer stem cells in solid tumors: an overview. Semin Radiat Oncol. 2009;19:71–7. doi: 10.1016/j.semradonc.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 41.Driessens G, Beck B, Caauwe A, et al. Defining the mode of tumour growth by clonal analysis. Nature. 2012 doi: 10.1038/nature11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schepers AG, Snippert HJ, Stange DE, et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012;337:730–5. doi: 10.1126/science.1224676. [DOI] [PubMed] [Google Scholar]
- 43.Chen J, Li Y, Yu TS, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012 doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu Y. A new role for p53 in maintaining genetic stability in embryonic stem cells. Cell Cycle. 2005;4:363–4. doi: 10.4161/cc.4.3.1529. [DOI] [PubMed] [Google Scholar]
- 45.Lin T, Chao C, Saito S, et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;7:165–71. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- 46•.Spike BT, Wahl GM. p53, Stem Cells, and Reprogramming: Tumor Suppression beyond Guarding the Genome. Genes Cancer. 2011;2:404–19. doi: 10.1177/1947601911410224. Provides a comprehensive overview about p53 regulation of stem cells, cellular reprogramming and the CSC hypothesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee KH, Li M, Michalowski AM, et al. A genomewide study identifies the Wnt signaling pathway as a major target of p53 in murine embryonic stem cells. Proc Natl Acad Sci U S A. 2010;107:69–74. doi: 10.1073/pnas.0909734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maimets T, Neganova I, Armstrong L, Lako M. Activation of p53 by nutlin leads to rapid differentiation of human embryonic stem cells. Oncogene. 2008;27:5277–87. doi: 10.1038/onc.2008.166. [DOI] [PubMed] [Google Scholar]
- 49.Krizhanovsky V, Lowe SW. Stem cells: The promises and perils of p53. Nature. 2009;460:1085–6. doi: 10.1038/4601085a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Puzio-Kuter AM, Levine AJ. Stem cell biology meets p53. Nat Biotechnol. 2009;27:914–5. doi: 10.1038/nbt1009-914. [DOI] [PubMed] [Google Scholar]
- 51.Utikal J, Polo JM, Stadtfeld M, et al. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009;460:1145–8. doi: 10.1038/nature08285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marion RM, Strati K, Li H, et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460:1149–53. doi: 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H, Collado M, Villasante A, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460:1136–9. doi: 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kawamura T, Suzuki J, Wang YV, et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460:1140–4. doi: 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hong H, Takahashi K, Ichisaka T, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460:1132–5. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tapia N, Scholer HR. p53 connects tumorigenesis and reprogramming to pluripotency. J Exp Med. 2010;207:2045–8. doi: 10.1084/jem.20101866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol. 2005;7:1074–82. doi: 10.1038/ncb1314. [DOI] [PubMed] [Google Scholar]
- 58.Sarig R, Rivlin N, Brosh R, et al. Mutant p53 facilitates somatic cell reprogramming and augments the malignant potential of reprogrammed cells. J Exp Med. 2010;207:2127–40. doi: 10.1084/jem.20100797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Flores I, Blasco MA. A p53-dependent response limits epidermal stem cell functionality and organismal size in mice with short telomeres. PLoS One. 2009;4:e4934. doi: 10.1371/journal.pone.0004934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou Z, Flesken-Nikitin A, Nikitin AY. Prostate cancer associated with p53 and Rb deficiency arises from the stem/progenitor cell-enriched proximal region of prostatic ducts. Cancer Res. 2007;67:5683–90. doi: 10.1158/0008-5472.CAN-07-0768. [DOI] [PubMed] [Google Scholar]
- 61.Meletis K, Wirta V, Hede SM, et al. p53 suppresses the self-renewal of adult neural stem cells. Development. 2006;133:363–9. doi: 10.1242/dev.02208. [DOI] [PubMed] [Google Scholar]
- 62.Mendrysa SM, Ghassemifar S, Malek R. p53 in the CNS: Perspectives on Development, Stem Cells, and Cancer. Genes Cancer. 2011;2:431–42. doi: 10.1177/1947601911409736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Armesilla-Diaz A, Bragado P, Del Valle I, et al. p53 regulates the self-renewal and differentiation of neural precursors. Neuroscience. 2009;158:1378–89. doi: 10.1016/j.neuroscience.2008.10.052. [DOI] [PubMed] [Google Scholar]
- 64.Hede SM, Nazarenko I, Nister M, Lindstrom MS. Novel Perspectives on p53 Function in Neural Stem Cells and Brain Tumors. J Oncol. 2011;2011:852970. doi: 10.1155/2011/852970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Akala OO, Park IK, Qian D, et al. Long-term haematopoietic reconstitution by Trp53-/-p16Ink4a-/-p19Arf-/- multipotent progenitors. Nature. 2008;453:228–32. doi: 10.1038/nature06869. [DOI] [PubMed] [Google Scholar]
- 66.Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6:309–22. doi: 10.1016/j.stem.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Korkaya H, Wicha MS. Selective targeting of cancer stem cells: a new concept in cancer therapeutics. BioDrugs. 2007;21:299–310. doi: 10.2165/00063030-200721050-00002. [DOI] [PubMed] [Google Scholar]
- 68.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–98. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 69.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–50. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 70.Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008;15:801–12. doi: 10.1016/j.devcel.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nickoloff BJ, Osborne BA, Miele L. Notch signaling as a therapeutic target in cancer: a new approach to the development of cell fate modifying agents. Oncogene. 2003;22:6598–608. doi: 10.1038/sj.onc.1206758. [DOI] [PubMed] [Google Scholar]
- 72.Bolos V, Grego-Bessa J, de la Pompa JL. Notch signaling in development and cancer. Endocr Rev. 2007;28:339–63. doi: 10.1210/er.2006-0046. [DOI] [PubMed] [Google Scholar]
- 73.de Sousa EM, Vermeulen L, Richel D, Medema JP. Targeting Wnt signaling in colon cancer stem cells. Clin Cancer Res. 2011;17:647–53. doi: 10.1158/1078-0432.CCR-10-1204. [DOI] [PubMed] [Google Scholar]
- 74.Pannuti A, Foreman K, Rizzo P, et al. Targeting Notch to target cancer stem cells. Clin Cancer Res. 2010;16:3141–52. doi: 10.1158/1078-0432.CCR-09-2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Merchant AA, Matsui W. Targeting Hedgehog--a cancer stem cell pathway. Clin Cancer Res. 2010;16:3130–40. doi: 10.1158/1078-0432.CCR-09-2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tao L, Roberts AL, Dunphy KA, et al. Repression of mammary stem/progenitor cells by p53 is mediated by Notch and separable from apoptotic activity. Stem Cells. 2011;29:119–27. doi: 10.1002/stem.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu Y, Elf SE, Miyata Y, et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4:37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cheng T, Rodrigues N, Shen H, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–8. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 79.van Os R, de Haan G, Dykstra BJ. Hematopoietic stem cell quiescence: yet another role for p53. Cell Stem Cell. 2009;4:7–8. doi: 10.1016/j.stem.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 80.Liu Y, Elf SE, Asai T, et al. The p53 tumor suppressor protein is a critical regulator of hematopoietic stem cell behavior. Cell Cycle. 2009;8:3120–4. doi: 10.4161/cc.8.19.9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hirschmann-Jax C, Foster AE, Wulf GG, et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A. 2004;101:14228–33. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bunting KD, Zhou S, Lu T, Sorrentino BP. Enforced P-glycoprotein pump function in murine bone marrow cells results in expansion of side population stem cells in vitro and repopulating cells in vivo. Blood. 2000;96:902–9. [PubMed] [Google Scholar]
- 83.Allen JE, Hart LS, Dicker DT, et al. Visualization and enrichment of live putative cancer stem cell populations following p53 inactivation or Bax deletion using non-toxic fluorescent dyes. Cancer Biol Ther. 2009;8:2194–205. doi: 10.4161/cbt.8.22.10450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vilgelm A, Wei JX, Piazuelo MB, et al. DeltaNp73alpha regulates MDR1 expression by inhibiting p53 function. Oncogene. 2008;27:2170–6. doi: 10.1038/sj.onc.1210862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Joseph B, Hermanson O. Molecular control of brain size: regulators of neural stem cell life, death and beyond. Exp Cell Res. 2010;316:1415–21. doi: 10.1016/j.yexcr.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 86.Nekulova M, Holcakova J, Coates P, Vojtesek B. The role of p63 in cancer, stem cells and cancer stem cells. Cell Mol Biol Lett. 2011;16:296–327. doi: 10.2478/s11658-011-0009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Godar S, Ince TA, Bell GW, et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Du Z, Li J, Wang L, et al. Overexpression of DeltaNp63alpha induces a stem cell phenotype in MCF7 breast carcinoma cell line through the Notch pathway. Cancer Sci. 2010;101:2417–24. doi: 10.1111/j.1349-7006.2010.01700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Van Maerken T, Speleman F, Vermeulen J, et al. Small-molecule MDM2 antagonists as a new therapy concept for neuroblastoma. Cancer Res. 2006;66:9646–55. doi: 10.1158/0008-5472.CAN-06-0792. [DOI] [PubMed] [Google Scholar]
- 90.Zhao Z, Zuber J, Diaz-Flores E, et al. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev. 2010;24:1389–402. doi: 10.1101/gad.1940710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mizuno H, Spike BT, Wahl GM, Levine AJ. Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proc Natl Acad Sci U S A. 2010;107:22745–50. doi: 10.1073/pnas.1017001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Paolella BR, Havrda MC, Mantani A, et al. p53 directly represses Id2 to inhibit the proliferation of neural progenitor cells. Stem Cells. 2011;29:1090–101. doi: 10.1002/stem.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zheng H, Ying H, Yan H, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129–33. doi: 10.1038/nature07443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Grinstein E, Wernet P. Cellular signaling in normal and cancerous stem cells. Cell Signal. 2007;19:2428–33. doi: 10.1016/j.cellsig.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 95.Pelicci PG. Do tumor-suppressive mechanisms contribute to organism aging by inducing stem cell senescence? J Clin Invest. 2004;113:4–7. doi: 10.1172/JCI200420750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–60. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 97.Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–83. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–71. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ji Q, Hao X, Zhang M, et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One. 2009;4:e6816. doi: 10.1371/journal.pone.0006816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu C, Kelnar K, Liu B, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17:211–5. doi: 10.1038/nm.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nalls D, Tang SN, Rodova M, et al. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS One. 2011;6:e24099. doi: 10.1371/journal.pone.0024099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ji Q, Hao X, Meng Y, et al. Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer. 2008;8:266. doi: 10.1186/1471-2407-8-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Amson R, Pece S, Lespagnol A, et al. Reciprocal repression between P53 and TCTP. Nat Med. 2011 doi: 10.1038/nm.2546. [DOI] [PubMed] [Google Scholar]
- 104.Koziol MJ, Garrett N, Gurdon JB. Tpt1 activates transcription of oct4 and nanog in transplanted somatic nuclei. Curr Biol. 2007;17:801–7. doi: 10.1016/j.cub.2007.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pece S, Tosoni D, Confalonieri S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 106.Ligon KL, Huillard E, Mehta S, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503–17. doi: 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kippin TE, Martens DJ, van der Kooy D. p21 loss compromises the relative quiescence of forebrain stem cell proliferation leading to exhaustion of their proliferation capacity. Genes Dev. 2005;19:756–67. doi: 10.1101/gad.1272305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Swanson KM, Hohl RJ. Anti-cancer therapy: targeting the mevalonate pathway. Curr Cancer Drug Targets. 2006;6:15–37. doi: 10.2174/156800906775471743. [DOI] [PubMed] [Google Scholar]
- 109.Fritz G. Targeting the mevalonate pathway for improved anticancer therapy. Curr Cancer Drug Targets. 2009;9:626–38. doi: 10.2174/156800909789057033. [DOI] [PubMed] [Google Scholar]
- 110.Freed-Pastor WA, Mizuno H, Zhao X, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244–58. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ginestier C, Monville F, Wicinski J, et al. Mevalonate metabolism regulates Basal breast cancer stem cells and is a potential therapeutic target. Stem Cells. 2012;30:1327–37. doi: 10.1002/stem.1122. [DOI] [PubMed] [Google Scholar]
- 112.Ginestier C, Charafe-Jauffret E, Birnbaum D. p53 and cancer stem cells: The mevalonate connexion. Cell Cycle. 2012:11. doi: 10.4161/cc.21092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin Cancer Biol. 2012 doi: 10.1016/j.semcancer.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Morel AP, Lievre M, Thomas C, et al. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen C, Wei Y, Hummel M, et al. Evidence for epithelial-mesenchymal transition in cancer stem cells of head and neck squamous cell carcinoma. PLoS One. 2011;6:e16466. doi: 10.1371/journal.pone.0016466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Asiedu MK, Ingle JN, Behrens MD, et al. TGFbeta/TNF(alpha)-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011;71:4707–19. doi: 10.1158/0008-5472.CAN-10-4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kong D, Banerjee S, Ahmad A, et al. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS One. 2010;5:e12445. doi: 10.1371/journal.pone.0012445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chang CJ, Chao CH, Xia W, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317–23. doi: 10.1038/ncb2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kogan-Sakin I, Tabach Y, Buganim Y, et al. Mutant p53(R175H) upregulates Twist1 expression and promotes epithelial-mesenchymal transition in immortalized prostate cells. Cell Death Differ. 2011;18:271–81. doi: 10.1038/cdd.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dong P, Karaayvaz M, Jia N, et al. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene. 2012 doi: 10.1038/onc.2012.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang Y, Yan W, Jung YS, Chen X. Mammary epithelial cell polarity is regulated differentially by p73 isoforms via epithelial-to-mesenchymal transition. J Biol Chem. 2012;287:17746–53. doi: 10.1074/jbc.M112.358143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124••.Li L, Wang L, Wang Z, et al. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell. 2012;21:266–81. doi: 10.1016/j.ccr.2011.12.020. Demonstrates targeting of CML leukemia stem cells by p53 restoration via SIRT inhibition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ito T, Zimdahl B, Reya T. aSIRTing control over cancer stem cells. Cancer Cell. 2012;21:140–2. doi: 10.1016/j.ccr.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 126• •.Cicalese A, Bonizzi G, Pasi CE, et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138:1083–95. doi: 10.1016/j.cell.2009.06.048. Clearly demonstrates regulation of mammary stem cell self-renewal by p53 along with the effects of p53 restoration on mammary CSCs. [DOI] [PubMed] [Google Scholar]
- 127.Huang C, Zhang XM, Tavaluc RT, et al. The combination of 5-fluorouracil plus p53 pathway restoration is associated with depletion of p53-deficient or mutant p53-expressing putative colon cancer stem cells. Cancer Biol Ther. 2009;8:2186–93. doi: 10.4161/cbt.8.22.10446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Janicke RU, Sohn D, Schulze-Osthoff K. The dark side of a tumor suppressor: anti-apoptotic p53. Cell Death Differ. 2008;15:959–76. doi: 10.1038/cdd.2008.33. [DOI] [PubMed] [Google Scholar]
- 129.Vousden KH. Outcomes of p53 activation--spoilt for choice. J Cell Sci. 2006;119:5015–20. doi: 10.1242/jcs.03293. [DOI] [PubMed] [Google Scholar]
- 130.Donehower LA. Does p53 affect organismal aging? J Cell Physiol. 2002;192:23–33. doi: 10.1002/jcp.10104. [DOI] [PubMed] [Google Scholar]
- 131.Gatza CE, Dumble M, Kittrell F, et al. Altered mammary gland development in the p53+/m mouse, a model of accelerated aging. Dev Biol. 2008;313:130–41. doi: 10.1016/j.ydbio.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]