

Abstract
Objectives
Raltegravir is the first integrase strand transfer inhibitor approved for treating HIV-1 infection. Although emerging data suggest that raltegravir may also be useful for HIV-2 treatment, studies addressing the in-vitro susceptibility of HIV-2 to raltegravir are scarce, and the genetic pathways leading to raltegravir resistance in HIV-2 have not been adequately characterized. Our objectives were to directly compare the susceptibilities of HIV-1 and HIV-2 to raltegravir and to examine the role of mutations in HIV-2 integrase in emergent raltegravir resistance.
Materials and methods
Single-cycle and spreading infection assays were used to quantify the sensitivities of wild-type HIV-1 and HIV-2 strains to raltegravir. HIV-2 integrase mutants were constructed by site-directed mutagenesis, and the replication capacities and raltegravir susceptibilities of the resultant variants were analyzed in single-cycle assays.
Results
Raltegravir showed comparable activity against wild-type HIV-1 and HIV-2 in both single-cycle and spreading infections, with EC50 values in the low nanomolar range. Amino acid changes Q148R and N155H individually conferred resistance to raltegravir (14-fold and seven-fold, respectively), whereas the Y143C replacement had no statistically significant effect on raltegravir sensitivity. The combination of Q148R with N155H resulted in high-level raltegravir resistance (>1000-fold). In addition, all HIV-2 integrase variants tested showed impairments in replication capacity.
Conclusion
Our data support clinical studies of raltegravir for treating HIV-2 infection and show that the Q148R and N155H changes alone are sufficient for raltegravir resistance in HIV-2. Further efforts are needed to improve access to HIV-2-active antiretrovirals, including raltegravir, in resource-limited areas where HIV-2 is endemic.
Keywords: HIV-1, HIV-2, integrase, N155H, Q148R, raltegravir, resistance, Y143C
Introduction
HIV-2 is endemic in West Africa and has achieved a limited prevalence in other locales [1–3]. Treatment of HIV-2 infection presents unique challenges; HIV-2 is intrinsically resistant to nonnucleoside reverse transcriptase inhibitors and the fusion inhibitor enfuvirtide [4], and limited data suggest that HIV-2 may also be resistant to certain protease inhibitors [5]. Furthermore, HIV-2 rapidly acquires resistance to multiple nucleoside analogue inhibitors through a small number of amino acid changes in reverse transcriptase [6,7]. These features highlight the pressing need to identify additional antiretroviral drugs that are active against HIV-2 and to evaluate their efficacy in HIV-2-infected patients.
Raltegravir (MK-0518, Isentress, Merck & Co. Inc., Whitehouse Station, New Jersey, USA) is the first integrase inhibitor approved by the US Food and Drug Administration for treating HIV-1 infection. Raltegravir exhibits potent activity against HIV-1 in culture and in cell-free reactions with purified HIV-1 integrase [8–10]. In addition, randomized clinical trials have shown that raltegravir suppresses HIV-1 viral loads when co-administered with other antiretroviral drugs (see [11,12] for review). Raltegravir was initially approved for clinical use based on its efficacy in highly antiretro-viral-experienced patients [13,14]. Further studies demonstrated the utility of raltegravir for first-line treatment [15] and as a substitute for boosted protease inhibitors in patients with therapeutically-suppressed viremia (i.e. ‘regimen simplification’) [16,17]. Collectively, these findings have led to the expanded use of raltegravir for antiretroviral therapy of HIV-1 infection.
In contrast, little is known regarding the efficacy of raltegravir for HIV-2 treatment. Roquebert et al. [18] reported that HIV-2 isolates from raltegravir-naive patients are susceptible to the drug in culture, and data from case reports and small-scale clinical studies suggest that raltegravir-containing regimens provide a favorable short-term response in HIV-2-infected patients [19–24]. Collectively, these findings suggest that raltegravir might be useful in West Africa and other resource-limited settings where HIV-2 is endemic, although additional clinical studies of raltegravir-based regimens for HIV-2 are needed.
As is the case for HIV-1, the emergence of raltegravir-resistant HIV-2 mutants will likely complicate treatment [19,21,24]. Specific amino acid changes that are known to confer raltegravir resistance in HIV-1 have been observed in integrase sequences from raltegravir-treated HIV-2 patients; these commonly include Q148R, N155H and, to a lesser extent, Y143C [19,21,22,24–26]. Ni et al. [26] recently reported that Q148R and N155H individually improve the ability of purified HIV-2 integrase to catalyze strand transfer in the presence of raltegravir, whereas the Y143C change alone had no impact on raltegravir sensitivity. Importantly, the biological effects of these changes in the HIV-2 integrase protein, as assessed by site-directed mutagenesis and culture-based drug susceptibility testing, have not been determined. Such experiments are necessary to delineate the genetic pathways leading to raltegravir resistance in HIV-2.
To examine the activity of raltegravir against HIV-2 and assess the mutational pathways leading to drug resistance, we initially compared the susceptibilities of wild-type HIV-1 and HIV-2 strains to raltegravir in a single cycle of viral replication. These experiments were supported by additional tests of drug sensitivity in spreading infections of immortalized T lymphocytes. Next, we used site-directed mutagenesis to construct HIV-2 variants containing specific amino acid replacements in the viral integrase and determined the replication capacities and raltegravir susceptibilities of the mutant strains in culture. Overall, our findings support the use of raltegravir for HIV-2 treatment and demonstrate that the Q148R and N155H changes alone are sufficient for phenotypic resistance to raltegravir in HIV-2.
Methods
HIV-1 MVP5180-91 and HIV-2 strains CBL20-H9, CBL23-H9, CDC77618, 7924A and CBL23-H9 were obtained from the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program. These virus strains were expanded in MT-2 lymphocyte cultures for 6–14 days of infection prior to phenotypic testing. Wild-type HIV-2 ROD9 (group A) and wild-type HIV-1 NL4-3 (group M, subtype B) were produced from plasmids pROD9 and pNL4-3, respectively, as described below. HIV-2 EHO (group B) was kindly provided by Jan McClure (University of Washington, Seattle, Washington, USA). The amino acid sequences of HIV-2 ROD9 and HIV-2 EHO are identical to each other and to HIV-2 group A and B consensus sequences from the Los Alamos HIV Sequence Database (http://www.hiv.lanl.gov) at integrase residues implicated in raltegravir resistance (i.e. E92, T97, Y143, Q148, A153 and N155). Although integrase sequences for CBL20-H9, CBL23-H9, CDC77618, 7924A and CBL23-H9 are not available, we note that in the Los Alamos dataset, sequences obtained from raltegravir-naive HIV-2 patients (n = 322, including 122 patient-derived samples from our previous study [27]) are completely invariant at positions Y143, Q148 and N155, and 98–99% conserved at positions E92, T97 and A153. Thus, resistance-associated mutations are exceedingly rare in wild-type HIV-2 integrase sequences from raltegravir-naive individuals.
Raltegravir was obtained from the NIH AIDS Research and Reference Reagent Program. Master stocks (5 mmol/l) and serial dilutions of the drug were prepared in sterile distilled water and stored at −80 °C.
MAGIC-5A indicator cells (CD4+ CCR5+ HeLa cells that express β-galactosidase under the control of an HIV-1 promoter) were provided by Michael Emerman (Fred Hutchinson Cancer Research Center, Seattle, Washington, USA) and were maintained as previously described [6]. MT-2 cells were acquired from Uta von Schwedler (University of Utah, Salt Lake City, Utah, USA) and were maintained in RPMI 1640 medium (Invitrogen, Carlsbad, California, USA) supplemented with 10% fetal bovine serum (Hyclone, Logan, Utah, USA).
Mutations were introduced into the integrase-encoding regions of pNL4-3 and pROD9 using the QuikChange II Site-directed Mutagenesis Kit (Stratagene, La Jolla, California, USA). Nucleotide sequences of the mutagenic primers are available upon request. The N155H HIV-1 replacement was initially constructed in an Apa I-Eco R1 subclone of pNL4-3 (pBS-pol, HIV-1 nucleotides 2010–5743). For HIV-2 Y143C, Q148R, N155H and Q148R with N155H, mutations were introduced into a pol-spanning Hind III subclone of pROD9 (pBS-RODH3, HIV-2 nucleotides 1457–5787). We then ligated the mutated pol genes back into their respective full-length plasmids. All full-length plasmids were purified using an EndoFree Plasmid Maxi Kit (Qiagen Inc., Valencia, California, USA) and were sequenced across the subcloned region to ensure that no additional nucleotide changes were introduced during the mutagenesis procedure.
Virus stocks were prepared by transfecting full-length HIV plasmid DNA into 293T-17 cells using a chloroquine-mediated, calcium phosphate coprecipitation method [6]. Single-cycle measurements of replication capacity and raltegravir susceptibility were performed in MAGIC-5A cells as previously described [6,28]. Culture monolayers were either stained with 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) and counted using a CTL Immunospot Analyzer (Cellular Technology Ltd, Shaker Heights, Ohio, USA) or by light microscopy, or were subjected to the β-Galactosidase Enzyme Assay System with Reporter Lysis Buffer (Promega Corp., Madison, Wisconsin, USA). For the β-galactosidase (β-gal) enzyme test, culture monolayers infected with serial dilutions of each virus strain, as well as standard curve reactions containing serial dilutions of purified β-gal, were included to ensure that HIV infection was scored within the linear range of the assay. Drug concentrations that inhibited focus formation or β-gal expression by 50% (EC50 values) were calculated from dose–response plots using the sigmoidal regression function of Prism (version 4.0a; GraphPad Software Inc., San Diego, California, USA). All three scoring methods (visual counting, CTL Immunospot counting and β-gal enzyme assay) yielded comparable EC50s for wild-type HIV-2 ROD9 (data not shown).
For the spreading infection assays, MT-2 cells were seeded in 48-well plates at a density of 1 × 105 cells in 400 μl of complete RPMI-1640 per well, treated with varying concentrations of raltegravir (or H2O only for no-drug controls, 50 μl per well) and incubated for 1 h at 37°C. Virus stocks then were adjusted to 104 MAGIC-5A focus-forming units per milliliter in RPMI complete, and 50 μl of each inoculum were added per well. On days 2 and 4 postinfection, 250 μl of supernatant were removed from each well (leaving the MT-2 cells undisturbed) and replaced with 200 μl of fresh RPMI complete with 50 μl of the appropriate raltegravir dilution. On the fifth day postinfection, the upper 300 μl of supernatant were removed from each well and frozen in two 150-μl aliquots at −80°C. Viral yields were quantified by plating the MT-2 culture supernatants onto MAGIC-5A cells, staining the monolayers with X-gal and visually counting β-gal-positive foci by light microscopy as described above.
Results
To determine the intrinsic susceptibility of HIV-2 to raltegravir, we tested the activity of the drug against wild-type strains of HIV-1 and HIV-2 in single-cycle assays with MAGIC-5A indicator cells. Raltegravir inhibited the formation of β-gal-positive foci by HIV-2 ROD9 in a dose-dependent manner, resulting in an EC50 value of 9.4 ± 2.7 nmol/l (mean ± SD, Fig. 1a). Raltegravir was also active against the five other wild-type HIV-2 strains tested, including four group A strains (CBL20-H9, CDC77618, 7924A and CBL23-H9) and one group B strain (EHO), with EC50 values ranging from 2.6 ± 1.1 nmol/l for 7924A to 20 ± 18 nmol/l for EHO (Fig. 1a). These results were comparable to the values obtained for two wild-type strains of HIV-1: NL4-3 (a group M strain, EC50 = 4.9 ± 2.4 nmol/l) and MVP5180-91 (group O, EC50 = 10.1 ± 6.2 nmol/l). The data obtained for MVP5180-91 are in agreement with a previous study of a patient-derived group O HIV-1 isolate [29]. In comparison, a site-directed mutant of HIV-1 NL4-3 encoding the N155H integrase replacement, which we constructed as a known raltegravir-resistant control for our assays, exhibited 15-fold resistance to the drug (EC50 = 73 ± 34 nmol/l, Fig. 1a). Collectively, these data demonstrate that wild-type HIV-1 and HIV-2 strains are comparably sensitive to raltegravir in a single cycle of viral replication.
Fig. 1. Susceptibilities of wild-type HIV-1 and HIV-2 strains to raltegravir.
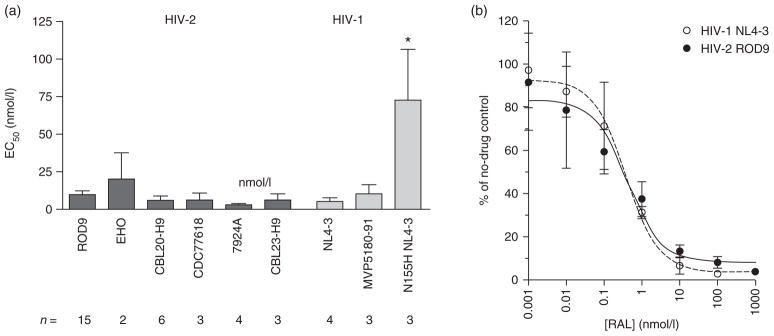
(a) Results of single-cycle assays with MAGIC-5A indicator cells. The number of independent determinations (n) performed for each strain is shown below the x-axis. *Significantly greater than wild-type HIV-1 NL4-3 [P <0.001, analysis of variance (ANOVA) of log10 (EC50) values with Tukey’s posttest]. (b) Representative results of spreading infection assays in MT-2 cells. Each datum point is the mean titer of infectious virus (expressed as the percentage of no-drug controls) from three independently maintained MT-2 cultures. In the panels (a and b), error bars indicate SDs.
We also measured the raltegravir susceptibilities of wild-type HIV-1 NL4-3 and wild-type HIV-2 ROD9 in spreading infections of an immortalized T-cell line (MT-2). In agreement with the single-cycle data, these prototypic strains yielded superimposable dose–response profiles when tested in the spreading infection assay (Fig. 1b). The mean EC50 values for HIV-1 NL4-3 and HIV-2 ROD9 were 0.50 ± 0.16 and 0.55 ± 0.19 nmol/l, respectively.
To examine the phenotypic effects of specific changes in HIV-2 integrase, we constructed individual full-length clones of HIV-2 ROD9 encoding the single amino acid replacements Y143C, Q148R and N155H. These replacements are known to confer raltegravir resistance in HIV-1 [11,12] and have been observed in HIV-2 sequences from patients receiving raltegravir therapy [19,21,22,24–26]. We also constructed a variant of HIV-2 ROD9 encoding the combination of Q148R and N155H on the same viral genome (Q148R with N155H). Although this particular genotype has not been observed in patient-derived HIV-1 or HIV-2 sequences, we sought to determine whether the combined effects of Q148R and N155H imparted higher levels of raltegravir resistance relative to the corresponding single amino acid changes.
We initially assessed the replication capacity of Y143C, Q148R, N155H and Q148R with N155H HIV-2 by transfecting full-length plasmids encoding these genotypes into 293T-17 cells. Titers of infectious virus produced from the plasmids were quantified by transferring serial dilutions of the 293T-17 supernatants onto MAGIC-5A indicator cells. In agreement with a previous report suggesting that the N155H change impairs HIV-2 replication capacity [22], plasmids encoding N155H HIV-2 ROD9 produced titers of infectious progeny that, on average, were 3.7-fold lower than the parental wild-type construct (Fig. 2). Y143C and Q148R also conferred statistically significant declines in replication capacity, with mean titers 4.9-fold and 17-fold lower than that of the parental ROD9 strain, respectively (Fig. 2). Q148R with N155H HIV-2 yielded the lowest level of infectious virus of all the mutants tested; the average titer for the double mutant was 53-fold lower than that of wild-type ROD9 (Fig. 2). These effects were not attributable to differences in transfection efficiency, as comparable titers were obtained from two independent preparations of plasmid DNA for each wild-type or mutant strain (black versus gray symbols, Fig. 2).
Fig. 2. Replication capacities of site-directed integrase mutants.

Each datum point is the infectious titer [MAGIC-5A, focus-forming unit (FFU)/ml] produced by an independent transfection of full-length HIV-1 or HIV-2 plasmid DNA into 293T-17 cells. Gray and black boxes represent the titers produced by two independent plasmid DNA preparations for each genotype. Mean titers for each strain are indicated with horizontal lines. The P values were determined by ANOVA of log10-transformed titers with Tukey’s posttest. WT, wild-type.
We also examined the effects of the aforementioned integrase mutations on raltegravir susceptibility. The Y143C change alone had no statistically significant effect with regard to raltegravir sensitivity in HIV-2 (Fig. 3a), although we note that EC50 values for the Y143C variant were on average 3.5-fold higher than the corresponding values for wild-type ROD9 (range = 1.7–6.1-fold higher, n = 4), potentially reflecting a low level of raltegravir resistance (Fig. 3b). In contrast, Q148R and N155H variants of HIV-2 ROD9 were both significantly resistant to raltegravir, with EC50 values of 130 ± 12 and 66 ± 31 nmol/l (14-fold and 7.0-fold higher than wild-type ROD9, respectively) (Fig. 3a). A similar level of resistance (15-fold) was observed for N155H HIV-1. Strikingly, the combination of Q148R with N155H in HIV-2 conferred a greater-than-additive increase in raltegravir resistance (Fig. 3a). At the highest concentration of drug tested, the mean number of β-gal-positive foci in cultures infected with the double mutant was 55–75% of untreated controls. Thus, the EC50 for Q148R with N155H HIV-2 was more than 10 μmol/l. Taken together, these findings indicate that integrase substitutions Q148R and N155H confer moderate levels of raltegravir resistance, and that the combination of these two changes produces more than 1000-fold resistance to the drug in HIV-2.
Fig. 3. Raltegravir susceptibilities of site-directed HIV-1 and HIV-2 integrase mutants in a single cycle of infection.
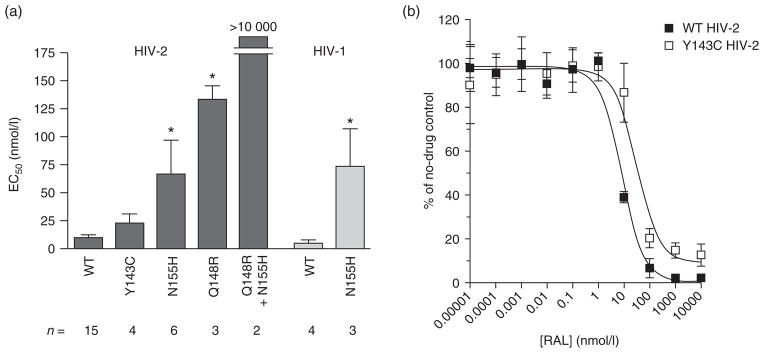
(a) EC50 values for wild-type and mutant HIV-2 ROD9 and HIV-1 NL4-3 strains. n, the number of independent determinations performed for each strain. *, significantly greater than the corresponding wild-type (WT) strain [P <0.001, ANOVA of log10 (EC50) values with Tukey’s posttest]. (b) Representative dose–response profiles for WT and Y143C HIV-2 ROD9 assayed in parallel. Data are the mean titers (expressed as percentages of untreated controls) from three cultures at each concentration of raltegravir (RAL) tested. EC50 values for the experiment shown were 8.0 nmol/l for WT HIV-2 and 32.7 nmol/l for Y143C HIV-2 ROD9. Additional assays yielded EC50s of 4.1, 7.8 and 8.3 nmol/l for WT HIV-2 versus 24.6, 13.6 and 19.3 nmol/l for the Y143C mutant (measurements paired in the order listed). Error bars in the panels (a and b) indicate SDs.
Discussion
Our analysis demonstrates that raltegravir is a potent inhibitor of wild-type HIV-2 strains in both single-cycle and spreading infection assays, with EC50 values comparable to those observed for wild-type HIV-1 (Fig. 1). These findings are consistent with previous analyses of the raltegravir sensitivity of HIV-2 in peripheral blood mononuclear cells [18,22]. In addition, we show that the Y143C, Q148R and N155H replacements in HIV-2 ROD9 integrase compromise viral replication capacity (Fig. 2) and that the Q148R and N155H changes individually confer moderate resistance to raltegravir in a single cycle of infection (Fig. 3a). For the latter two variants, the levels of resistance observed in our single-cycle assays were on par with those reported for Q148R and N155H mutants of HIV-1 (Fig. 3a) [30–33]. Thus, Q148R and N155H individually confer similar effects on raltegravir susceptibility in HIV-1 and HIV-2, despite the fact that these viruses differ at approximately one third of the 109 amino acid sites in the integrase catalytic core domain [34].
In contrast, the Y143C replacement in HIV-2 integrase had little or no effect on raltegravir sensitivity (Fig. 3a and b). This result agrees with findings from a biochemical analysis of purified HIV-2 integrase proteins [26], but differs from the outcomes observed in previous studies of HIV-1. For example, Delelis et al. [35] reported that Y143C alone is sufficient for high-level raltegravir resistance in HIV-1, as measured in HeLa-P4 indicator cell assays (EC50 approximately 500-fold greater than wild-type). Similarly, Fransen et al. [30] found that Y143C conferred 16-fold resistance to raltegravir in the Phenosense HIV-1 single-cycle test. These data suggest that HIV-1 and HIV-2 might differ with regard to the role of Y143C in raltegravir resistance, although it should be noted that other studies of HIV-1 mutants using multicycle assays reported relatively modest effects for Y143C (i.e., three-fold to four-fold raltegravir resistance) [33,36]. Additional, head-to-head comparisons of the phenotypic effects of Y143C in HIV-1 and HIV-2 are needed to resolve this issue.
In HIV-1, the development of high-level (>100-fold) resistance to raltegravir involves an accumulation of ‘primary’ and ‘secondary’ amino acid changes in the integrase protein [11,12]. Studies of HIV-1 patients have identified three principal mutational patterns that emerge in response to raltegravir treatment: Q148H/K/R with or without G140S/A, N155H with or without E92Q and Y143C/R with or without T97A [12]. Generally, the amino acid changes that appear in conjunction with Q148H/K/R, N155H and Y143C/R augment the level of raltegravir resistance in HIV-1 and, in some cases, mitigate the fitness costs incurred by primary resistance-associated mutations [11,12]. These findings, together with our in-vitro analysis (Fig. 3) and the sequence data currently available from HIV-2-infected patients [19,21,22,24–26], suggest that additional changes in HIV-2 integrase are required in combination with Y143C, Q148R or N155H to achieve high-level raltegravir resistance. In support of this view, Ni et al. [26] recently showed that the combination of G140S with Q148R confers a more than 100-fold loss of raltegravir sensitivity in cell-free assays with purified integrase proteins. Cooperative effects were also observed between the E92Q and Y143C replacements in HIV-2 integrase; although these changes individually had no measurable effect on raltegravir sensitivity, the E92Q with Y143C enzyme was approximately 10-fold resistant to the inhibitor [26]. Further studies of the phenotypic effects of resistance-associated changes in HIV-2 integrase should include these and other mutational combinations observed in patient-derived sequences.
Finally, we note that the combination of two ‘primary’ resistance changes in HIV-2 (Q148R with N155H) confers a dramatic loss of raltegravir sensitivity (Fig. 3a), albeit at a substantial cost to replication capacity (Fig. 2). These findings are comparable to the data obtained in a previous analysis of Q148H with N155H mutants of HIV-1 [37]. The relatively low level of viral infectivity observed in our experiments potentially explains why the combination of Q148R and N155H has not been observed in sequences from raltegravir-treated HIV-2 patients [19,21,22,24–26]. Although we cannot exclude the possibility that additional changes in the HIV-2 integrase protein play a compensatory role, our data suggest that the Q148R and N155H replacements define mutually exclusive pathways to raltegravir resistance in HIV-2. A similar conclusion has been reached for HIV-1 based on the aforementioned culture-based study [37] and the absence of Q148 with N155 double mutants in sequences from raltegravir-treated HIV-1 patients [30].
Taken together, our findings support the use of raltegravir as a component of combination antiretroviral therapy for HIV-2 infection. Although randomized controlled trials are ultimately needed to determine optimal therapy for HIV-2, given the limited range of drugs available for HIV-2 treatment, our data should encourage efforts to improve patient access to raltegravir in the resource-limited areas where HIV-2 is endemic.
Acknowledgments
The authors would like to thank Charlotte Pan and Alexandra Hernandez (University of Washington) for excellent technical assistance and the National Institutes of Health AIDS Research and Reference Reagent Program for HIV strains and raltegravir.
R.A.S. and G.S.G. were responsible for conceiving and designing the experiments, analyzing the data and preparing the manuscript. R.A.S. and D.N.R. were responsible for performing the experiments. N.B.K., S.E.H., J.I.M. and P.S.S. assisted in interpreting the data and preparing the final manuscript for publication.
The UW-Dakar HIV-2 Study Group also includes Macoumba Toure, Selly Ba, Ndeye M.D. Badiane, Louise Fortes, Cheikh T. Ndour, Jacques Ndour, Fatou Niasse, Fatima Sall, Balla Tall, Fatou Traore, Habibatou D. Agne, Ndeye R. Fall, Sophie Chablis, Marie P. Sy, Mame Dieumba, Bintou Diaw, Mbaye Ndoye, Khady Diop, Amadou B. Diop, Cheikh Gueye, Boubacar Diamanka, Marianne Ndiaye, Marie C. Thioye, Fatou Cisse, Madeleine Mbow, Marianne F. Diome, Marie Diedhiou, Donna Kenny, Steve Cherne, Joshua Stern, John Lin, Charlotte Pan, Beruk Asfaw, Brad Church, Matthew Coyne and Alexandra Hernandez.
These studies were supported by grants to GSG from the National Institutes of Health/National Institute of Allergy and Infectious Diseases (2R01-AI060466), the University of Washington Center for AIDS Research, and the University of Washington Royalty Research Fund. Additional support was provided through a New Investigator Award to R.A.S. from the University of Washington Center for AIDS Research (P30 AI27757).
Footnotes
Conflicts of interest
The authors report no conflict of interest.
Presented in part at the 6th IAS Conference on HIV Pathogenesis, Treatment and Prevention; 17 July 2011; Rome, Italy [abstract #TUPE091].
References
- 1.de Silva TI, Cotten M, Rowland-Jones SL. HIV-2: the forgotten AIDS virus. Trends Microbiol. 2008;16:588–595. doi: 10.1016/j.tim.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Chiara M, Rony Z, Homa M, Bhanumati V, Ladomirska J, Manzi M, et al. Characteristics, immunological response and treatment outcomes of HIV-2 compared with HIV-1 and dual infections (HIV-1/2) in Mumbai. Indian J Med Res. 2010;132:683–689. [PMC free article] [PubMed] [Google Scholar]
- 3.Schim van der Loeff MF, Aaby P. Towards a better understanding of the epidemiology of HIV-2. AIDS. 1999;13 (Suppl A):S69–S84. [PubMed] [Google Scholar]
- 4.Witvrouw M, Pannecouque C, Switzer WM, Folks TM, De Clercq E, Heneine W. Susceptibility of HIV-2, SIV and SHIV to various anti-HIV-1 compounds: implications for treatment and postexposure prophylaxis. Antivir Ther. 2004;9:57–65. [PubMed] [Google Scholar]
- 5.Menendez-Arias L, Tozser J. HIV-1 protease inhibitors: effects on HIV-2 replication and resistance. Trends Pharmacol Sci. 2008;29:42–49. doi: 10.1016/j.tips.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 6.Smith RA, Anderson DJ, Pyrak CL, Preston BD, Gottlieb GS. Antiretroviral drug resistance in HIV-2: three amino acid changes are sufficient for classwide nucleoside analogue resistance. J Infect Dis. 2009;199:1323–1326. doi: 10.1086/597802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gottlieb GS, Badiane NM, Hawes SE, Fortes L, Toure M, Ndour CT, et al. Emergence of multiclass drug-resistance in HIV-2 in antiretroviral-treated individuals in Senegal: implications for HIV-2 treatment in resource-limited West Africa. Clin Infect Dis. 2009;48:476–483. doi: 10.1086/596504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, et al. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J Med Chem. 2008;51:5843–5855. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 9.Bar-Magen T, Sloan RD, Faltenbacher VH, Donahue DA, Kuhl BD, Oliveira M, et al. Comparative biochemical analysis of HIV-1 subtype B and C integrase enzymes. Retrovirology. 2009;6:103. doi: 10.1186/1742-4690-6-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Low A, Prada N, Topper M, Vaida F, Castor D, Mohri H, et al. Natural polymorphisms of human immunodeficiency virus type 1 integrase and inherent susceptibilities to a panel of integrase inhibitors. Antimicrob Agents Chemother. 2009;53:4275–4282. doi: 10.1128/AAC.00397-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McColl DJ, Chen X. Strand transfer inhibitors of HIV-1 integrase: bringing IN a new era of antiretroviral therapy. Antiviral Res. 2010;85:101–118. doi: 10.1016/j.antiviral.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 12.Blanco JL, Varghese V, Rhee SY, Gatell JM, Shafer RW. HIV-1 integrase inhibitor resistance and its clinical implications. J Infect Dis. 2011;203:1204–1214. doi: 10.1093/infdis/jir025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grinsztejn B, Nguyen BY, Katlama C, Gatell JM, Lazzarin A, Vittecoq D, et al. Safety and efficacy of the HIV-1 integrase inhibitor raltegravir (MK-0518) in treatment-experienced patients with multidrug-resistant virus: a phase II randomised controlled trial. Lancet. 2007;369:1261–1269. doi: 10.1016/S0140-6736(07)60597-2. [DOI] [PubMed] [Google Scholar]
- 14.Cooper DA, Steigbigel RT, Gatell JM, Rockstroh JK, Katlama C, Yeni P, et al. Subgroup and resistance analyses of raltegravir for resistant HIV-1 infection. N Engl J Med. 2008;359:355–365. doi: 10.1056/NEJMoa0708978. [DOI] [PubMed] [Google Scholar]
- 15.Lennox JL, DeJesus E, Lazzarin A, Pollard RB, Madruga JV, Berger DS, et al. Safety and efficacy of raltegravir-based versus efavirenz-based combination therapy in treatment-naive patients with HIV-1 infection: a multicentre, double-blind randomised controlled trial. Lancet. 2009;374:796–806. doi: 10.1016/S0140-6736(09)60918-1. [DOI] [PubMed] [Google Scholar]
- 16.Eron JJ, Young B, Cooper DA, Youle M, Dejesus E, Andrade-Villanueva J, et al. Switch to a raltegravir-based regimen versus continuation of a lopinavir-ritonavir-based regimen in stable HIV-infected patients with suppressed viraemia (SWITCHMRK 1 and 2): two multicentre, double-blind, randomised controlled trials. Lancet. 2010;375:396–407. doi: 10.1016/S0140-6736(09)62041-9. [DOI] [PubMed] [Google Scholar]
- 17.Martinez E, Larrousse M, Llibre JM, Gutierrez F, Saumoy M, Antela A, et al. Substitution of raltegravir for ritonavir-boosted protease inhibitors in HIV-infected patients: the SPIRAL study. AIDS. 2010;24:1697–1707. doi: 10.1097/QAD.0b013e32833a608a. [DOI] [PubMed] [Google Scholar]
- 18.Roquebert B, Damond F, Collin G, Matheron S, Peytavin G, Benard A, et al. HIV-2 integrase gene polymorphism and phenotypic susceptibility of HIV-2 clinical isolates to the integrase inhibitors raltegravir and elvitegravir in vitro. J Antimicrob Chemother. 2008;62:914–920. doi: 10.1093/jac/dkn335. [DOI] [PubMed] [Google Scholar]
- 19.Roquebert B, Blum L, Collin G, Damond F, Peytavin G, Leleu J, et al. Selection of the Q148R integrase inhibitor resistance mutation in a failing raltegravir containing regimen. AIDS. 2008;22:2045–2046. doi: 10.1097/QAD.0b013e32830f4c7d. [DOI] [PubMed] [Google Scholar]
- 20.Damond F, Lariven S, Roquebert B, Males S, Peytavin G, Morau G, et al. Virological and immunological response to HAART regimen containing integrase inhibitors in HIV-2-infected patients. AIDS. 2008;22:665–666. doi: 10.1097/QAD.0b013e3282f51203. [DOI] [PubMed] [Google Scholar]
- 21.Garrett N, Xu L, Smit E, Ferns B, El-Gadi S, Anderson J. Raltegravir treatment response in an HIV-2 infected patient: a case report. AIDS. 2008;22:1091–1092. doi: 10.1097/QAD.0b013e3282f9b165. [DOI] [PubMed] [Google Scholar]
- 22.Salgado M, Toro C, Simon A, Garrido C, Blanco F, Soriano V, Rodes B. Mutation N155H in HIV-2 integrase confers high phenotypic resistance to raltegravir and impairs replication capacity. J Clin Virol. 2009;46:173–175. doi: 10.1016/j.jcv.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 23.Armstrong-James D, Stebbing J, Scourfield A, Smit E, Ferns B, Pillay D, Nelson M. Clinical outcome in resistant HIV-2 infection treated with raltegravir and maraviroc. Antiviral Res. 2010;86:224–226. doi: 10.1016/j.antiviral.2010.02.324. [DOI] [PubMed] [Google Scholar]
- 24.Charpentier C, Roquebert B, Delelis O, Larrouy L, Matheron S, Tubiana R, et al. Hot spots of integrase genotypic changes leading to HIV-2 resistance to raltegravir. Antimicrob Agents Chemother. 2011;55:1293–1295. doi: 10.1128/AAC.00942-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu L, Anderson J, Garrett N, Ferns B, Wildfire A, Cook P, et al. Dynamics of raltegravir resistance profile in an HIV type 2-infected patient. AIDS Res Hum Retroviruses. 2009;25:843–847. doi: 10.1089/aid.2009.0039. [DOI] [PubMed] [Google Scholar]
- 26.Ni XJ, Delelis O, Charpentier C, Storto A, Collin G, Damond F, et al. G140S/Q148R and N155H mutations render HIV-2 integrase resistant to raltegravir whereas Y143C does not. Retrovirology. 2011;8:68. doi: 10.1186/1742-4690-8-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gottlieb GS, Smith RA, Dia Badiane NM, Ba S, Hawes SE, Toure M, et al. HIV-2 integrase variation in integrase inhibitor-naive adults in Senegal, West Africa. PLoS One. 2011;6:e22204. doi: 10.1371/journal.pone.0022204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith RA, Gottlieb GS, Miller AD. Susceptibility of the human retrovirus XMRV to antiretroviral inhibitors. Retrovirology. 2010;7:70. doi: 10.1186/1742-4690-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Briz V, Garrido C, Poveda E, Morello J, Barreiro P, de Mendoza C, Soriano V. Raltegravir and etravirine are active against HIV type 1 group O. AIDS Res Hum Retroviruses. 2009;25:225–227. doi: 10.1089/aid.2008.0222. [DOI] [PubMed] [Google Scholar]
- 30.Fransen S, Gupta S, Danovich R, Hazuda D, Miller M, Witmer M, et al. Loss of raltegravir susceptibility by human immunodeficiency virus type 1 is conferred via multiple nonoverlapping genetic pathways. J Virol. 2009;83:11440–11446. doi: 10.1128/JVI.01168-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Baelen K, Rondelez E, Van Eygen V, Arien K, Clynhens M, Van den Zegel P, et al. A combined genotypic and phenotypic human immunodeficiency virus type 1 recombinant virus assay for the reverse transcriptase and integrase genes. J Virol Methods. 2009;161:231–239. doi: 10.1016/j.jviromet.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 32.Goethals O, Clayton R, Van Ginderen M, Vereycken I, Wagemans E, Geluykens P, et al. Resistance mutations in human immunodeficiency virus type 1 integrase selected with elvitegravir confer reduced susceptibility to a wide range of integrase inhibitors. J Virol. 2008;82:10366–10374. doi: 10.1128/JVI.00470-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kobayashi M, Yoshinaga T, Seki T, Wakasa-Morimoto C, Brown KW, Ferris R, et al. In vitro antiretroviral properties of S/GSK1349572;a next-generation HIV integrase inhibitor. Antimicrob Agents Chemother. 2011;55:813–821. doi: 10.1128/AAC.01209-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu L, Anderson J, Ferns B, Cook P, Wildfire A, Workman J, et al. Genetic diversity of integrase (IN) sequences in antiretroviral treatment-naive and treatment-experienced HIV type 2 patients. AIDS Res Hum Retroviruses. 2008;24:1003–1007. doi: 10.1089/aid.2007.0303. [DOI] [PubMed] [Google Scholar]
- 35.Delelis O, Thierry S, Subra F, Simon F, Malet I, Alloui C, et al. Impact of Y143 HIV-1 integrase mutations on resistance to raltegravir in vitro and in vivo. Antimicrob Agents Chemother. 2010;54:491–501. doi: 10.1128/AAC.01075-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goethals O, Vos A, Van Ginderen M, Geluykens P, Smits V, Schols D, et al. Primary mutations selected in vitro with raltegravir confer large fold changes in susceptibility to first-generation integrase inhibitors, but minor fold changes to inhibitors with second-generation resistance profiles. Virology. 2010;402:338–346. doi: 10.1016/j.virol.2010.03.034. [DOI] [PubMed] [Google Scholar]
- 37.Quercia R, Dam E, Perez-Bercoff D, Clavel F. Selective-advantage profile of human immunodeficiency virus type 1 integrase mutants explains in vivo evolution of raltegravir resistance genotypes. J Virol. 2009;83:10245–10249. doi: 10.1128/JVI.00894-09. [DOI] [PMC free article] [PubMed] [Google Scholar]