

Abstract
RNA granules are structures within cells that play major roles in gene expression and homeostasis. Two principle kinds of RNA granules are conserved from yeast to mammals: stress granules (SGs), which contain stalled translation initiation complexes, and processing bodies (P‐bodies, PBs), which are enriched with factors involved in RNA turnover. Since RNA granules are associated with silenced transcripts, viruses subvert RNA granule function for replicative advantages. This review, focusing on RNA viruses, discusses mechanisms that manipulate stress granules and P‐bodies to promote synthesis of viral proteins. Three main themes have emerged for how viruses manipulate RNA granules; (1) cleavage of key host factors, (2) control of protein kinase R (PKR) activation, and (3) redirecting RNA granule components for new or parallel roles in viral reproduction, at the same time disrupting RNA granules. Viruses utilize one or more of these routes to achieve robust and productive infection. WIREs RNA 2013, 4:317–331. doi: 10.1002/wrna.1162
This article is categorized under:
-
1
RNA in Disease and Development > RNA in Disease
INTRODUCTION
RNA granules, typified by stress granules (SGs) and processing bodies (P‐bodies, PBs), contain concentrations of translationally silenced host mRNPs and are important for mRNA cycling and gene regulation. Because RNA granules regulate the mRNA cycle, metabolism, and gene expression, they comprise an important point of manipulation for viruses. The schemes of viral manipulation of RNA granules are quite variable, reflecting the diversity of viral replication strategies, and the impact of SGs on virus replication is wide‐ranging. Virus infection produces many types of stresses in cells, even during nonlytic infections. These perturbations of cellular homeostasis are detected in many ways in pathways that feed directly into stress responses. An emerging concept is that general stress responses and innate immune responses are both primordial, intimately linked, and interface at many levels. Typically outcomes of stress responses serve to restrict or reprogram host gene expression patterns, usually to the disadvantage of a virus. Thus, a common tendency of viruses is to block and/or co‐opt stress responses to foster more productive replication rates. This review covers the range of interactions between RNA viruses and cytoplasmic RNA granules, but focuses mostly on information from virus systems, where some details of the mechanisms are known. SG and PB interactions with viruses are grouped into classes according to current understanding and will require revision as further research emerges.
RNA GRANULES
Viruses must control cellular gene expression to provide conditions conducive for replication. Eukaryotic genes are regulated post‐transcriptionally by constantly altering the total assembled mRNP components bound on transcripts. These constantly changing mRNP compositions in turn regulate splicing, export, translation, subcellular localization, and mRNA turnover. Often these events are interconnected and the processes share proteins, for example, mRNA translation is linked to poly(A) shortening and decay.1, 2 The composition of proteins in mRNPs also determines if the mRNA constituents are translationally competent and able to recruit ribosomes, or translationally silenced and unable to recruit active ribosomal machinery. Both nuclear and cytoplasmic mRNP granules exist. Nuclear granules include cajal bodies, histone locus bodies, nuclear speckles, nuclear stress bodies, and paraspeckles (reviewed in Refs 3 and 4). The function of nuclear mRNP granules is diverse, ranging from stress responsive granules to granules controlling processing of mRNAs (e.g., histone locus bodies, nuclear speckles and paraspeckles) and noncoding RNAs. This review will focus on cytoplasmic RNA granules and the tendency for RNA viruses to modify these granules and implications linking cytoplasmic RNA granules in innate immunity. A more comprehensive review that includes DNA viruses has been recently published.5
There are two major classes of cytoplasmic RNA granules known as SGs and PBs, both of which contain translationally silenced mRNPs. Emerging evidence supports the existence of a cytoplasmic mRNA cycle where mRNPs are in dynamic equilibrium between active polysomes and silenced compartments, which are mostly comprised of PBs and SGs.6, 7, 8 SGs and PBs transiently dock with each other, they rapidly exchange protein constituents with surrounding cytoplasm and can share many protein components and specific mRNA moieties.1, 2, 9, 10, 11 These findings and others suggest that SGs and PBs can rapidly exchange mRNP cargo.
Stress granules are defined as foci enriched in translation initiation factors and 40S ribosome subunits, whereas PBs are enriched for RNA decay machinery. Each type RNA of granule has unique defining marker proteins, however, many proteins have been described in both SGs and PBs such as Ago2, eIF4E, APOBEC3, PCBP2, TTP, and others.9, 12 Several other types of RNA granules have been described in Caenorhabditis elegans, Drosophila, and neurons that contain various levels of proteins uniquely found in either SG or PBs. Thus, a continuum of RNA granules has been suggested to exist in eukaryotic cells with degrees of similarity to either SGs or PBs.10
Stress Granules
SGs are reversible dynamic structures that rapidly form when cells encounter environmental stress that reduces global translation rates. SG form from concentration of stalled assembled 43S and 48S ribosomal preinitiation complexes and serve as temporary repositories for these complexes. Thus, these translation complexes can be rapidly released to resume protein synthesis when stress conditions end. The most commonly described trigger of SG formation starts with oxidative, nutrient deprivation, or heat stress activation one of the eIF2α kinases (heme‐regulated kinase, HRI; general control nondepressible 2 kinase, GCN2; double‐stranded RNA (dsRNA)‐activated protein kinase R (PKR); and PKR‐like endoplasmic reticulum kinase, PERK), which phosphorylate the α subunit of translation initiation factor eIF2 and block translation (Figure 1). Virus infection commonly activates PKR via triggering its dsRNA recognition domain. Alternatively, translation inhibition through restriction of eIF4G or eIF4A function can also drive SG formation13 and some mechanisms of SG formation can proceed without eIF2α phosphorylation7, 14, 15 (Figure 1). Translation blockage forces accumulation of the stalled 43S and 48S ribosomal preinitiation complexes that are then concentrated by poorly understood mechanisms that actually form SGs.
Figure 1.
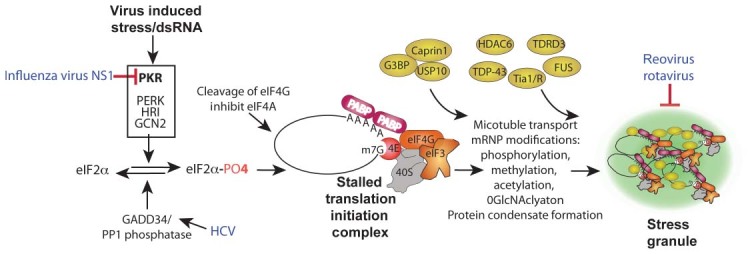
Stress granule assembly and interference by RNA viruses modulating PKR. Virus infection causes stress at multiple levels that reduces host translation through activation of eIF2 kinases, principally PKR, cleavage or inactivation of other initiation factors or other mechanisms. These translation insults convert active polysome mRNPs into stalled translation initiation complex mRNPs containing 40S ribosome subunits, initiation factors and mRNAs. A complex series of events (not depicted) involving nucleation of multiple stress granule proteins such as G3BP1, Tia‐1/TIAR, TDRD3, FUS, TDP43, and HDAC6 plus transport of mRNP complexes on microtubules leads to aggregates of translation initiation complex mRNPs in stress granules. Reovirus and rotavirus can repress SGs, but mechanisms are not known. Note that many viruses control PKR activation; only those discussed in the text are indicated. Also note that stress granule triggers by virus infection may operate at other levels and feed into this scheme.
The molecular mechanism(s) by which SGs condense involves several steps that are thought to include the (1) self‐oligomerization of key constituent RNA‐binding proteins (e.g., G3BP1, TIA‐1, TIAR), (2) post‐translational modifications of proteins, and (3) mRNP transport on microtubules (Figure 1). SGs contain hundreds of RNA‐interacting proteins and an siRNA screen indicates more than 100 genes are involved in SG assembly, so the mechanism of SG formation is multifactorial and quite complex.16 Yet simple viruses with limited genes have evolved efficient means to control their formation and function.
Variables in SG Composition
SGs form from condensation of stalled translation initiation complexes, and canonical SGs are defined by the presence of high concentrations of key translation initiation factors (e.g., eIF4E, eIF4G, eIF4A, eIF4B, eIF3, eIF2, PABP), mRNA and the 40S ribosome subunit.8, 9, 17 Additionally there are many RNA‐binding proteins such as caprin1, FMRP, YB1, HuR, and TTP and presumably any protein that binds mRNA or interacts strongly with mRNPs may also be found in SGs. Many of these are passenger proteins unlikely to have significant functions in SG biology. However, SGs contain key marker proteins that are linked to their formation, notably G3BP1, TIA1 and TIAR, TDRD3, HDAC6, and Caprin118, 19, 20, 21, 22 (Figure 1). Critically, in studying virus interactions with SGs, it is clear that cytoplasmic aggregates containing some of these marker proteins are not necessarily SGs23 and that a thorough evaluation of functional constituents of aggregates is required to distinguish SGs from unique virus‐induced foci.
The composition of SGs can vary depending on the type of stress that induced them, though the majority of markers that define SG function as depots of stalled translation complexes are consistent among all types of SG. For instance, heat shock induced stress granules (HS‐SGs) uniquely contain heat shock protein 27 (hsp27), which is absent in arsenite (Ars)‐induced SGs.8, 19, 24 Selenite‐induced SGs contain most typical translation factors, but are conspicuously lacking initiation factor eIF3b.25 Virus infection produces unique types of cell stresses and often induces SG to form (V‐SG) and some V‐SGs uniquely contain Sam68 which is not found in HS‐SGs.24
While many transcripts are thought to enter SGs; ER‐associated mRNAs are generally excluded,26 heat shock protein mRNAs do not enter HS‐SGs27 and certain IRES‐containing mRNAs of stress activated proteins may be preferentially excluded from SGs. Overall, aggregation of mRNPs into SGs likely promotes increased cell survival during stress conditions and rapid return to homeostasis at stress termination.28 Emerging evidence shows SGs may be linked to signaling pathways (discussed below).
Mechanism of Stress Granule Formation
For SG formation, multiple steps are required after the initial translation inhibition occurs. These include mobilization/activation of a series of RNA‐binding proteins involved in nucleation of SGs, such as G3BP, TIA1/TIAR, TDRD3; mRNP movement on microtubules and post‐translational modifications of factors.16, 29, 30 Virus interference in any of these steps may inhibit SG formation.
The actual molecular mechanism of mRNP aggregation into granules remains elusive but recent advances provide clues how RNA granules assemble components into coherent structures while simultaneously facilitating dynamic molecular exchange they are noted for. In particular, this may entail formation of liquid droplets or condensed gel phases that can better be envisioned as dynamic RNA/protein droplets. Such phase transitions from dispersed to condensed phases have been described to exhibit characteristic liquid droplet behavior and solutions of purified protein can also condense into droplet phases.31 Evidence suggests weak, but multivalent binding interactions between RNP proteins that involve repeated SRC homology 3 (SH3) domains, proline‐rich motifs (PRM) or other low complexity amino acid sequences are capable of assembling liquid phase droplets.32 Fused in sarcoma (FUS) is one example of an abundant RNA‐binding protein containing 27 repeats of a tripeptide that participates in hydrogels. FUS has been proposed to function in RNA neuronal granules with G3BP and TDP‐43,33, 34, 35 though TDP‐43 may play a more prominent functional role in SGs.36 G3BP, TIA1, and many other RNA‐binding proteins have domains suitable for this type of weak interaction. These protein interactions can also be affected by post‐translational modifications that can shift the equilibrium between the soluble and condensed phases.32, 34 For instance, G3BP is both phosphorylated and arginine methylated in association with its functional regulation.18, 37 Deacetylation of other SG target proteins may also be key for granule condensation.21 Consistent with a role of methylation in the process, increased polyamines repress SG formation, though it is not clear if this affects pathways or directly interferes with nucleation.38 Also, protein arginine methyltransferase 1 (PRMT1) is required for RAP55 inclusion in PBs39 and RGG motif methylation and ubiquitination of TDP‐43 modulate coaggregation with the SG‐nucleating protein G3BP.40
Processing Bodies
PBs are constitutively present in cells but increase in size and number when translational arrest occurs. PBs contain deadenylases, decapping enzymes, exonucleases, RNA‐binding proteins involved in nonsense‐mediated decay and microRNA‐mediated silencing (Figure 2). This enrichment of RNA decay machinery implies significant RNA decay occurs within PBs, but this is controversial.41 Recruitment of mRNA to PBs requires active silencing via miRNA or RNAi mechanisms and is not just an occurrence of nontranslation, perhaps providing a mechanistic distinction from SGs.42 Like SGs, the mechanism of PB formation is unclear but thought to involve condensation of RNA‐binding proteins, as well as the mRNA itself as an organizing structure.42 The human DEAD box helicase RCK/p54 (also called DDX6) may coat mRNAs and relax mRNA secondary structures before entry into PBs.43 Similar to SGs, PBs include variable protein constituents in both mammalian cells and Drosophila, with proteins like PCBP2, Hedls, Xrn1, defining subsets of PBs.42, 44 PBs dynamically exchange mRNP cargo with SGs and have been proposed to serve as nucleation sites for SG formation.9
Figure 2.
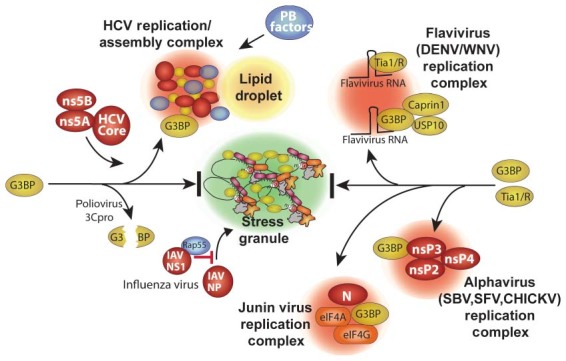
Virus blockade and co‐opting of stress granule responses. Specific points/proteins where viruses interact with and inhibit or divert the RNA granule assembly pathway are shown. Poliovirus 3Cproteinase cleaves the critical SG‐nucleating protein G3BP1. Several viruses co‐opt G3BP and divert it into novel virus‐induced foci. HCV diverts G3BP1 into replication/assembly complexes together with HCV core, ns5A and ns5B proteins that also associate with lipid droplets. HCVcomplexes also contain many PB components detailed in Figure 3. Flaviviruses divert G3BP1 (with USP10 and caprin1) and TIA1/TIAR to replication complexes by binding the host proteins on virus RNAs. Alphaviruses recruit G3BP1 into viral replication complexes via direct interaction viral protein nsP3. Junin virus (possibly N and G proteins) recruits G3BP1 into replication complexes that also contain translation factors eIF4G and eIF4A.
VIRAL MECHANISMS TO REGULATE STRESS GRANULE RESPONSES
Relationships between RNA Granules and Viruses
Virus infection will activate cell stress responses on many levels as various host processes are interrupted. Indeed, some viruses initially induce SGs early in infection, however, most viruses typically suppress SG formation at some point in the infection cycle and few examples are known where functional SGs (defined as containing stalled translation complexes) co‐exist within virus infected cells when levels of virus gene expression are high. Virus proteins can also disperse PBs. This implies an overall antagonistic relationship between viruses and RNA granules, which is not surprising given the established roles of RNA granules in translation silencing and RNA decay. For broad understanding of readers, the discussion below are categorized into classes based on mechanisms of virus interaction with the RNA granule machinery, namely (1) cleavage of RNA granule factors, (2) control of PKR function, and (3) co‐opting of RNA granule proteins. Because interactions are only provisionally probed in most virus systems and some reports conflict, this review is not comprehensive and these groupings will likely require future revision.
Cleavage of SG Components
Many plus strand RNA viruses express viral proteinases that often cleave key host proteins to modify the cellular environment. To date, only enteroviruses such as poliovirus have been shown to utilize proteinases to cleave factors in RNA granules. Poliovirus infection allows V‐SG to form during the very early phase of infection but then SGs are dispersed and viral functions actively block stress granule assembly during the mid to late‐phase of infection. The mechanism of SG disassembly involves cleavage of the key stress granule nucleating protein G3BP1 by the viral 3C proteinase (3Cpro) (Figure 2). G3BP1 cleavage separates the N‐terminal protein‐interacting domain from the C‐terminal RNA recognition motif and presumably disrupts its SG aggregation function. Expression of a 3Cpro cleavage‐resistant mutant of G3BP1 rescues stress granules at late times post infection, demonstrating the importance of G3BP in the SG formation process.45 A contradicting study showed that some V‐SG containing the SG marker TIA1 linger late into the infection cycle,24 but a follow‐up study showed those granules were lacking the translation factors eIF3, eIF4G and eIF4E, and mRNA. This indicated that TIA1 granules remaining after G3BP cleavage are remnants of normal SGs and do not correlate with translational repression and accumulation of stalled translation initiation complexes that define functional SGs.17 Therefore, poliovirus unlinks TIA1 aggregation from condensation of translation initiation factors in stress granules, likely through cleavage of G3BP1. These findings emphasize reports that SG can differ in composition and function in virus infected cells and cannot be reliably stereotyped based on analysis of limited SG markers. Poliovirus infection induces eIF2α phosphorylation, which simultaneously drives SG formation and inhibits cellular protein synthesis machinery required for viral RNA translation. Interestingly, poliovirus avoids this translation restriction due to 3Cpro‐mediated cleavage of eIF5B, which bypasses the need for eIF2α during translation initiation on the viral IRES but is not thought to play a role in SG formation.46
Two other viruses in the picornavirus superfamily that express a 3C proteinase (with different cleavage specificities) also restrict SGs but have not been reported to cleave key SG proteins. Theiler's murine encephalomyelitis virus blocks SG formation through an undetermined function of the viral leader protein, yet retains intact G3BP1 during infection.47 Cricket paralysis virus, a member of the picornavirus subgroup, also blocks stress granule formation at early times post infection (2 h) without cleavage of G3BP and TIA‐1 paralogs (Rin‐8 and Rox). Viral 3C proteinase is sequestered in SG during cell stress but not during infection, suggesting other viral proteins influence its subcellular location,48 and leaving open the possibility that other unknown important SG proteins are cleaved.
Manipulation of PKR
PKR is a critical sensor of cell stress and virus infection that activates stress responses and innate immunity. Most animal viruses trigger activation of PKR at some level and a plethora of viral mechanisms exist to counteract its activation, which will also influence SG formation. PKR activation and resulting translation inhibition strongly induces SG formation but SG formation through G3BP‐induced mRNP aggregation also induces PKR activation.15 It is possible that some viruses may also activate PERK through an unfolded protein response, which should result in similar downstream SG formation, but direct linkage has not yet been reported.
Influenza A virus (IAV) prevents stress granule formation throughout normal infections through the activity of viral protein NS1, which is a recognized antagonist of PKR phosphorylation and activation through its own dsRNA‐binding domain. IAV expressing an NS1 mutant that does not bind dsRNA allows eIF2α phosphorylation and SG accumulation. When SG form, virus replication is repressed, measured by expression of viral NP protein.49 Further, PKR knockout cells do not form stress granules during infection with NS1 mutant virus suggesting that repression of PKR activity by NS1 is critical for inhibition of SGs.49 Similarly, infection with an IAV NS1 deletion mutant (IAV ΔNS1) also results in SG formation.50 The mechanism of NS1 repression of SG formation involves its interaction in a complex containing cellular RNA associated protein 55 (RAP55). RAP55 is a component of both SGs and PBs and may facilitate shuttling of mRNP cargo between them. Overexpression of RAP55 induced SGs and blocked virus replication. The portion of NS1 interacting within RAP55 complexes maps to the PKR‐interacting domain.51 Viral nucleoprotein (NP) colocalizes with SGs in the absence of NS1 but switches and colocalizes with PBs during wild type virus infection. A partly conflicting report found levels of IAV NP (hence replication) were not altered in infections with IAV NS1 deletion mutant (IAV ΔNS1), despite pronounced PKR activation, eIF2α phosphorylation and SG formation.50 These results suggest that not only do SGs differ in composition depending on the context (discussed above), but also the same virus can interact with SGs and/or the translational apparatus in a cell type‐dependent manner (A549 cells vs HeLa).
Rotavirus actively takes over host translation partly by actively promoting phosphorylation of eIF2α through activities of viral proteins VP2, NSP2, and NSP5.52 This provides the virus transcripts with a translational edge over endogenous mRNAs but should also strongly induce SG formation. However, rotavirus can control SG formation since it actively blocks SG formation induced by exogenous stressors, but the mechanism remains unknown. eIF2α phosphorylation still partly restricts rotavirus translation and/or replication since virus replicates more efficiently in eIF2α S51A mutant mouse embryonic fibroblasts where eIF2α cannot be phosphorylated.
Mammalian orthoreovirus (MRV) induces stress granules early in infection (6 hpi), which correlates with increased phosphorylation of eIF2α and translation restriction of both cellular and viral mRNAs.53 eIF2α phosphorylation is required for uncoating of the virus, with MRV induction of stress granules as a consequence. However, PKR and the other individual eIF2α kinases are not solely required for SG induction by MRV, suggesting SGs arise by signaling through multiple eIF2α kinases or another mechanism.54 Later in infection MRV‐induced SGs are depleted and preferential translation of viral mRNAs occurs despite continued eIF2α phosphorylation.53, 54 It is unclear whether MRV translation persists through an alternate eIF2‐independent translation mechanisms similar to poliovirus, Hepatitis C virus (HCV), and Sindbis virus.46, 55, 56 MRV restricts formation of SGs at a point downstream of eIF2α phosphorylation at late times post infection (24 hpi). This occurs at some fundamental level, since even eIF4A inhibitors, which act independently of eIF2α phosphorylation, cannot induce SGs late in infection.53
Other investigators using different strains of reovirus (Dearing, c8 and c87) showed virus propagation declined in knock‐in cells expressing S51A eIF2α mutant, as well as in PERK and ATF4 knockout cells.57 This correlated the initial induction of stress granules with increased virus production but also with variations in virus production of p58IPK, an inhibitor of eIF2α kinases PKR and PERK. High level expression of p58IPK correlated with reduced eIF2α phosphorylation and stress granule persistence later in infection (19.5 hpi).57 ATF4 is a transcription factor whose expression is translationally regulated by eIF2α and it was proposed that reovirus replication is enhanced by expression of ATF4‐induced genes.
HCV also induces stress granules in a manner dependent on eIF2α phosphorylation.58, 59 HCV replicates very slowly and induces oscillating SG assembly/disassembly over hours and days during long infections. Oscillations temporarily dissassemble SGs and relieve translation repression to enable translation of virus proteins and to maintain prolonged cell survival to support chronic infection. The induction of SGs is dependent on PKR activation and disassembly is dependent on GADD34 which regulates dephosphorylation of eIF2α 58 (Figure 1).
The coronaviruses Transmissible Gastroenteritis (TGEV) and Mouse Hepatitis Coronavirus (MHC) both form TIAR‐containing granules as infection progresses60, 61 that correlate with an increase in eIF2α phosphorylation early in infection (MHC)60 or later (TGEV).61 There is no evidence yet that these granules disassemble during the infection for either virus though published data is incomplete in this regard. SGs may restrict TGEV infection, since depletion of the SG component PTB resulted in increased virus replication and PTB induction negatively correlated with virus output. MHV also replicated better in PKR S51A mutant mouse embryonic fibroblasts60 that are defective in triggering SGs. Both MHC and TGEV induced TIAR foci but the presence of stalled translation complexes was not determined in these foci, and TIA1/TIAR can be unreliable SG markers in virus infection.17 Thus it has not been demonstrated that bona fide SGs accumulate and persist in coronavirus infection. Further research may eventually reveal that coronaviruses can co‐opt SG components as discussed below.
Viruses Co‐opt SG Components
Most cellular stress responses regulate gene expression at multiple levels, most notably translation and RNA decay. After initial synthesis of virus proteins has occurred, plus strand RNA viruses must convert individual genomes from a state that recruits ribosomes to a state of translational repression in order to clear ribosomes off the template to allow RNA replication. Thus, it is not surprising that many cellular RNA regulatory proteins are linked to virus replication schemes. If key SG factors like G3BP1 and TIA1 must aggregate to nucleate SG formation, viral sequestration of these host factors and/or redirection of their aggregation/condensation tendencies may inhibit their intended host functions in favor of new roles in virus replication. It is interesting that with alphavirus, flavivirus and HCV, and coronavirus systems discussed below, no cleavage of SG proteins has been documented, unlike picornaviruses, despite the fact that each of these viruses produces viral proteinases.
Nonstructural proteins of several alphaviruses interact in complexes together with G3BP1. G3BP1 can enter complexes containing nsP3,62, 63, 64, 65 nsP266 and nsP4, the viral polymerase62 (Figure 2). Semliki Forest Virus (SFV) inhibits stress granule formation after an initial phase of eIF2α phosphorylation and stress granule induction.67 SFV nsP3 sequesters G3BP1 into viral replication complexes and simultaneously inhibits SG formation.68 Similarly, Chikungunya virus nsP3 also represses stress granules by recruiting G3BP1 to novel cytoplasmic foci.65 The G3BP‐interacting domains of the nsP3s of both of these viruses were mapped to C‐terminal regions, but were not congruent. The G3BP1/nsP3‐containing foci appearing later in infection are not canonical stress granules as they lack the SG marker eIF3.65, 68 A viral translational enhancer near the initiating AUG codon allows SFV RNA to escape translational repression induced by eIF2α phosphorylation,67 however, efficient translation of viral RNAs with this motif also helps disassemble SGs during infection.68 The mechanistic benefit of co‐opting G3BP1 to replicase complexes is unclear but deletions of the G3BP1‐intereacting sequence in nsP3 reduces replication of viral replicons or virus.65
Sindbis virus (SBV) is another alphavirus whose RNA‐dependent RNA polymerase nsP4 is found in immunoprecipitation complexes with G3BP1 and 2.62 Since G3BP1 also complexes with nsP2 and nsP3, this may reflect overlapping interactions within a large viral replicase complex.63, 64 G3BP1 may not affect RNA replication as much as virus translation since depletion of G3BP1 only slightly altered SV RNA levels, but significantly increased SV polyprotein production.62 Furthermore, G3BP1 has been shown to regulate translation of some cellular mRNAs.69 Since G3BP1 is critical in SG assembly,45 depletion of G3BP1 and 2 abrogates SGs during infection and promotes virus production by eliminating the translation block from SG formation.
Flaviviruses such as West Nile virus (WNV) and Dengue (DENV) also prevent arsenite‐induced SG formation, however, co‐opting mechanisms involve multiple key SG‐nucleating proteins, for example, TIA1 and TIAR, in addition to G3BP1 (Figure 2). WNV can suppress arsenite‐induced SG formation,70 likely in order to divert TIAR to new functions that promote viral RNA replication.71, 72 The 3′ stem loop that is a promoter for minus strand RNA synthesis binds TIA1 and TIAR, and both proteins colocalize with replicase components in perinuclear regions of cells during WNV and DENV infections.70 Proteomic studies indicate both G3BP1 and 2 bind the 3′ UTR of DENV genomic RNA, as well as G3BP1‐interacting proteins USP10 and Caprin1.73 However, it is unclear what role these proteins have on Dengue virus replication because functional studies have not yet been performed. It is possible that stress granule proteins in new contexts promote virus translation or RNA replication, or alternatively flaviviruses may recruit stress granule proteins to prevent a strong innate immune responses induced by SG assembly (discussed below).
HCV as mentioned above induces stress granules via eIF2α phosphorylation but also co‐opts SG factors and can induce SGs at low multiplicity of infection in an eIF2‐independent manner.74 HCV induces novel foci containing HCV core protein near cytoplasmic lipid droplets (Figure 2). Several PB and stress granule proteins are redistributed to lipid droplets during the course of infection, including DDX6, G3BP1, RCK/p54, and Xrn1.23, 74 Further interactions are complex since depletion of G3BP1, TIA‐1, TIAR, PABP, USP10, and HuR affect different steps of the HCV lifecycle.23, 59, 74, 75 G3BP1 also colocalizes and interacts with NS5A and NS5B, two components of the HCV replication complex, suggesting a role for G3BP1 in HCV RNA replication,75 though it may also restrict assembly and release of HCV virions.74
The Arenavirus Junin virus does not cause induction of stress granules since expression of viral proteins N and glycoprotein precursor inhibits stress granule formation.76 Junin virus infection subverts some components of stress granules into replication‐transcription complexes. G3BP1 colocalizes with the viral protein N in novel foci that augment the infection and may also sequester it and disassemble stress granules to enhance Junin virus translation77 (Figure 2). The virus‐induced G3BP1‐containing foci are not normal SG since they contain neither PABP nor TIA1. Interestingly, some initiation factors including eIF4G and eIF4A and large and small ribosomal subunit proteins L10a and S6 are also present in replication–transcription complexes and may participate in translation of the viral RNA in replication–transcription complexes. It is unclear if G3BP functions in replication.
Finally, although HTLV Tax protein may not co‐opt SG factors in the manner discussed above, it inhibits stress granule assembly when transiently expressed.78 Tax was shown to interact with HDAC6, whose activity was earlier shown to be required for stress granule assembly,21 thus potentially blocking deacetylation‐mediated protein condensates. However, it is unclear if Tax directly inhibits HDAC6 activity, or whether Tax expression during infection was even required to inhibit stress granules.
VIRUS REGULATION OF PBS
Like stress granules, viruses can also disrupt PBs and co‐opt their components. As one might expect, relationships between viruses and PBs appear as varied and complex as with they are with stress granules. The relationships are loosely grouped into categories below that will likely require revision as more research findings are produced. Figure 3 illustrates some of the documented virus–PB relationships.
Figure 3.
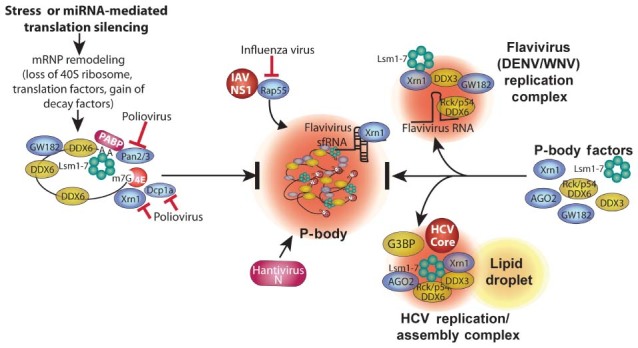
Pathways of PB disruption by viruses. PBs form via a complex series of events involving remodeling mRNPs by stripping of initiation factors and ribosome subunits, association with GW182, undergoing Pan2/3‐mediated deadenylation,microtubule transport, and association with other RNA decay factors (e.g., Xrn1, Dcp1a, DDX6 (Rck/p54), GW182 and Lsm components of the exosome), and final concentration in P‐bodies. The order of association of factors with mRNPs in PBs is arbitrary. HCV subverts many PB components into novel viral replication/assembly foci with viral core protein that also contain some SG components (e.g., G3BP, Figure 2). Flaviviruses also divert PB factors into replication foci, likely bound with viral RNA through interaction with DDX6(Rck/p54). Poliovirus induces cleavage of Dcp1a and rapid degradation of Xrn1 and Pan3. Rap55 is a critical PB factor that IAV protein NS1 diverts from normal association with PBs. Bunyavirus Junin virus incorporates viral N protein into PBs to interfere with cellular Dcp1a/2 decapping function and facilitate viral cap‐snatching.
Disruption of PBs
Several viruses disrupt PBs during infection, but details of mechanisms involved are limited and in some cases disruption and co‐opting PB components are coincident. The enteroviruses poliovirus and Coxsackievirus B3 cause total disruption of PB foci by the mid‐phase of the replication cycle. Viral and cell proteinases are involved since three PB factors involved in mRNA turnover are degraded simultaneously, Xrn1 and Pan3, and Dcp1a. The latter may be directly cleaved by viral proteinase 3C (Figure 2).79 Partial deadenylation of mRNA by the Pan2/Pan3 deadenylase complex is a requirement for mRNP inclusion in PBs,80 thus loss of Pan3 may be sufficient to disrupt PBs. Dcp1a has also been linked to regulation of PBs and its cleavage may also trigger PB dispersal,81 but further work is required to test these hypotheses. Also, the insect dicistrovirus Cricket paralysis virus moderately disrupts PB in insect cells by late times in infection. Granules tagged with GFP‐GW182 and GFP‐DCP1 diminished; however, those tagged with GFP fusion of AGO1 or AGO2 did not, suggesting that PB constituents are modified during infection resulting in alternate PB‐like foci of undetermined function.48
Influenza virus slowly disperses PBs during replication by forming a complex containing viral protein NS1 and cellular RAP55, which is otherwise required for PB formation. NS1 enters PBs, but overexpression of NS1 reduced RAP55‐associated PBs in cells.51 A viral benefit of NS1–RAP55 complex formation is to prevent viral nucleoprotein (with viral RNP) from entering PBs, where it is sequestered from viral translation or virus replication in the nucleus.
Co‐opting of PB Components
Compared to frank disruption of PBs and destruction of their components by enteroviruses, more viruses appear to co‐opt PB components during infection, usually in conjuction with a moderate decrease in PB numbers in cells. The flavivirus West Nile virus, which sequesters Tia1 on viral RNA, also sequesters several PB components, including Lsm1, GW182, DDX3, DDX6, and Xrn1, to viral replication centers82 while the numbers of PBs in cells diminish70 (Figure 3). Viral genomic RNA may directly interact with some PB components to recruit them to WNV replication centers. Some of these proteins may support RNA replication because their depletion via siRNA knockdown lowers viral RNA output.82 Genomic RNA of another flavivirus, Dengue virus, binds DDX6 (Rck/p54) at a conserved stem loop structure in the 3′ UTR adjacent to the unstructured region that binds SG proteins G3BP1and USP10.73 Similar to WNV, DDX6 knockdown reduced virus replication. Thus, Dengue and West Nile virus can co‐opt PB proteins for virus replication, and may interfere with their role in PB assembly and function. As DDX6 is proposed to coat mRNAs and organize structures within PBs43 it is possible that it plays some organizational role in RNA replication or packaging.
Flaviviruses also generate sfRNA, a fragment of the 3′ UTR of the genomic transcript produced by stalling of 5′–3′ exonucleolytic decay by Xrn1 at a highly structured pseudoknot.83, 84 sfRNA can colocalize with Xrn1 in some PB, and it is important for cytopathogenicity of Kunjin Virus.84 sfRNA's ability to inhibit Xrn1 activity via sequestration forces accumulation of uncapped cellular mRNAs in cells.85 sfRNA also exhibits RNAi suppressor activity and can inhibit Dicer cleavage activity.86 Thus, sfRNA has emerging roles in inhibiting host nucleases involved in gene regulation and innate immunity that may indirectly affect RNA granule function.
HCV also interacts with PBs or PB proteins. In this case, PBs also slowly decline throughout infection74 and the HCV core protein forms complexes with DDX3 and colocalizes in cytoplasmic foci87, 88 that are likely lipid droplets,74 although HCV core may not directly interact with DDX3.89 Additional work expanded the scope of PB components co‐opted to HCV core‐containing assembly sites at lipid droplets, and now includes DDX6 (Rck/p54), Lsm1, PATL1, Ago2, and Xrn123, 74 (Figure 3). Knockdown of PB proteins DDX3, DDX6, Lsm1, and PatL123, 90 or RCK/p54 and Ge‐174 reduced HCV replication, implying some factors play functional roles, however, these may be more in viral assembly than viral RNA replication.74 DDX6, PatL1, and the Lsm1‐7 heptameric ring play pivotal roles in the HCV life cycle at the translational and RNA replication levels.91, 92 But do PB foci per se inhibit or influence HCV replication? Recent work suggests they do not, as PB knockdown by siRNA depletion of RAP55 did not influence HCV RNA or protein levels.93 HCV may hijack DDX6 for RNA packaging, which has been observed previously for the spumaretrovirus foamy virus.94 Together these data suggest that HCV co‐opts certain PB constituents for replicative functions and others for assembly; however, there is no requirement for PB foci in HCV replication. Finally, unlike more rapidly growing and lytic enteroviruses, HCV does not seem to rely on cleavage and degradation of PB components by the virus protease.74
The ambisense segmented RNA virus family Bunyaviridae, initiate viral transcription by ‘cap‐snatching,’ to acquire 5′ m7‐guanosine capped oligonucleotides from cellular mRNAs in this process. Hantavirus nucleocapsid protein (N) binds tightly to the 5′cap of cellular mRNAs. N accumulates in PBs to inhibit Dcp1a/Dcp2‐mediated decapping (Figure 3) and also to provide ‘snatched’ 5′ caps to prime virus mRNA synthesis.95 Hantavirus transcripts must flux out of PBs to reenter the soluble cytoplasmic milieu to engage ribosomes to translate virus proteins,96 subverting PB function, rather than co‐opting PB components, for generating capped viral mRNAs that can be translated.
The trends described above were echoed in studies of replication of the plant virus Brome mosaic virus in a yeast system. Lsm1p–7p complex, Pat1p, and Dhh1p (Rck/p54, DDX6) were all required for entry of viral RNA into replication complexes on membranes.97 Interestingly, viral RNA and the viral RNA‐dependent RNA polymerase, which complexes with Lsm1p, colocalized with PBs.98 Also, some PBs can associate with membranes where viral replication complexes are built.98, 99
LINKAGE BETWEEN RNA GRANULES AND INNATE IMMUNITY
Virus infections interface with cells and induce host stress responses at multiple levels and sensors of cellular stress may be part of virus sentinel systems used to activate innate immune functions. Emerging evidence supports this notion; that innate immunity and cell stress responses, even SG and PB function, are linked at many levels. PKR, a classic interferon response protein, can coordinate pathogen sensing with cellular stress and metabolic homeostasis100 and helps regulate c‐Jun N‐terminal kinase (JNK) activation that is involved in stress responses. Further, PKR plays roles in insulin activity and metabolism by phosphorylating the insulin receptor substrate IRS1.101 PKR also functions in inflammasome activation in macrophages in response to dsRNA and bacterial infections that provoke release of cytokines IL‐1b and HMGB1.102 PKR is also a key activator of innate immune transcription responses.103, 104, 105 Thus, multiple nutrient and pathogen response systems may be integrated through PKR. G3BP1 can induce stress granules to form in the absence of applied stress or infection, which activates PKR and downstream eIF2‐dependent translational repression. This indicates PKR may sense formation of stress granules per se through a unique mechanism of activation.15 However, once activated, PKR may signal downstream to several innate immune effectors. The transcription factor nuclear factor κB (NF‐κB) functions in many immune and inflammatory responses.106 PKR interacts with the IκB kinase complex, promoting dissociation of IκB from NF‐κB and NF‐κB transcriptional activation.107, 108
In a new mechanistic aspect of innate immune activation, it was recently demonstrated that cells normally concentrate proteins that activate interferon responses together with stress granule proteins. RIG‐I like receptors (RIG‐I, MDA‐5, LGP2) that sense viral RNA can enter SGs after arsenite induction or after virus infection with an NS1 deletion mutant influenza virus.50 Functional interaction of the SG‐based sentinel mechanism with interferon activation was shown by a loss of IFN β mRNA production after depletion of PKR or G3BP1, the latter of which depletes SGs. SGs did not form in PKR knockout MEFs, a phenotype observed with many other viruses.49, 50 Virus RNA also entered the SG, but this only occurred with IAV ΔNS1 virus.50 PKR also enters stress granules, thus concentrating and colocalizing many components of the innate immune response (RIG‐I, MDA, PKR) with the mutant form of viral IAV RNA. It is interesting that PKR also enters PBs during human papilloma virus infection.109 Conversely, poliovirus RNA does not enter V‐SGs24 thus, inclusion of viral RNA in SGs is variable depending on the WT versus mutant forms of virus and is likely counteracted by viral proteins. Together, these studies suggest that SGs mediate activation of other stress signaling pathways. Indeed, the stress‐responsive MAPK JNK is activated in a noncanonical manner during stress granule formation.110
CONCLUSION
The field of virus–RNA granule interactions is still very young. While broad descriptive outlines of viral manipulation of RNA granule responses have emerged, the molecular details of mechanisms are mostly sparse.
When cells confront stressful situations, the responses are extensive and complex as cells optimize resources and mobilize many components to manage the pools of active and silenced mRNAs that are continually in flux. Viruses, as powerful inducers of cell stress, have always coped with an environment of rapid mobilization of these cellular changes, and evolved many mechanisms to either block them or subvert and redirect them for viral gain so that viral RNA expression is maintained. Details of how viruses control these responses remain sketchy in all cases, partly because the underlying mechanisms of RNA granule formation are very complex and poorly understood.
The importance of post‐transcriptional and post‐translational regulation in mobilizing stress responses is gaining recognition and may play dominant roles in mechanisms of liquid droplet condensation that actually form the RNA granules. Several key questions remain as to how this occurs, what are the major proteins that drive condensation and what specific post‐translational modifications on these proteins are responsible. Questions remain as to the impact of actual RNA granule condensation on virus replication and if viral proteins directly modulate condensation of host proteins. Since RNA granules cannot be purified from cells for analysis, major questions remain as to the identity and scope of mRNAs that become included in these foci and the extent to which they can trap viral transcripts. This is especially true for the various types of V‐SGs as opposed to SGs induced by overwhelming insults such as arsenite or heat shock. For viruses that redirect the SG response to concentrate subsets of SG components in novel virus‐induced foci, the roles of these proteins, if any, in virus replication and package require investigation. This will take significant investigative effort since the roles of many of these proteins in RNA granule assembly and function is unclear. Of course viruses are excellent probes of cellular biology and regulation of RNA granules by viruses poses an opportunity to understand more about basic mechanisms that govern RNA granule biology.
As these investigations unfold it will be important for distinctions in the compositions of RNA granules to be thoroughly characterized. The persistence of one type of granule containing one or two marker proteins during infection does not mean that a functional SG or PB is present in the cell, and fortunately there is a growing trend for investigators to examine many more marker proteins. It is also important to examine the functional consequences of RNA granule persistence, for example, translational repression for SGs and RNA stability for PBs. In the case of novel virus foci containing translation factors, the determination of whether active in situ translation occurs in these granules should be examined with modern assays.
Finally, the emerging concept that SG formation signals downstream stress signals that activate innate antiviral mechanisms as part of an integrated stress response should receive more attention. As stress responses and innate immunity likely crosstalk at multiple levels, it is possible that aspects of RNA granule biology could be exploited in the future as a broad spectrum antiviral strategy.
Acknowledgements
This work was supported by National Institutes of Health (NIH) grant R01 AI AI50237 to R.E.L. and an NCI Cancer Center Support Grant (P30CA125123).
Conflict of interest: The author has declared no conflicts of interest for this article.
REFERENCES
- 1. Chang T‐C, Yamashita A, Chen C‐YA, Yamashita Y, Zhu W, Durdan S, Kahvejian A, Sonenberg N, Shyu A‐B. UNR, a new partner of poly(A)‐binding protein, plays a key role in translationally coupled mRNA turnover mediated by the c‐fos major coding‐region determinant. Genes Dev 2004, 18:2010–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shyu A‐B, Wilkinson MF, van Hoof A. Messenger RNA regulation: to translate or to degrade. EMBO J 2008, 27:471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mao YS, Zhang B, Spector DL. Biogenesis and function of nuclear bodies. Trends Genet 2011, 27: 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caudron‐Herger M, Rippe K. Nuclear architecture by RNA. Curr Opin Genet Dev 2012, 22:179–187. [DOI] [PubMed] [Google Scholar]
- 5. Reineke LC, Lloyd RE. Diversion of stress granules and P‐bodies during viral infection. Virology 2013, 436:255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mokas S, Mills JR, Garreau C, Fournier M‐J, Robert F, Arya P, Kaufman RJ, Pelletier J, Mazroui R. Uncoupling stress granule assembly and translation initiation inhibition. Mol Biol Cell 2009, 20:2673–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dang Y, Kedersha N, Low W‐K, Romo D, Gorospe M, Kaufman R, Anderson P, Liu JO. Eukaryotic initiation factor 2α‐independent pathway of stress granule induction by the natural product pateamine A. J Biol Chem 2006, 281:32870–32878. [DOI] [PubMed] [Google Scholar]
- 8. Kedersha NL, Gupta M, Li W, Miller I, Anderson P. RNA‐binding proteins TIA‐1 and TIAR link the phosphorylation of eIF‐2 α to the assembly of mammalian stress granules. J Cell Biol 1999, 147:1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke‐Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 2005, 169:871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buchan JR, Parker R. Eukaryotic stress granules: the ins and outs of translation. Mol Cell 2009, 36:932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci 2008, 33:141–150. [DOI] [PubMed] [Google Scholar]
- 12. Kedersha N, Anderson P. Mammalian stress granules and processing bodies. MethEnzymol 2007, 431:61–81. [DOI] [PubMed] [Google Scholar]
- 13. Mazroui R, Sukarieh R, Bordeleau M‐E, Kaufman RJ, Northcote P, Tanaka J, Gallouzi I, Pelletier J. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2α phosphorylation. Mol Biol Cell 2006, 17:4212–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Emara MM, Fujimura K, Sciaranghella D, Ivanova V, Ivanov P, Anderson P. Hydrogen peroxide induces stress granule formation independent of eIF2α phosphorylation. Biochem Biophys Res Commun 2012, 423:763–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reineke LC, Dougherty JD, Pierre P, Lloyd RE. Large G3BP‐induced granules trigger eIF2α phosphorylation. Mol Biol Cell 2012, 23:3499–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ohn T, Kedersha N, Hickman T, Tisdale S, Anderson P. A functional RNAi screen links O‐GlcNAc modification of ribosomal proteins to stress granule and processing body assembly. Nat Cell Biol 2008, 10:1224–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. White JP, Lloyd RE. Poliovirus unlinks TIA1 aggregation and mRNA stress granule formation. J Virol 2011, 85:12442–12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tourrière H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, Tazi J. The RasGAP‐associated endoribonuclease G3BP assembles stress granules. J Cell Biol 2003, 160:823–831. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19. Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, Anderson P. Stress granule assembly is mediated by prion‐like aggregation of TIA‐1. Mol Biol Cell 2004, 15:5383–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goulet I, Boisvenue S, Mokas S, Mazroui R, Côté J. TDRD3, a novel Tudor domain‐containing protein, localizes to cytoplasmic stress granules. Human Molec Genet 2008, 17:3055–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kwon S, Zhang Y, Matthias P. The deacetylase HDAC6 is a novel critical component of stress granules involved in the stress response. Genes Dev 2007, 21:3381–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Solomon S, Xu Y, Wang B, David MD, Schubert P, Kennedy D, Schrader JW. Distinct structural features of Caprin‐1 mediate its interaction with G3BP‐1 and its induction of phosphorylation of eukaryotic translation initiation factor 2, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Mol Cell Biol 2007, 27:2324–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ariumi Y, Kuroki M, Kushima Y, Osugi K, Hijikata M, Maki M, Ikeda M, Kato N. Hepatitis C virus hijacks P‐body and stress granule components around lipid droplets. J Virol 2011, 85:6882–6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Piotrowska J, Hansen SJ, Park N, Jamka K, Sarnow P, Gustin KE. Stable formation of compositionally unique stress granules in virus‐infected cells. J Virol 2010, 84:3654–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujimura K, Sasaki AT, Anderson P. Selenite targets eIF4E‐binding protein‐1 to inhibit translation initiation and induce the assembly of non‐canonical stress granules. Nucl Acids Res 2012, 40:8099–8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Unsworth H, Raguz S, Edwards HJ, Higgins CF, Yagüe E. mRNA escape from stress granule sequestration is dictated by localization to the endoplasmic reticulum. FASEB J 2010, 24:3370–3380. [DOI] [PubMed] [Google Scholar]
- 27. Nover L, Scharf KD, Neumann D. Cytoplasmic heat shock granules are formed from precursor particles and are associated with a specific set of mRNAs. Mol Cell Biol 1989, 9:1298–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eisinger‐Mathason TSK, Andrade J, Groehler AL, Clark DE, Muratore‐Schroeder TL, Pasic L, Smith JA, Shabanowitz J, Hunt DF, Macara IG, et al. Codependent functions of RSK2 and the apoptosis‐promoting factor TIA‐1 in stress granule assembly and cell survival. Mol Cell 2008, 31:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Loschi M, Leishman CC, Berardone N, Boccaccio GL. Dynein and kinesin regulate stress‐granule and P‐body dynamics. J Cell Sci 2009, 122:3973–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ohn T, Anderson P. The role of posttranslational modifications in the assembly of stress granules. WIREs RNA 2010, 1:486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weber SC, Brangwynne CP. Getting RNA and protein in phase. Cell 2012, 149:1188–1191. [DOI] [PubMed] [Google Scholar]
- 32. Li P, Banjade S, Cheng H‐C, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483:336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, et al. Cell‐free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149:753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J. Cell‐free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 2012, 149:768–779. [DOI] [PubMed] [Google Scholar]
- 35. Bentmann E, Neumann M, Tahirovic S, Rodde R, Dormann D, Haass C. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA‐binding protein of 43 kDa (TDP‐43). J Biol Chem 2012, 287:23079–23094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aulas A, Stabile S, Vande Velde C. Endogenous TDP‐43, but not FUS, contributes to stress granule assembly via G3BP. Mol Neurodegener 2012, 7:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bikkavilli RK, Malbon CC. Arginine methylation of G3BP1 in response to Wnt3a regulates β‐catenin mRNA. J Cell Sci 2011, 124:2310–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zou T, Rao JN, Liu L, Xiao L, Cui Y‐H, Jiang Z, Ouyang M, Donahue JM, Wang J‐Y. Polyamines inhibit the assembly of stress granules in normal intestinal epithelial cells regulating apoptosis. Am J Physiol, Cell Physiol 2012, 303:C102–C111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Matsumoto K, Nakayama H, Yoshimura M, Masuda A, Dohmae N, Matsumoto S, Tsujimoto M. PRMT1 is required for RAP55 to localize to processing bodies. RNA Biol 2012, 9:610–623. [DOI] [PubMed] [Google Scholar]
- 40. Dammer EB, Fallini C, Gozal YM, Duong DM, Rossoll W, Xu P, Lah JJ, Levey AI, Peng J, Bassell GJ, et al. Coaggregation of RNA‐binding proteins in a model of TDP‐43 Proteinopathy with selective RGG motif methylation and a role for RRM1 ubiquitination. PLoS ONE 2012, 7:e38658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arribere JA, Doudna JA, Gilbert WV. Reconsidering movement of eukaryotic mRNAs between polysomes and P bodies. Mol Cell 2011, 44:745–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eulalio A, Behm‐Ansmant I, Schweizer D, Izaurralde E. P‐body formation is a consequence, not the cause, of RNA‐mediated gene silencing. Mol Cell Biol 2007, 27:3970–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ernoult‐Lange M, Baconnais S, Harper M, Minshall N, Souquere S, Boudier T, Bénard M, Andrey P, Pierron G, Kress M, Standart N, et al. Multiple binding of repressed mRNAs by the P‐body protein Rck/ p54. RNA 2012, 18:1702–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Teixeira D, Sheth U, Valencia‐Sanchez MA, Brengues M, Parker R. Processing bodies require RNA for assembly and contain nontranslating mRNAs. RNA 2005, 11:371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. White JP, Cardenas AM, Marissen WE, Lloyd RE. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2007, 2:295–305. [DOI] [PubMed] [Google Scholar]
- 46. White JP, Reineke LC, Lloyd RE. Poliovirus switches to an eIF2‐independent mode of translation during infection. J Virol 2011, 85:8884–8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Borghese F, Michiels T. The leader protein of cardioviruses inhibits stress granule assembly. J Virol 2011, 85:9614–9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Khong A, Jan E. Modulation of stress granules and P bodies during dicistrovirus infection. J Virol 2011, 85:1439–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Khaperskyy DA, Hatchette TF, McCormick C. Influenza A virus inhibits cytoplasmic stress granule formation. FASEB J 2012, 26:1629–1639. [DOI] [PubMed] [Google Scholar]
- 50. Onomoto K, Jogi M, Yoo J‐S, Narita R, Morimoto S, Takemura A, Sambhara S, Kawaguchi A, Osari S, Nagata K, et al. Critical role of an antiviral stress granule containing RIG‐I and PKR in viral detection and innate immunity. PLoS ONE 2012, 7:e43031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mok BW‐Y, Song W, Wang P, Tai H, Chen Y, Zheng M, Wen X, Lau S‐Y, Wu WL, Matsumoto K, et al. The NS1 protein of influenza a virus interacts with cellular processing bodies and stress granules through RNA‐associated protein 55 (RAP55) during virus infection. J Virol 2012, 86:12695–12707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Montero H, Rojas M, Arias CF, López S. Rotavirus infection induces the phosphorylation of eIF2α but prevents the formation of stress granules. J Virol 2008, 82:1496–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Qin Q, Carroll K, Hastings C, Miller CL. Mammalian Orthoreovirus escape from host translational shutoff correlates with stress granule disruption and is independent of eIF2α phosphorylation and PKR. J Virol 2011, 85:8798–8810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Qin Q, Hastings C, Miller CL. Mammalian orthoreovirus particles induce and are recruited into stress granules at early times postinfection. J Virol 2009, 83:11090–11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ventoso I, Sanz MA, Molina S, Berlanga JJ, Carrasco L, Esteban M. Translational resistance of late alphavirus mRNA to eIF2α phosphorylation: a strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev 2006, 20:87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Terenin IM, Dmitriev SE, Andreev DE, Shatsky IN. Eukaryotic translation initiation machinery can operate in a bacterial‐like mode without eIF2. Nat Struct Mol Biol 2008, 15:836–841. [DOI] [PubMed] [Google Scholar]
- 57. Smith JA, Schmechel SC, Raghavan A, Abelson M, Reilly C, Katze MG, Kaufman RJ, Bohjanen PR, Schiff LA. Reovirus induces and benefits from an integrated cellular stress response. J Virol 2006, 80:2019–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ruggieri A, Dazert E, Metz P, Hofmann S, Bergeest J‐P, Mazur J, Bankhead P, Hiet M‐S, Kallis S, Alvisi G, et al. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe 2012, 12:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Garaigorta U, Heim MH, Boyd B, Wieland S, Chisari FV. Hepatitis C virus induces the formation of stress granules whose proteins regulate HCV RNA replication, virus assembly and egress. J Virol 2012, 86:11043–11056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Raaben M, Groot Koerkamp MJA, Rottier PJM, de Haan CAM. Mouse hepatitis coronavirus replication induces host translational shutoff and mRNA decay, with concomitant formation of stress granules and processing bodies. Cell Microbiol 2007, 9:2218–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sola I, Galán C, Mateos‐Gómez PA, Palacio L, Zúñiga S, Cruz JL, Almazán F, Enjuanes L. The polypyrimidine tract‐binding protein affects coronavirus RNA accumulation levels and relocalizes viral RNAs to novel cytoplasmic domains different from replication‐transcription sites. J Virol 2011, 85:5136–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cristea IM, Rozjabek H, Molloy KR, Karki S, White LL, Rice CM, Rout MP, Chait BT, MacDonald MR. Host factors associated with the Sindbis virus RNA‐dependent RNA polymerase: role for G3BP1 and G3BP2 in virus replication. J Virol 2010, 84: 6720–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Frolova E, Gorchakov R, Garmashova N, Atasheva S, Vergara LA, Frolov I. Formation of nsP3‐specific protein complexes during Sindbis virus replication. J Virol 2006, 80:4122–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gorchakov R, Garmashova N, Frolova E, Frolov I. Different types of nsP3‐containing protein complexes in Sindbis virus‐infected cells. J Virol 2008, 82:10088–10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fros JJ, Domeradzka NE, Baggen J, Geertsema C, Flipse J, Vlak JM, Pijlman GP. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J Virol 2012, 86: 10873–10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Atasheva S, Gorchakov R, English R, Frolov I, Frolova E. Development of Sindbis viruses encoding nsP2/ GFP chimeric proteins and their application for studying nsP2 functioning. J Virol 2007, 81:5046–5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McInerney GM, Kedersha NL, Kaufman RJ, Anderson P, Liljeström P. Importance of eIF2α phosphorylation and stress granule assembly in alphavirus translation regulation. Mol Biol Cell 2005, 16:3753–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Panas MD, Varjak M, Lulla A, Eng KE, Merits A, Karlsson Hedestam GB, McInerney GM. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol Biol Cell 2012, 23:4701–4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ortega AD, Willers IM, Sala S, Cuezva JM. Human G3BP1 interacts with β‐F1‐ATPase mRNA and inhibits its translation. J Cell Sci 2010, 123: 2685–2696. [DOI] [PubMed] [Google Scholar]
- 70. Emara MM, Brinton MA. Interaction of TIA‐1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc Natl Acad Sci U S A 2007, 104:9041–9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Emara MM, Liu H, Davis WG, Brinton MA. Mutation of mapped TIA‐1/TIAR binding sites in the 3′ terminal stem‐loop of West Nile virus minus‐strand RNA in an infectious clone negatively affects genomic RNA amplification. J Virol 2008, 82:10657–10670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Li W, Li Y, Kedersha N, Anderson P, Emara M, Swiderek KM, Moreno GT, Brinton MA. Cell proteins TIA‐1 and TIAR interact with the 3′ stem‐loop of the West Nile virus complementary minus‐strand RNA and facilitate virus replication. J Virol 2002, 76: 11989–12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ward AM, Bidet K, Yinglin A, Ler SG, Hogue K, Blackstock W, Gunaratne J, Garcia‐Blanco MA Quantitative mass spectrometry of DENV‐2 RNA‐interacting proteins reveals that the DEAD‐box RNA helicase DDX6 binds the DB1 and DB2 3′ UTR structures. RNA Biol 2011, 8:1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pager CT, Schütz S, Abraham TM, Luo G, Sarnow P. Modulation of hepatitis C virus RNA abundance and virus release by dispersion of processing bodies and enrichment of stress granules. Virology 2013, 435: 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yi Z, Pan T, Wu X, Song W, Wang S, Xu Y, Rice CM, MacDonald MR, Yuan Z. Hepatitis C virus co‐opts ras‐GTPase‐activating protein‐binding protein 1 for its genome replication. J Virol 2011, 85:6996–7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Linero FN, Thomas MG, Boccaccio GL, Scolaro LA. Junin virus infection impairs stress‐granule formation in Vero cells treated with arsenite via inhibition of eIF2α phosphorylation. J Gen Virol 2011, 92:2889–2899. [DOI] [PubMed] [Google Scholar]
- 77. Baird NL, York J, Nunberg JH. Arenavirus infection induces discrete cytosolic structures for RNA replication. J Virol 2012, 86:11301–11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Legros S, Boxus M, Gatot JS, Van Lint C, Kruys V, Kettmann R, Twizere JC, Dequiedt F. The HTLV‐1 tax protein inhibits formation of stress granules by interacting with histone deacetylase 6. Oncogene 2011, 30:4050–4062. [DOI] [PubMed] [Google Scholar]
- 79. Dougherty JD, White JP, Lloyd RE. Poliovirus‐mediated disruption of cytoplasmic processing bodies. J Virol 2011, 85:64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zheng D, Ezzeddine N, Chen C‐YA, Zhu W, He X, Shyu A‐B. Deadenylation is prerequisite for P‐body formation and mRNA decay in mammalian cells. J Cell Biol 2008, 182:89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rzeczkowski K, Beuerlein K, Müller H, Dittrich‐Breiholz O, Schneider H, Kettner‐Buhrow D, Holtmann H, Kracht M. c‐Jun N‐terminal kinase phosphorylates DCP1a to control formation of P bodies. J Cell Biol 2011, 194:581–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chahar HS, Chen S, Manjunath N. P‐body components LSM1, GW182, DDX3, DDX6 and XRN1 are recruited to WNV replication sites and positively regulate viral replication. Virology 2013, 436:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Silva PAGC, Pereira CF, Dalebout TJ, Spaan WJM, Bredenbeek PJ. An RNA pseudoknot is required for production of yellow fever virus subgenomic RNA by the host nuclease XRN1. J Virol 2010, 84: 11395–11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Pijlman GP, Funk A, Kondratieva N, Leung J, Torres S, van der Aa L, Liu WJ, Palmenberg AC, Shi P‐Y, Hall RA, et al. A highly structured, nuclease‐resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 2008, 4:579–591. [DOI] [PubMed] [Google Scholar]
- 85. Moon SL, Anderson JR, Kumagai Y, Wilusz CJ, Akira S, Khromykh AA, Wilusz J. A noncoding RNA produced by arthropod‐borne flaviviruses inhibits the cellular exoribonuclease XRN1 and alters host mRNA stability. RNA 2012, 18:2029–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Schnettler E, Sterken MG, Leung JY, Metz SW, Geertsema C, Goldbach RW, Vlak JM, Kohl A, Khromykh AA, Pijlman GP. Noncoding flavivirus RNA displays RNA interference suppressor activity in insect and Mammalian cells. J Virol 2012, 86:13486–13500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mamiya N, Worman HJ. Hepatitis C virus core protein binds to a DEAD box RNA helicase. J Biol Chem 1999, 274:15751–15756. [DOI] [PubMed] [Google Scholar]
- 88. You LR, Chen CM, Yeh TS, Tsai TY, Mai RT, Lin CH, Lee YH. Hepatitis C virus core protein interacts with cellular putative RNA helicase. J Virol 1999, 73:2841–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Angus AGN, Dalrymple D, Boulant S, Mcgivern DR, Clayton RF, Scott MJ, Adair R, Graham S, Owsianka AM, Targett‐Adams P, et al. Requirement of cellular DDX3 for hepatitis C virus replication is unrelated to its interaction with the viral core protein. J Gen Virol 2010, 91:122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ariumi Y, Kuroki M, Abe K‐I, Dansako H, Ikeda M, Wakita T, Kato N. DDX3 DEAD‐box RNA helicase is required for hepatitis C virus RNA replication. J Virol 2007, 81:13922–13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Scheller N, Mina LB, Galão RP, Chari A, Giménez‐Barcons M, Noueiry A, Fischer U, Meyerhans A, Díez J. Translation and replication of hepatitis C virus genomic RNA depends on ancient cellular proteins that control mRNA fates. Proc Natl Acad Sci U S A 2009, 106:13517–13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jangra RK, Yi M, Lemon SM. DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not for internal ribosome entry site‐directed translation. J Virol 2010, 84:6810–6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Pérez‐Vilaró G, Scheller N, Saludes V, Díez J. Hepatitis C virus infection alters P‐body composition but is independent of P‐body granules. J Virol 2012, 86:8740–8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yu SF, Lujan P, Jackson DL, Emerman M, Linial ML. The DEAD‐box RNA helicase DDX6 is required for efficient encapsidation of a retroviral genome. PLoS Path 2011, 7:e1002303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mir MA, Duran WA, Hjelle BL, Ye C, Panganiban AT. Storage of cellular 5′ mRNA caps in P bodies for viral cap‐snatching. Proc Natl Acad Sci U S A 2008, 105:19294–19299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mir MA, Panganiban AT. A protein that replaces the entire cellular eIF4F complex. EMBO J 2008, 27: 3129–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Noueiry AO, Díez J, Falk SP, Chen J, Ahlquist P. Yeast Lsm1p‐7p/Pat1p deadenylation‐dependent mRNA‐decapping factors are required for brome mosaic virus genomic RNA translation. Mol Cell Biol 2003, 23:4094–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Beckham CJ, Light HR, Nissan TA, Ahlquist P, Parker R, Noueiry A. Interactions between brome mosaic virus RNAs and cytoplasmic processing bodies. J Virol 2007, 81:9759–9768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wang X, Lee W‐M, Watanabe T, Schwartz M, Janda M, Ahlquist P. Brome mosaic virus 1a nucleoside triphosphatase/helicase domain plays crucial roles in recruiting RNA replication templates. J Virol 2005, 79:13747–13758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Nakamura T, Furuhashi M, Li P, Cao H, Tuncman G, Sonenberg N, Gorgun CZ, Hotamisligil GS. Double‐stranded RNA‐dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 2010, 140:338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Yang X, Nath A, Opperman MJ, Chan C. The double‐stranded RNA‐dependent protein kinase differentially regulates insulin receptor substrates 1 and 2 in HepG2 cells. Mol Biol Cell 2010, 21:3449–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundbäck P, Valdes‐Ferrer SI, Olofsson PS, Kalb T, Roth J, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012, 488:670–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Taghavi N, Samuel CE. Protein kinase PKR catalytic activity is required for the PKR‐dependent activation of mitogen‐activated protein kinases and amplification of interferon β induction following virus infection. Virology 2012, 427:208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Steele L, Errington F, Prestwich R, Ilett E, Harrington K, Pandha H, Coffey M, Selby P, Vile R, Melcher A. Pro‐inflammatory cytokine/chemokine production by reovirus treated melanoma cells is PKR/NF‐κB mediated and supports innate and adaptive anti‐tumour immune priming. Mol Cancer 2011, 10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 2006, 70:1032–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ghosh S, May MJ, Kopp EB. NF‐κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 1998, 16:225–260. [DOI] [PubMed] [Google Scholar]
- 107. Gil J, Rullas J, Garcia MA, Alcamí J, Esteban M. The catalytic activity of dsRNA‐dependent protein kinase, PKR, is required for NF‐κB activation. Oncogene 2001, 20:385–394. [DOI] [PubMed] [Google Scholar]
- 108. Bonnet MC, Weil R, Dam E, Hovanessian AG, Meurs EF. PKR stimulates NF‐κB irrespective of its kinase function by interacting with the IκB kinase complex. Mol Cell Biol 2000, 20:4532–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hebner CM, Wilson R, Rader J, Bidder M, Laimins LA. Human papillomaviruses target the double‐stranded RNA protein kinase pathway. J Gen Virol 2006, 87:3183–3193. [DOI] [PubMed] [Google Scholar]
- 110. Wasserman T, Katsenelson K, Daniliuc S, Hasin T, Choder M, Aronheim A. A novel c‐Jun N‐terminal kinase (JNK)‐binding protein WDR62 is recruited to stress granules and mediates a nonclassical JNK activation. Mol Biol Cell 2010, 21:117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]