

Abstract
Acute stress reduces pain sensitivity by engaging an endocannabinoid signaling circuit in the midbrain. The neural mechanisms governing this process and molecular identity of the endocannabinoid substance(s) involved are unknown. We combined behavior, pharmacology, immunohistochemistry, RNA interference, quantitative RT-PCR, enzyme assays, and lipidomic analyses of endocannabinoid content to uncover the role of the endocannabinoid 2-arachidonoyl-sn-glycerol (2-AG) in controlling pain sensitivity in vivo. Here, we show that footshock stress produces antinociception in rats by activating type 5 metabotropic glutamate receptors (mGlu5) in the dorsolateral periaqueductal gray (dlPAG) and mobilizing 2-AG. Stimulation of mGlu5 in the dlPAG with DHPG [(S)-3,5-dihydroxyphenylglycine] triggered 2-AG formation and enhanced stress-dependent antinociception through a mechanism dependent upon both postsynaptic diacylglycerol lipase (DGL) activity, which releases 2-AG, and presynaptic CB1 cannabinoid receptors. Pharmacological blockade of DGL activity in the dlPAG with RHC80267 [1,6-bis(cyclohexyloximinocarbonylamino)hexane] and (−)-tetrahydrolipstatin (THL), which inhibit activity of DGL-α and DGL-β isoforms, suppressed stress-induced antinociception. Inhibition of DGL activity in the dlPAG with THL selectively decreased accumulation of 2-AG without altering levels of anandamide. The putative 2-AG-synthesizing enzyme DGL-α colocalized with mGlu5 at postsynaptic sites of the dlPAG, whereas CB1 was confined to presynaptic terminals, consistent with a role for 2-AG as a retrograde signaling messenger. Finally, virally mediated silencing of DGL-α, but not DGL-β, transcription in the dlPAG mimicked effects of DGL inhibition in suppressing both endocannabinoid-mediated stress antinociception and 2-AG formation. The results indicate that activation of the postsynaptic mGlu5–DGL-α cascade triggers retrograde 2-AG signaling in vivo. This pathway is required for endocannabinoid-mediated stress-induced analgesia.
Introduction
Endocannabinoids are phospholipid-derived messengers that regulate central neurotransmission by activating presynaptic CB1-type cannabinoid receptors (Wilson and Nicoll, 2001; Alger, 2002; Gerdeman et al., 2002; Carey et al., 2011). One of these neuromodulatory lipids, the arachidonic acid derivative 2- arachidonoyl-sn-glycerol (2-AG) (Mechoulam et al., 1995; Sugiura et al., 1995), is thought to act as a retrograde signal at excitatory and inhibitory synapses throughout the brain (Robbe et al., 2002; Chevaleyre and Castillo, 2003; Uchigashima et al., 2007; Gao et al., 2010; Tanimura et al., 2010). Evidence suggests that 2-AG is released in a stimulus-dependent manner by the consecutive action of two enzymes found at postsynaptic spines, phospholipase C-β (PLC-β) and diacylglycerol lipase-α (DGL-α) (Stella et al., 1997; Hashimotodani et al., 2005, 2008, 2011; Lafourcade et al., 2007). PLC-β hydrolyzes phosphatidylinositol 4,5-bisphosphate to generate 1,2-diacylglycerol (DAG), while DGL-α cleaves newly formed DAG to generate 2-AG (Piomelli, 2003; Jung et al., 2007). At central synapses, DGL-α often colocalizes with type 5 metabotropic glutamate receptors (mGlu5), combining into a multiprotein complex that may initiate retrograde endocannabinoid signaling (Maejima et al., 2001; Uchigashima et al., 2007; Nyilas et al., 2009). Indeed, mGlu5 activation stimulates DGL-α to trigger the biosynthesis of 2-AG in vitro (Jung et al., 2005, 2007) and depresses synaptic transmission through a CB1-dependent mechanism (Uchigashima et al., 2007). However, direct evidence that this pathway controls 2-AG formation in vivo has remained elusive. Genetic deletion of DGL-α decreases 2-AG levels in mouse brain and suppresses retrograde endocannabinoid signaling (Gao et al., 2010; Tanimura et al., 2010). Nevertheless, DGL-α deletion is accompanied by substantial alterations in the steady-state levels of other signaling lipids, including arachidonic acid and the endocannabinoid anandamide (Gao et al., 2010; Tanimura et al., 2010). Thus, alternative approaches are required to unequivocally identify the role of 2-AG in behavior. The physiological mechanisms that control 2-AG signaling in vivo and conditions under which such signaling is engaged remain unknown.
Exposure to an acute environmental stressor causes a profound antinociceptive response in rats that is partially dependent on endocannabinoid signaling (Hohmann et al., 2005; Suplita et al., 2005, 2006; Nyilas et al., 2009). The endocannabinoid-mediated component of this stress-induced antinociceptive response is enhanced by blocking the enzymatic degradation of 2-AG in the dorsolateral periaqueductal gray (dlPAG) of the midbrain (Hohmann et al., 2005). These results provide an experimental framework in which to investigate the functional significance of 2-AG signaling in vivo. Here, we used a multidisciplinary approach to elucidate the molecular mechanisms that govern 2-AG formation in the PAG and identify the resulting effects on behavior. These studies combined behavior, pharmacology, immunohistochemistry (at light- and electron-microscopic levels), RNA interference, quantitative RT-PCR, enzyme assays, and targeted lipidomic analyses to uncover the role of 2-AG signaling in controlling pain sensitivity in vivo. Our findings indicate that exposure to an environmental stressor modulates nociception by triggering a postsynaptic mGlu5–DGL-α–2-AG cascade in neurons of the dlPAG. This process initiates local retrograde signaling that is specifically mediated by 2-AG acting at presynaptic CB1 receptors.
Materials and Methods
Subjects.
Male Sprague Dawley rats (275–350 g; Harlan) were used for in vivo studies. Procedures were approved by the Institutional Animal Care and Use Committee and followed guidelines of the International Association for the Study of Pain (Zimmermann, 1983). The experimenter was blinded to experimental conditions in all studies.
Cannulae implantation.
Rats were anesthetized (intraperitoneally) with Nembutal (25 mg/kg) and ketamine (40 mg/kg). Stainless-steel guide cannulae (24 g; Small Parts) were implanted unilaterally 2 mm above the dlPAG [+1.6 mm anteroposterior (AP), +0.67 mm lateromedial (LM), −5.35 mm dorsoventral (DV), relative to lambda, the midline suture, and the skull surface, respectively] or deliberately off-site using a rat brain atlas (Paxinos and Watson, 1998). Stainless-steel insect pins prevented occlusion of cannulae. On test days, the microinjection needle was inserted 2 mm beyond the cannulae to hit the PAG at the stereotaxic coordinate (DV) designated above. Animals were allowed to recover 5–7 d before testing.
Drug preparation and administration.
(S)-3,5-Dihydroxyphenylglycine (DHPG) was from Tocris Bioscience. N-(Piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (SR141716) (rimonabant) was from NIDA. N-Formyl-l-leucine (1S)-1-[[(2S,3S)-3-hexyl-4-oxo-2-oxetanyl]methyl]dodecyl ester [(−)-tetrahydrolipstatin (THL)] and 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP) were from Sigma-Aldrich. 1,6-bis(cyclohexyloximinocarbonylamino)hexane (RHC80267) was from Calbiochem. Drugs were dissolved in a DMSO vehicle and delivered intracranially using our previously published methods (Hohmann et al., 2005). Doses were those used previously (Hohmann et al., 2005; Nyilas et al., 2009) or selected based upon pilot studies performed in our laboratory. Animals received a single intracranial injection, administered at a speed of 30 nl/s with a microinfusion pump (UltraMicroPump II; World Precision Instruments). To ensure that injection spread did not differ between studies, multiple compounds were always coinjected in a single injection volume (1 μl). The vehicle and injection volume was constant in all studies using pharmacological manipulations. Effects of pharmacological manipulations were always compared with vehicle controls tested concurrently.
Preparation of small hairpin RNA expressing adenoassociated virus.
We generated small hairpin RNA (shRNA) constructs containing both a cytomegalovirus promoter-driven green fluorescent protein (GFP) and a U6 promoter-driven shRNA expression system (Jung et al., 2007). shRNAs were designed using BLOCK-iT RNAi Designer (https://rnaidesigner.invitrogen.com/rnaiexpress/), and corresponding oligonucleotides were synthesized. Sense and antisense oligonucleotides were annealed and ligated into pENTR entry vector (Invitrogen) to generate U6 promoter-shRNA-Pol III terminator cassette. The oligonucleotides used were as follows: LacZi, 5′-CACCGCTACACAAATCAGCGATTTCGAAAAATCGCTGATTTGTGTAG-3′ (top) and 5′-AAAACTACACAAATCAGCGATTTTTCGAAATCGCTGATTTGTGTAGC-3′ (bottom); DGL-αi, 5′-CACCGCTACAAAGAAGTCTGCTACTCGAAAGTAGCAGACTTCTTTGTAGC-3′ (top) and 5′-AAAAGCTACAAAGAAGTCTGCTACTTTCGAGTAGCAGACTTCTTTGTAGC-3′ (bottom); DGL-βi, 5′-CACCGGATCGCCAGTCTGACGTTATCGAAATAACGTCAGACTGGCGATCC-3′ (top) and 5′-AAAAGGATCGCCAGTCTGACGTTATTTCGATAACGTCAGACTGGCGATCC-3′ (bottom). shRNA plasmids were tested for silencing activity in cultured cells, and shRNA sequences were moved into an adenoassociated virus (AAV) vector (pAAV2-hrGFP; Stratagene) as described previously (Jung et al., 2007). Adenoassociated viruses were prepared and purified, and viral genome titers were determined (Applied Viromics) (Zi-AAV, 7.6e11 GC/ml; αi, 1.2e12 GC/ml; βi, 5.3e11 GC/ml).
Behavioral testing.
Stress antinociception was quantified behaviorally by measuring the latency for a rat to remove its tail from a radiant heat source (IITC; model 33A) (Hohmann et al., 2005; Nyilas et al., 2009). A cutoff latency (10 s) was used to prevent tissue damage. Stable baseline withdrawal responses were established before drug manipulations. Animals received a single intracranial injection, administered at a speed of 30 nl/s, with a microinfusion pump (UltraMicroPump II; World Precision Instruments). The microinjection needle remained in place for 60 s following infusion. In separate studies, groups of rats received intra-dlPAG injections of one of the following: (1) vehicle or the mGlu5 antagonist MPEP (2.3 × 10−5 μg or 2.3 μg) (n = 7–8 per group); (2) vehicle or the group I mGlu agonist DHPG (0.02 or 0.2 μg) (n = 6 per group); (3) vehicle, DHPG (0.2 μg), or DHPG (0.2 μg) coadministered with MPEP (2.3 × 10−5 μg) (n = 5–8 per group); (4) vehicle, DHPG (0.2 μg), the CB1 antagonist rimonabant (0.1 μg) or DHPG (0.2 μg) coadministered with rimonabant (0.1 μg) (n = 6 per group); (5) vehicle or the diacylglycerol lipase inhibitor RHC80267 (1, 10 or 100 μg) (n = 6–7 per group); (6) vehicle or the diacylglycerol lipase inhibitor THL (0.1, 1, or 10 μg) (n = 7–9 per group); and (7) vehicle, DHPG (0.2 μg), or DHPG (0.2 μg) coadministered with THL (0.1 μg) (n = 9–11 per group). In control experiments, active compounds and the vehicle were also injected deliberately off-site of the dlPAG to control for site specificity (n = 4–8 per group). Immediately following microinjection, tail-flick latencies were assessed at 2 min intervals and averaged to assess changes in basal nociceptive thresholds induced by pharmacological manipulations before footshock. Stress antinociception was induced 5 min following a single unilateral microinjection as described previously (Hohmann et al., 2005). The use of unilateral microinjections enabled us to maintain consistency with our previously published work while also minimizing possible stress associated with performing multiple injections that could otherwise alter endogenous analgesic tone. The experimenter was blinded to experimental conditions in all studies. Tail-flick latencies were measured following footshock at 2 min intervals over 40–60 min. Consecutive measurements were averaged for each rat before (baseline) and after (postshock) exposure to the footshock stressor to create two-point blocks, used for repeated-measure statistical analyses, as described previously (Hohmann et al., 2005).
In vivo RNA silencing.
Viruses were injected bilaterally into the dlPAG (n = 12 per group) or deliberately off-site of the dlPAG (n = 5–6 per group) under isoflurane anesthesia. Separate groups of rats received infusions (0.4 μl per side) of either the DGL-α silencing virus (DGL-αi), the DGL-β silencing virus (DGL-βi), or the LacZ control virus (LacZi) in a single survival surgery. Naive animals (n = 9) were left intact. Two weeks following surgery (or no surgery), tail-flick latencies were assessed before (baseline) and after exposure to footshock (3 min, 3 mA; MED Associates) at 2 min intervals. Tissue samples were obtained from separate groups of rats killed at the time point (1 min) and postsurgical delay (2 weeks) associated with maximal suppression of stress antinociception in pilot studies (compared with animals killed 1 or 4 weeks after viral infection) (data not shown).
Histology.
Injection sites were confirmed in Nissl-stained sections (40 μm) by an experimenter blinded to experimental conditions. GFP localization was assessed using a Leica DMLB fluorescence microscope to confirm viral infection and map injection sites.
Ex vivo studies: tissue generation.
To generate tissue for ex vivo experiments, animals with chronic indwelling cannulae were habituated to handling and the guillotine for at least 5 d before the experiment. Behaviorally active doses of DHPG (0.2 μg), THL (10 μg), or vehicle were microinjected into the dlPAG. Rats were decapitated at behaviorally relevant time points following termination of footshock (i.e., when effects of pharmacological manipulations were maximal; 15 min for DHPG; 2 min for THL; 1 min for RNA silencing). Whole brains were rapidly dissected, snap frozen in isopentane (−30°C), and stored (−80°C) until use. Punches (1 mm deep) containing the entire PAG were taken with a 2-mm-diameter sample corer between plate 50 and plate 44 of the rat brain atlas (Paxinos and Watson, 1998). Coronal sections were obtained with a cryostat before and after the punch to verify that the level of each punch was consistent between animals. To control for site specificity, equivalently sized punches were obtained deliberately off-site from the midbrain reticular formation in the same brains.
Lipid extractions.
Punches obtained from frozen brains were homogenized in methanol (1 ml per punch) containing heptadecenoylethanolamide and [2H8]-2-AG (Cayman Chemical) as internal standards. Lipids were extracted with methanol/chloroform/water (1:2:1, by vol). Organic phases were collected and dried under N2. Endocannabinoids were fractionated by open-bed silica gel column chromatography, as described previously (Giuffrida et al., 2000). Lipids were reconstituted in chloroform and loaded onto small glass columns packed with Silica Gel G (60 Å 230–400 Mesh ASTM; Whatman). Anandamide and 2-AG were eluted with 9:1 chloroform/methanol (v/v). Eluates were dried under N2 and reconstituted in 70 μl of chloroform/methanol (1:3, v/v) for liquid chromatography/mass spectrometry (LC/MS) analysis.
In vitro DGL assay.
DGL activity assay was performed as described previously (Jung et al., 2007). Brain punches were homogenized in 50 mm Tris-HCl, pH 7.0, and homogenates were centrifuged (800 × g for 5 min at 4°C). DGL activity was measured using the supernatant (100 μg proteins) at 37°C for 30 min in 50 mm Tris-HCl, pH 7.0, containing 0.1% Triton X-100, and the substrate diheptadecanoylglycerol (50 μm). Reactions were stopped by adding chloroform/methanol (1:1) containing [2H8]-2-AG. Lipids were extracted, and the levels of reaction product monoheptadecanoylglycerol [M+Na]+ (m/z = 367) were quantified by LC/MS using [2H8]2-AG (m/z = 409) as an internal standard.
mRNA quantitation.
We used a quantitative real-time PCR method to analyze mRNAs. We extracted RNA from brain punches using a TRIzol (Invitrogen)/RNeasy (QIAGEN) hybrid protocol. First-strand complementary DNAs were synthesized from 2 μg of the total RNA using SuperScript II RNase H reverse transcriptase (Invitrogen) and oligo-dT12–18 primers, for 50 min at 42°C. Quantitative PCR was conducted using Mx3000P system (Stratagene) by a TaqMan method. DGL mRNA levels were normalized using GAPDH (glyceraldehyde-3-phosphate dehydrogenase) as an internal standard. The primer/probe set were as follows: for rat DGL-α (GI: 54312093), forward, 5′-CCAGGCCTTTGGGCG-3′; reverse, 5′-GCCTACCACAATCAGGCCAT-3′; TaqMan probe, 5′-ACCTGGGCCGTGGAACCAAACA-3′, for rat DGL-β (GI: 34870416), forward, 5′-AGGACTGCGTGGCCCAC-3′; reverse, 5′-CGTTGACCAGTCTTCGATAAATGT-3′; TaqMan probe, 5′-AGGGAATTGCTCAAGCGGCCAGA-3′.
LC/MS analyses.
We measured endocannabinoids using an 1100-LC system coupled to a 1946A-MS detector (Agilent Technologies) equipped with an electrospray ionization interface in punches obtained from each frozen sample. Lipids were separated using an XDB Eclipse C18 column (50 × 4.6 mm; inner diameter, 1.8 μm; Zorbax), eluted with a gradient of methanol in water (from 75 to 85% in 2.5 min, 85 to 90% in 5 min, and then 100% in 0.5 min) at a flow rate of 1.0 ml/min. Column temperature was kept at 40°C. MS detection was in the positive ionization mode, capillary voltage was set at 3 kV, and fragmentor voltage was varied from 120 V. N2 was used as drying gas at a flow rate of 13 L/min and a temperature of 350°C. Nebulizer pressure was set at 60 psi. Quantifications were conducted using an isotope-dilution method, monitoring Na+ adducts of the molecular ions ([M+Na]+).
Immunohistochemistry for mGlu5.
Adult male Sprague Dawley rats (n = 5; ∼70 d of age) were perfused transcardially under deep Nembutal (50 mg/ml) anesthesia with 100 ml of cold heparinized 0.1 m PBS followed by 300 ml of cold 4% paraformaldehyde. Brains were collected, postfixed (4% paraformaldehyde at 4°C for 24 h), and cryoprotected in 30% sucrose (4°C for at least 72 h). Free-floating alternating sections (30 μm) were kept in wells of 0.1 m PBS. Endogenous peroxidases were inactivated by incubation in 0.1 m PBS containing 0.3% H2O2. Tissues were blocked (1.5% goat serum) and incubated for 1 h at 20°C and 48 h at 4°C in polyclonal 21 residue synthetic peptide rabbit anti-mGlu5 directed against the C-terminal of mGlu5 with a lysine added to the N-terminal (Millipore; 1:10,000). Antibodies were diluted in 0.4% Triton PBS. Biotinylated goat anti-rabbit IgG (1:600; Vector Laboratories) served as the secondary antibody in which the tissues incubated for 1 h at 20°C. Tissues were incubated with Elite ABC kit (1:200; Vector Laboratories) for 1 h, followed by 5 min incubation in diaminobenzidine solution (0.5 mg/ml; KPL) containing 0.1% H2O2, and intensified with 2% Ni SO46H2O. Sections were mounted onto gelatin-subbed slides, air-dried, dehydrated in ascending concentrations of ethanol, cleared in xylene, and coverslipped. Preabsorption controls performed previously (Alvarez et al., 2000) with the same commercially available anti-mGlu5 antibody showed specificity. Additional controls included primary and secondary antibody omissions. Staining in our hands was absent in all control conditions and was consistent with expression patterns reported previously (Azkue et al., 1997). Images were captured using a Leica DMLB light microscope and a Retiga 1300 digital camera. Brightness/contrast was adjusted for the entire image in Adobe Photoshop, so that backgrounds appeared similar.
Light and electron microscopy for CB1, DGL-α, and mGlu5.
Experiments were performed according to institutional guidelines of ethical code and the Hungarian Act of Animal Care and Experimentation (1998. XXVIII. Section 243/1998). Brains were fixed in 4% paraformaldehyde and processed for immunoperoxidase and preembedding immunogold labeling as described previously (Katona et al., 2006). For light- and electron-microscopic analyses, sections from adult C57BL/6J wild-type mice and their CB1 knock-out (Zimmer et al., 1999) littermates (four males, 50 and 58 d of age; two females, 76 d of age) and three male Wistar rats (47–80 d of age) were used. For primary antibodies, we used affinity-purified guinea pig or goat anti-CB1 antibodies [1:200–1:500 (Fukudome et al., 2004); or 1:2000, ∼1 or 0.225 μg/ml, respectively], a rabbit anti-DGL-α antibody [1:1000–1:3000; ∼0.3–1 μg/ml; termed “INT” (Katona et al., 2006)], and an affinity-purified guinea pig anti-mGlu5 antibody [1:300; ∼1 μg/ml (Uchigashima et al., 2007)]. In immunoperoxidase stainings, primary antibody incubations were followed by application of the appropriate biotinylated secondary antibody (1:300–1:500; Vector Laboratories) and incubation with avidin–biotinylated-horseradish peroxidase complex (1:500; Elite-ABC; Vector Laboratories); the immunoperoxidase reaction was developed using 3,3′-diaminobenzidine (DAB) as the chromogen. In the double-immunostaining experiments, sections were developed first for immunogold and then for immunoperoxidase labeling. In the immunogold staining procedure, sections were incubated in 0.8 nm gold-conjugated secondary antibody (1:50; AURION), and then sections were silver intensified using the R-GENT SE-EM (AURION). Sections for electron microscopy were treated with OsO4 and uranyl acetate, dehydrated in an ascending series of ethanol and acetonitrile, and embedded in Durcupan (ACM; Fluka). Areas of interest were reembedded and resectioned, serial ultrathin sections (60 nm thickness) were collected on Formvar-coated single-slot grids, and they were analyzed using electron microscopy.
Statistical analysis.
Data were analyzed by repeated-measures ANOVA, ANOVA, and planned comparison t tests, one- or two-tailed, as appropriate. The Greenhouse–Geisser correction was applied to repeated factors. Post hoc comparisons were performed using Fisher's protected least-squares difference (PLSD) test (in the case of multiple comparisons) or Dunnett's test (in the case of comparisons to control) with p < 0.05 considered significant. SPSS (version 16.0; SPSS, Inc.) and GraphPad Prism (version 5) statistical software were used. Every two consecutive tail-flick latencies were averaged for each animal before (baseline) and after (postshock) exposure to footshock to form two-point blocks used in data analyses. Postinjection tail-flick latencies, determined immediately before footshock, were averaged into a single block for each animal and averaged across animals for each drug treatment as described previously (Hohmann et al., 2005).
Results
mGlu5 initiates stress antinociception and 2-AG mobilization
To examine the role of mGlu5 in the expression of endocannabinoid-mediated stress antinociception, we first mapped the distribution of mGlu5 in the rat PAG. We observed dense and specific immunoreactivity for mGlu5 in the dorsolateral (dl) columns of the rat PAG (Fig. 1A–D), a midbrain structure implicated in nociceptive signaling (Walker et al., 1999; Hohmann et al., 2005) and defensive behavior (Bandler et al., 2000). Pharmacological experiments subsequently revealed that mGlu5 signaling in the dlPAG contributes to stress antinociception. Microinjection of the mGlu5 antagonist MPEP into the dlPAG suppressed (F(1,19) = 8.50; p = 0.002) the antinociceptive effect produced by a brief acute stress (3 min continuous electric footshock), and these effects were time dependent (F(45,285) = 109.7; p < 0.0001) (Fig. 1E,F). The high dose of MPEP suppressed stress antinociception relative to treatment with either the vehicle (p = 0.001) or the low dose of MPEP (p = 0.038), whereas the low dose of MPEP did not reliably alter stress antinociception (p = 0.09). By contrast, basal nociceptive thresholds, assessed immediately before exposure to the stressor, were unaltered by pharmacological manipulations (Fig. 1E). Consistent with the findings outlined above, microinjections of DHPG, a group I (mGlu5 and mGlu1) agonist, into the dlPAG increased the magnitude (F(2,15) = 8.11; p = 0.004) and altered the time course (F(30,225) = 2.51; p = 0.006) of stress antinociception without changing basal nociceptive thresholds (Fig. 2A). Post hoc comparisons revealed that both the high (p = 0.002) and low (p = 0.005) doses of DHPG enhanced stress antinociception. The effect of DHPG was also confined to the dlPAG because off-site injections of DHPG (0.2 μg) failed to alter stress antinociception (p = 0.402; Fig. 2B). Moreover, microinjection of DHPG (0.2 μg) into the dlPAG potentiated both the duration (F(30,240) = 3.36; p = 0.004) and magnitude (F(2,16) = 6.47; p = 0.009) of stress antinociception relative to vehicle (p = 0.01). This effect was prevented (p = 0.05) by treatment with the mGlu5 antagonist MPEP, used at a dose (2.3 × 10−5 μg; Fig. 1E) that did not influence nociception when administered alone (Fig. 2C). These results suggest that activation of mGlu5 in the dlPAG is both necessary and sufficient for the expression of endocannabinoid-mediated stress-induced antinociception.
Figure 1.
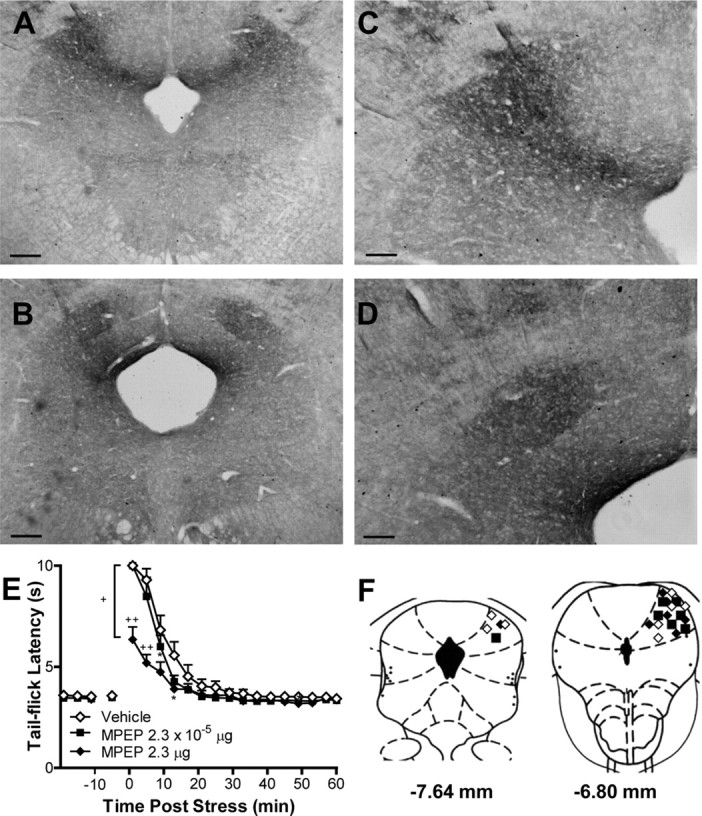
mGlu5 in the dlPAG is involved in stress-induced analgesia. A–D, Light micrographs show dense and specific immunoreactivity for mGlu5 in the dlPAG. Scale bars: A, B, 100 μm; C, D, 200 μm. E, The mGlu5 antagonist MPEP (2.3 μg) suppressed stress antinociception (p < 0.01, ANOVA) in the tail-flick test relative to vehicle (p < 0.001) or groups receiving an inactive dose of MPEP (2.3 × 10−5 μg) (p < 0.05). MPEP (2.3 μg) produced a time-dependent suppression (p < 0.0001, ANOVA) of stress antinociception without altering the basal nociceptive threshold. Antinociception was measured as the tail-flick latency. F, Microinjection sites, plotted on coronal diagrams relative to bregma, for groups receiving 2.3 × 10−5 μg (■), 2.3 μg (♦) MPEP or vehicle (♢). Data are expressed as the mean ± SEM. *p < 0.05 versus control; ++p < 0.001, +p < 0.05 versus all conditions (ANOVA, Fisher's PLSD test; n = 7–8 per group).
Figure 2.
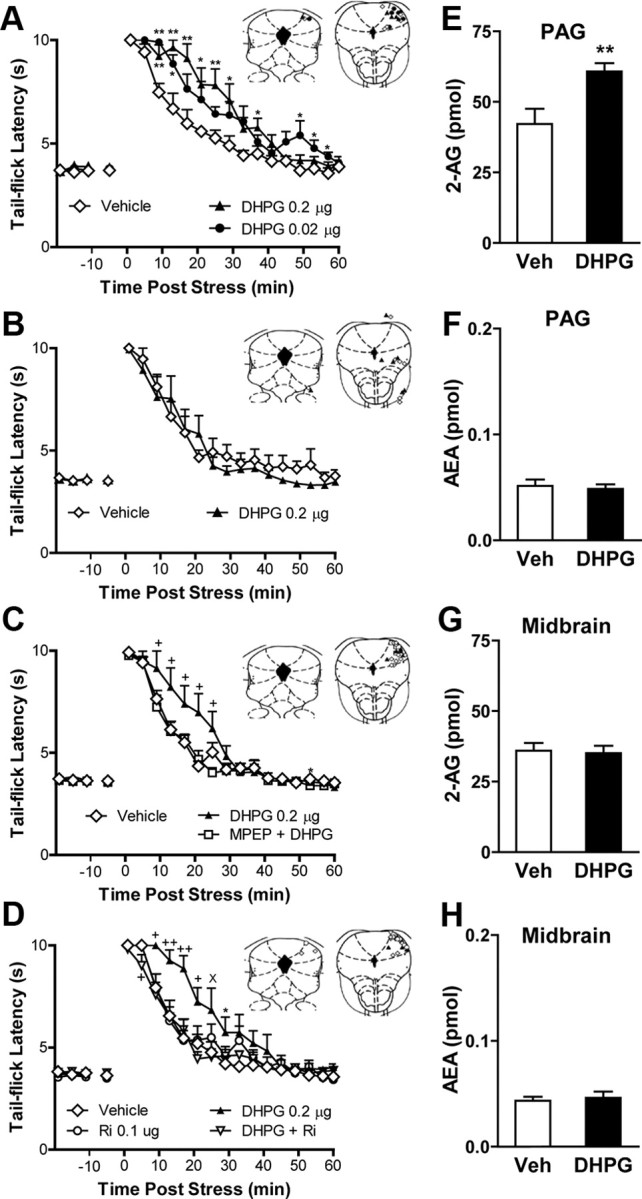
Activation of mGlu5 in the dlPAG enhances endocannabinoid-mediated stress antinociception by mobilizing the endocannabinoid 2-AG. A, Intra-dlPAG microinjection of the group I mGlu agonist DHPG (0.02 or 0.2 μg) enhanced (p < 0.01, ANOVA) stress antinociception relative to vehicle (p < 0.01 vs all comparisons). B, Off-site microinjection of DHPG (0.2 μg) did not alter stress antinociception. C, D, DHPG-induced enhancement of stress antinociception (p < 0.01, ANOVA) was blocked by behaviorally inactive doses of MPEP (2.3 × 10−5 μg; p < 0.01 vs DHPG) (C) or the CB1 antagonist rimonabant (0.1 μg; p = 0.01, ANOVA; p < 0.01 vs DHPG) (D). E, F, Intra-dlPAG microinjection of DHPG (0.2 μg) increased 2-AG (p < 0.01, one-tailed t test) (E) but not anandamide (F) accumulation in the PAG relative to vehicle (Veh). Intra-dlPAG microinjection of DHPG (0.2 μg) did not alter 2-AG (G) or anandamide (H) accumulation in the midbrain reticular formation. Data are expressed as mean ± SEM. Insets, Microinjection sites following 0.02 μg of DHPG (●), 0.2 μg of DHPG (▴), MPEP plus DHPG (□), Rimonabant (Ri) (○), DHPG plus Rimonabant (▿), and vehicle (♢) injections in the PAG (A, C, D) or off-site (B). A–D, **p < 0.01, *p < 0.05 versus control; +p < 0.05 versus all conditions; ×p < 0.05 versus antagonist coadministration (ANOVA, Fisher's PLSD test; n = 6–8 per group). E–H, **p < 0.01 (t test; n = 7–8 per group).
If mGlu5 activation in the dlPAG mobilizes an endocannabinoid messenger, then (1) DHPG microinjection into the dlPAG should cause on-demand mobilization of endocannabinoid mediators, and (2) local blockade of CB1 receptors should prevent the ability of DHPG to enhance stress-induced antinociception. In agreement with these predictions, liquid chromatography/mass spectrometry analyses of midbrain punches containing the PAG showed that DHPG elicits a local accumulation of 2-AG (t(13) = 2.93; p = 0.006) (Fig. 2E), but not anandamide (p = 0.727; Fig. 2F), in the PAG of rats subjected to footshock and killed at the peak of the DHPG-induced potentiation of stress antinociception. This effect was restricted to the PAG as no changes in either 2-AG (p = 0.818) or anandamide (p = 0.675) content were observed when DHPG was administered in the adjacent midbrain reticular formation (Fig. 2G,H). These findings suggest that acute footshock stress modulates nociceptive signaling by activating mGlu5 and stimulating 2-AG mobilization within the PAG. Consistent with these observations, DHPG increased both the duration (F(45,300) = 2.53; p = 0.003) and magnitude (F(3,20) = 4.59; p = 0.01) of stress antinociception and this effect was blocked (p = 0.005) by coinjection of the CB1 antagonist rimonabant [at a dose that did not depress nociceptive responding relative to vehicle (p = 0.609) when administered alone] (Fig. 2D). Post hoc analyses revealed that DHPG reliably increased stress antinociception relative to groups receiving vehicle (p = 0.005), rimonabant (p = 0.016), or rimonabant coadministered with DHPG (p = 0.005). Stress antinociception in groups receiving rimonabant alone (p = 0.609) or coadministered with DHPG (p = 0.981) was similar to that observed in groups receiving vehicle. Thus, DHPG microinjection in the dlPAG triggers 2-AG mobilization to enhance stress antinociception through a mechanism that requires cannabinoid CB1 receptor activation.
Presynaptic CB1 receptors and postsynaptic DGL-α is present in dlPAG
We next probed the molecular architecture of the sites of action of 2-AG within the PAG. With light microscopy, we observed strong labeling of CB1 in rat PAG, especially in its dorsal aspect (Fig. 3A). At higher magnification, localization of CB1 immunoreactivity to putative varicosities and thin axonal segments was apparent in the dlPAG and other PAG regions (Fig. 3B). Notably, cell bodies in the PAG were immunonegative for CB1, but were surrounded by basket-like axons exhibiting robust CB1 immunoreactivity (Fig. 3B). A comparable pattern of CB1 immunolabeling was seen in PAG derived from wild-type mice (Fig. 3C), whereas, as expected, CB1 immunoreactivity was absent in the PAG of mutant CB1-deficient mice (Fig. 3D).
Figure 3.
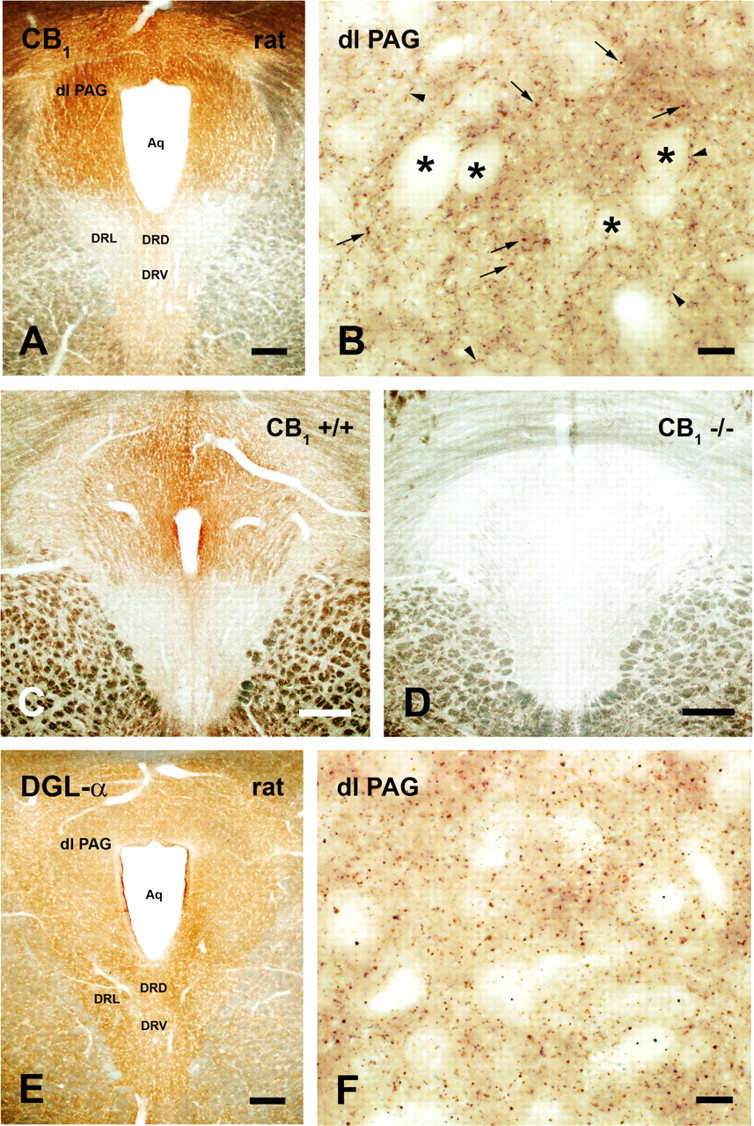
Widespread distribution of CB1 receptors and DGL-α in the PAG. A, B, Light micrographs of CB1 immunostaining in the rat PAG. A, The immunoperoxidase reaction revealed strong labeling, especially in the dorsal part of the PAG. Modest immunostaining was observed in the dorsal (DRD) and ventral (DRV) parts of the dorsal raphe nucleus, whereas CB1 immunoreactivity was below detection in the lateral part (DRL). B, In high-power micrograph, CB1-immunopositive varicosities (small arrows) and thin axonal segments (arrowheads) are visible in the dlPAG. Note basket-like axons showing strong CB1 immunostaining around immunonegative cell bodies (asterisks). C, D, CB1 immunolabeling in sections from wild-type and CB1-deficient mice. C, Immunoperoxidase staining for CB1 receptors in midbrain sections derived from a wild-type (CB1+/+) mouse shows identical CB1 receptor localization pattern to that observed in rats, with highest density of CB1 immunostaining located in the PAG. D, Antibody specificity is confirmed by the lack of immunostaining in sections derived from a CB1-deficient animal (CB1−/−). E, F, Light micrographs of DGL-α immunoreactivity in the rat PAG. E, High density of DGL-α staining is visible throughout the PAG, including the dlPAG, and the dorsal, ventral, and lateral parts of the dorsal raphe nucleus (DRD, DRV, and DRL, respectively). F, At higher magnification, a very dense punctate staining pattern is found in the dlPAG, indicating accumulation of the DGL-α protein within specific subcellular compartments. Abbreviation: Aq, cerebral aqueduct. Scale bars: A, E; C, D, 250 μm; B, F, 10 μm.
We next compared the distribution of CB1 immunoreactivity in the PAG with that of DGL-α, the enzyme isoform postulated to control 2-AG formation in the adult CNS. We observed a high density of DGL-α immunoreactivity throughout the PAG, including, but not limited to, the dlPAG (Fig. 3E). Staining for DGL-α was homogeneously distributed in the PAG, but was also present in dorsal, ventral, and lateral parts of the dorsal raphe nucleus (Fig. 3E). At higher magnification, a punctate staining pattern was observed, which is consistent with accumulation of the enzyme protein within specific subcellular compartments (Fig. 3F). Based upon these observations, we probed the specific subcellular compartments containing CB1 and DGL-α in the rodent dlPAG. We performed peroxidase immunolabeling followed by high-resolution electron microscopy to establish the precise anatomical distribution of these markers in the dlPAG. Our staining demonstrated that CB1 was found presynaptically on axon terminals (Fig. 4A–D) that were in synaptic connection with dendritic shafts (Fig. 4A), spines (Fig. 4D), or cell bodies (Fig. 4C). A strikingly high density of CB1 immunoreactivity also occurred on preterminal fibers (Fig. 4A), as described previously in other midbrain areas (Mátyás et al., 2006). By contrast, DGL-α was situated in a striking postsynaptic position (Fig. 5A–C), where it was exclusively localized to dendritic shafts (Fig. 5A,B) and spine heads (Fig. 5C). Postsynaptic DGL-α and presynaptic CB1 were observed at both excitatory (Figs. 5A,C, 4A,D) as well as putative inhibitory (Figs. 5B, 4B,C) synaptic specializations of the dlPAG.
Figure 4.
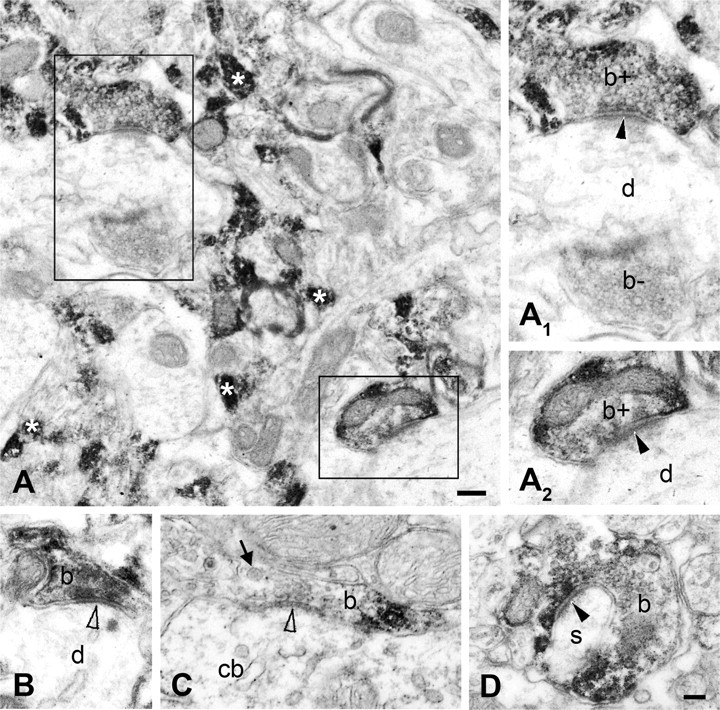
CB1 receptors are present presynaptically on both excitatory and inhibitory axon terminals in the mouse and rat dlPAG. A–D, High-resolution electron-microscopic analysis of immunoperoxidase staining for CB1 confirms presynaptic localization of the receptor (visualized by DAB precipitate) both in the mouse (A, B) and the rat (C, D) PAG. The white asterisks in A identify densely immunostained putative preterminal axonal fibers. The boxed areas are enlarged as A1 and A2. CB1 immunolabeling was found in both excitatory axon terminals (b+ in A1, A2; b in D), forming asymmetric synapses (marked by black arrowheads), and in putative inhibitory boutons (b in B, C), giving symmetric synapses (depicted by empty arrowheads). In synaptic connections with CB1-immunoreactive axon terminals, dendritic shafts (d in A1, A2), dendritic spines (s in D), and cell bodies (cb in C) are observed. The thick arrow indicates dense core vesicles. b− in A1 labels an immunonegative axon ending. Scale bars: A, 0.2 μm; A1–D, 0.1 μm.
Figure 5.
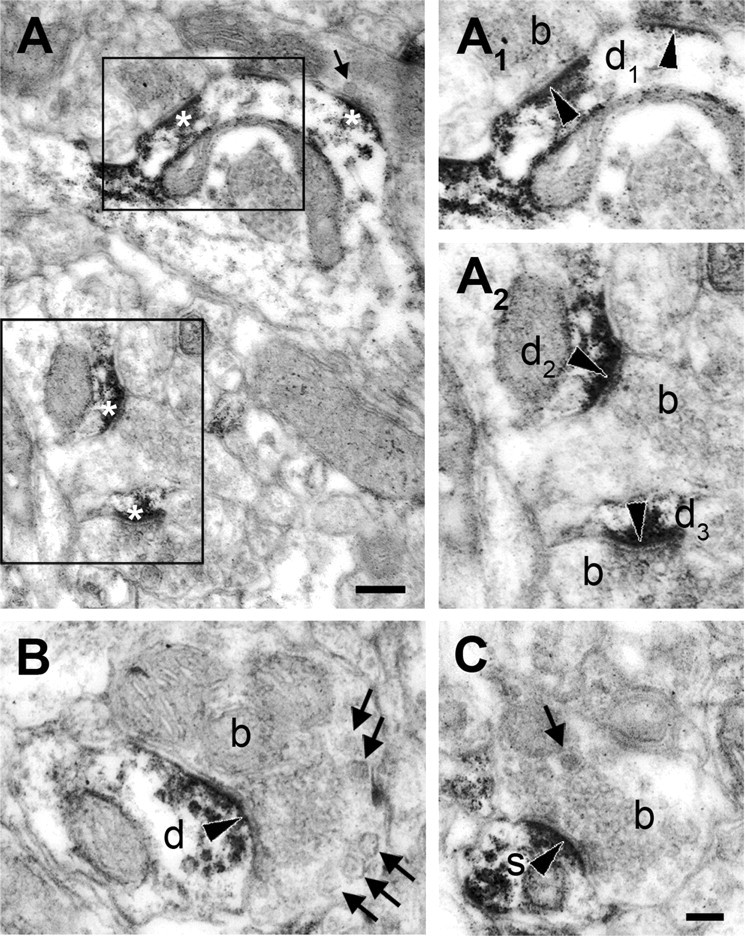
DGL-α is located in dendritic shafts and spine heads situated postsynaptically to axon terminals in the rat dlPAG. A–C, High-power electron micrographs of peroxidase-based DGL-α immunostaining demonstrate that DGL-α, represented by the dense reaction end product (DAB), is located postsynaptically in dendritic shafts (d1–3 and d in A1–B) and in the heads of dendritic spines (s in C). The boxed areas in A are enlarged as A1 and A2. Note that most of the synapses contain DGL-α (indicated by white asterisks in A). Among the DGL-α-negative axon terminals from which the DGL-α-containing dendrites receive synapses (labeled with black arrowheads), there are terminals containing round vesicles and others with flat vesicles indicating both excitatory boutons (b in A2, C) and putative inhibitory axon terminals (b in B), respectively. The thick arrows in A–C depict dense core vesicles. Scale bars: A, 0.2 μm; A1–C, 0.1 μm.
DGL-α drives 2-AG accumulation in the PAG and endocannabinoid-mediated antinociception through activation of the postsynaptic mGlu5–DGL-α signalosome
DGL-mediated hydrolysis of DAG may represent the first committed step in 2-AG biosynthesis (Piomelli, 2003). To probe the molecular mechanisms responsible for 2-AG mobilization in vivo, we examined the impact of pharmacological blockade of DGL activity on both stress antinociception and 2-AG formation. We microinjected RHC80267 and THL, two structurally distinct compounds that block 2-AG production in vitro by inhibiting the activity of the two known DGL isoforms, DGL-α and DGL-β (Bisogno et al., 2003). Injections of either of the two inhibitors into the dlPAG reduced stress-induced antinociception (Fig. 6A,B), whereas off-site injections had no effect (p = 0.26; Fig. 6C). RHC80267 (F(12,87) = 3.19; p = 0.001) and THL (F(16,151) = 6.15; p < 0.0001) suppressed stress antinociception in a time-dependent manner, and this suppression was maximal immediately following termination of the stressor. The high dose of THL (10 μg) suppressed stress antinociception (F(3,28) = 4.413; p = 0.01) relative to vehicle (p = 0.004) or the lowest dose of THL (0.1 μg; p = 0.004), which failed to alter stress antinocicepion (p = 0.90). We also tested the ability of THL, the more potent of the two inhibitors, to block DGL activity in situ. THL microinjections into the dlPAG reduced 2-AG (t(15) = 2.12; p = 0.026) levels in midbrain punches containing the PAG (Fig. 6E). The injections did not alter anandamide levels (p = 0.657) in the same punches (Fig. 6F). Moreover, 2-AG and anandamide levels in punches obtained from the surrounding midbrain reticular formation were not affected (p > 0.93 for each comparison; Fig. 6G,H). We asked, therefore, whether the ability of mGlu5 activation to potentiate stress antinociception in our studies was dependent upon DGL. The ability of locally administered DHPG (p = 0.001 vs vehicle) to potentiate stress-induced antinociception (F(30,360) = 5.17; p < 0.0009; Fig. 6D) was completely blocked (p = 0.0009 vs THL coadministered with DHPG) by a behaviorally inactive dose of THL (0.1 μg), microinjected into the dlPAG.
Figure 6.
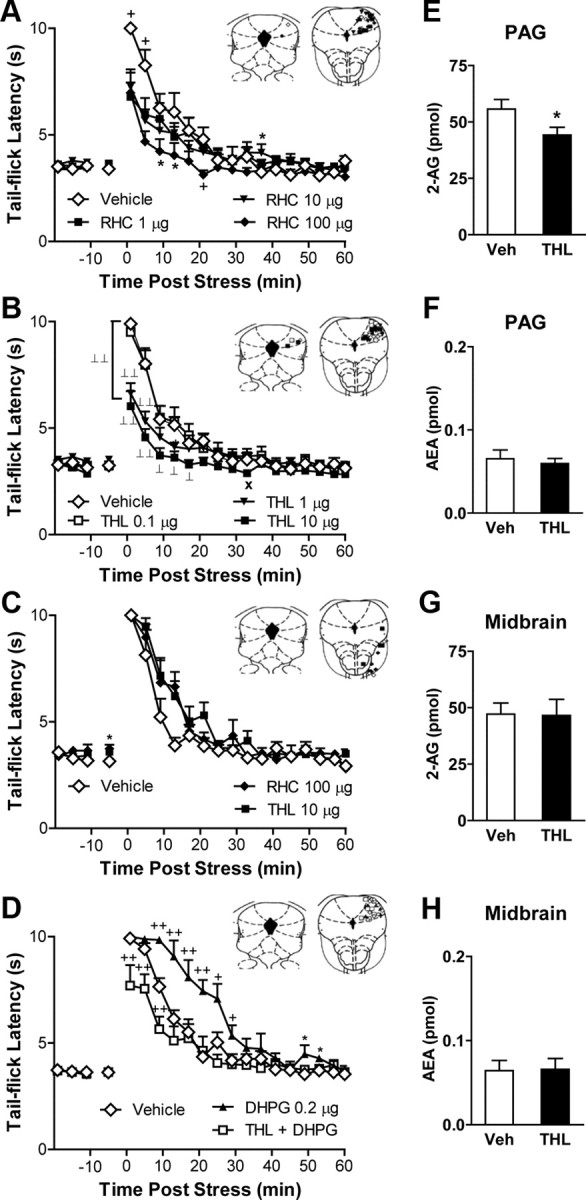
Inhibition of DGL activity in the dlPAG suppresses both endocannabinoid-mediated stress antinociception and 2-AG accumulation. A, B, Intra-dlPAG microinjection of RHC80267 (p = 0.001, ANOVA; RHC, 1, 10, or 100 μg) (A) or tetrahydrolipstatin (p < 0.0001, ANOVA; THL, 1 or 10 μg) (B) produced time-dependent suppressions of stress antinociception. THL (10 μg) suppressed stress antinociception relative to vehicle (p < 0.01) or a behaviorally inactive dose of THL (0.1 μg; p < 0.01). C, Off-site RHC (100 μg) or THL (10 μg) microinjection failed to alter stress antinociception. D, THL (0.1 μg) blocked the DHPG (0.2 μg)-induced potentiation of stress antinociception (p < 0.001, ANOVA). E–H, Intra-dlPAG microinjection of THL (10 μg) decreased 2-AG (p < 0.05, one-tailed t test; n = 8–9 per group) (E) but not anandamide (F) accumulation in the PAG without altering 2-AG (G) or anandamide (H) accumulation in the midbrain reticular formation. Data are expressed as mean ± SEM. Insets, Microinjection sites for groups receiving 1 μg (■), 10 μg (▾), 100 μg (♦) of RHC80267, 0.1 μg (□), 1 μg (▾), 10 μg (■) of THL, THL plus DHPG (□), 0.2 μg of DHPG (▴) and vehicle (♢) in the dlPAG (A, B, D) or off-site (C). *p < 0.05 versus control; +p < 0.05 versus all conditions; ×p < 0.05 versus low dose; ⊥⊥p < 0.01, ⊥p < 0.05 versus control and low dose (ANOVA, Fisher's PLSD test; n = 6–11 per group).
Based upon these observations, we asked whether the mGlu5–DGL-α signaling complex (“signalosome”) is present in dlPAG neurons, where it could control on-demand 2-AG production. To assess colocalization of mGlu5 and DGL-α, electron microscopy studies combined immunogold labeling for DGL-α with immunoperoxidase staining for mGlu5. Silver-intensified immunogold particles showed the precise subcellular localization of DGL-α on the cytosolic side of the plasma membrane within dendritic shafts expressing immunoreactivity for mGlu5, which was identified by the diffusible, electron-dense immunoreaction end product diaminobenzidine (Fig. 7A). Moreover, DGL-α and mGlu5 colocalized within the same dendritic spine heads receiving synapses with postsynaptic asymmetrical membrane thickenings (Fig. 7A), indicating the putative excitatory nature of these synapses. The results suggest that DGL-α colocalizes with mGlu5 at postsynaptic sites receiving excitatory synaptic contacts from axon terminals in the dlPAG. However, they do not tell us whether DGL-α is the enzyme that controls stimulation-dependent 2-AG formation and endocannabinoid-mediated analgesia in vivo. We, therefore, tested this hypothesis directly by using virally mediated mRNA silencing.
Figure 7.

Colocalization of DGL-α and mGlu5 in postsynaptic structures of the rat dlPAG. A1–A3, High-resolution serial electron micrographs of preembedding immunogold labeling for DGL-α combined with immunoperoxidase staining for mGlu5 in the rat dorsolateral PAG. A1, A2, Silver-intensified immunogold particles (thin arrows), representing the precise subcellular position of DGL-α, are localized perisynaptically in an mGlu5-immunopositive dendritic shaft (d), indicated by the diffusible immunoreaction end product DAB. Presynaptically to the dendrite, putative excitatory boutons (labeled with b1, b2, and b3) form asymmetrical synapses (black arrowheads). The thick arrows represent dense core vesicles in b2. A3, DGL-α (thin arrow) and mGlu5 (DAB precipitate) are present in the same dendritic spine head (s) receiving an asymmetrical synapse (black arrowhead) from a fourth axon terminal (labeled with b4). Note that the gold particles are attached to the cytosolic surface of the plasma membrane, consistent with the predicted position of the epitope of DGL-α protein. Scale bar: (in A3) A1–A3, 0.1 μm.
To identify the specific DGL isoform(s) involved in 2-AG formation and stress-induced antinociception, we injected bilaterally into the dlPAG viral vectors directing the expression of shRNAs engineered to silence expression of either DGL-α or DGL-β (Jung et al., 2007). Separate groups of rats received identical bilateral injections of either the DGL-α silencing virus (DGL-αi), the DGL-β silencing virus (DGL-βi), or the LacZ control virus (LacZi) in the dlPAG. Punches containing either the entire PAG (Fig. 8A,B) or off-site control tissue (i.e., midbrain reticular formation) were obtained from animals killed 2 weeks following viral infection, when effects of RNA silencing on behavior were maximal. Silencing the expression of either DGL-α or DGL-β in the dlPAG was accompanied by a reduction in DGL activity (F(3,24) = 4.58; p = 0.011) in midbrain punches containing the entire PAG (Fig. 8C, left), but not in punches prepared from the adjacent midbrain reticular formation (p = 0.66; Fig. 8C, right). DGL-αi lowered DGL activity relative to LacZi (p = 0.014) or off-target injections of DGL-αi (p = 0.021). DGL-βi also reduced (t(12) = 2.49; p = 0.029) DGL activity relative to LacZi treatment in a manner that was indistinguishable from that of DGL-αi (p = 0.456). Notably, DGL-α silencing in the dlPAG was associated with localized decreases in 2-AG (t(14) = 2.41; p = 0.015; this test was one-tailed based upon the a priori prediction that DGL-α silencing would reduce 2-AG production), but not anandamide (p = 0.394), levels (Fig. 8D) in PAG punches. By contrast, DGL-β silencing in the dlPAG failed to alter either 2-AG (p = 0.904) or anandamide (p = 0.511) levels (Fig. 8E), despite the fact that DGL-αi and DGL-βi produced comparable reductions in DGL activity. No changes in endocannabinoid content were observed off-site of the PAG (i.e., in punches derived from the midbrain reticular formation) following RNA silencing of either DGL-α or DGL-β (data not shown). The samples used in these analyses were obtained from animals killed at the peak of stress-induced antinociception, 1 min after footshock, when both 2-AG accumulation and antinociception were maximal (Hohmann et al., 2005; Hohmann and Suplita, 2006). RNA silencing of DGL-α in the dlPAG, which reduced 2-AG accumulation in our studies (Fig. 8D), was also associated with localized decreases (F(2,7) = 5.21; p = 0.041) in DGL-α, but not DGL-β (p = 0.859), mRNA (Fig. 8F), further documenting the specificity of virally mediated RNA silencing in downregulating DGL-α mRNA. Moreover, DGL-αi suppressed DGL-α mRNA relative to LacZi controls (p = 0.0269; this test was one-tailed based upon the a priori prediction that silencing DGL-α transcription would reduce DGL-α mRNA) or off-target injections of DGL-αi (p = 0.03), suggesting that effects of RNA silencing were localized to the PAG. It is likely that our measures underestimate the level of DGL-α silencing induced in dlPAG neurons because microinjections were confined to the dlPAG, whereas lipid and mRNA measurements were made in punches comprising the entire PAG.
Figure 8.

Virally mediated RNA silencing of DGL-α, but not DGL-β, transcription suppresses both endocannabinoid-mediated stress antinociception and 2-AG accumulation in the PAG. A, B, Diagram representing the middle of the tissue punch (A), encompassing the level −6.80 mm from bregma, which represented the center of the injection sites. B, GFP within the expression cassette marks cells in the dlPAG containing DGL-αi shRNA. Inset, Schematic representation of injection sites expressing GFP. C, DGL-αi (p = 0.01, Dunnett's test) and DGL-βi (p < 0.05, two-tailed t test) shRNAs suppressed (p = 0.01, ANOVA) DGL activity in PAG (left panel) but not off-site of PAG in midbrain reticular formation (right panel) (n = 6–8). D, DGL-αi in the dlPAG suppresses 2-AG (p < 0.05, one-tailed t test; left panel) but not anandamide (right panel) accumulation in the PAG (n = 8–9). E, DGL-βi in the dlPAG did not alter either 2-AG (left panel) or anandamide (right panel) levels in the PAG (n = 9). F, Intra-PAG DGL-αi suppressed DGL-α mRNA (p < 0.05, ANOVA; p < 0.05 for each comparison one-tailed t test; n = 3–4) relative to intra-PAG or off-target (left panel) LacZi. DGL-αi shRNA did not alter DGL-β mRNA (n = 3–4; right panel). C–F, Data are expressed as mean ± SEM. *p < 0.05 versus control conditions (ANOVA and Student's t test). G, Intra-dlPAG DGL-αi shRNA injection suppressed (p = 0.01, ANOVA) stress antinociception relative to naive or LacZi-injected controls (p < 0.05 for each comparison). H, DGL-αi suppressed (p < 0.01, ANOVA) stress antinociception relative to DGL-βi (p < 0.01) or LacZi (p < 0.05) shRNA. I, DGL-αi suppressed (p < 0.05, ANOVA) stress antinociception relative to off-target conditions (p < 0.01 vs Off-DGL-αi; p < 0.05 vs Off-LacZi). DGL-αi produced time-dependent suppressions (p ≤ 0.01 in G-I, respectively) of stress antinociception; control conditions did not differ (G–I). **p < 0.01, *p < 0.05 versus all groups; ++p < 0.01, +p < 0.05 versus DGL-βi; ××p < 0.01, ×p < 0.05 versus Off-DGL-αi (ANOVA, Fisher's PLSD test; n = 5–12).
To identify a role for DGL-α in behavior, we examined the impact of virally mediated RNA silencing of DGL-α and DGL-β in the dlPAG on endocannabinoid-mediated stress antinociception. Separate groups of rats received either identical bilateral injections of viruses engineered to silence either DGL-α (DGL-αi) or DGL-β (DGL-βi), the LacZ control virus (LacZi), or no injections (naive rats) (Fig. 8G–I). Microinjection of the DGL-α silencing virus (DGL-αi) into the dlPAG reduced stress-induced antinociception in a time-dependent manner (F(20,300) = 2.42; p = 0.011) (Fig. 8G). This effect was maximal 14 d after viral infection, when DGL activity, DGL-α mRNA and 2-AG accumulation was also selectively reduced in PAG punches (Fig. 8A–F and data not shown). As observed with DGL inhibitors (Fig. 6A,B), the effect of mRNA silencing was greatest immediately following termination of the stressor (Fig. 8G). Moreover, neither virus altered the basal nociceptive threshold (assessed immediately before administration of the stressor) relative to naive groups (Fig. 8G). The DGL-αi virus markedly lowered stress antinociception, producing a 41.6% decrease in the maximal possible antinociceptive effect (% MPE) elicited by the stressor at the peak of stress-induced analgesia, which occurred 1 min after termination of footshock (% MPE, mean ± SEM: 49.8 ± 5 vs 85.3 ± 3.5% in DGL-α and LacZ groups, respectively; p < 0.0001, one-tailed t test). A statistically robust reduction in stress antinociception was observed relative to naive groups (p = 0.0009) or groups receiving bilateral injections of LacZ control virus (p = 0.0009) directed at the same dlPAG site when stress antinociception was maximal (Fig. 8G). Stress antinociception did not differ between LacZi-treated and naive groups (p = 0.515) throughout the observation interval. Moreover, DGL-αi selectively suppressed stress antinociception (F(2,41) = 5.76; p = 0.006). Stress antinociception was lower in groups receiving the DGL-α silencing virus compared with either the DGL-β silencing virus (p = 0.002) or the lacZ control virus (p = 0.025) (Fig. 8H). By contrast, microinjection of the DGL-β silencing virus (DGL-βi) into the dlPAG did not alter endocannabinoid-mediated analgesia relative to LacZi-treated controls (p = 0.13; Fig. 8H). Microinjection of DGL-αi bilaterally into the dlPAG suppressed (F(2,20) = 5.15; p = 0.016) stress antinociception relative to either off-target injections of DGL-αi (p = 0.037) or the LacZ control virus (p = 0.009; Fig. 8I); this effect was also time dependent (F(20,200) = 3.03; p = 0.002). Stress antinociception was similar in groups receiving deliberate off-site bilateral injections of either DGL-αi or LacZi (p = 0.64; Fig. 8I), further documenting the site specificity of virally mediated DGL-α silencing in the PAG. Our results thus provide definitive evidence that DGL-α is the DGL isoform required for both in vivo 2-AG formation and endocannabinoid-mediated stress antinociception. These studies collectively demonstrate that 2-AG is synthesized on demand by activation of an mGlu5–DGL-α pathway in the dlPAG to produce 2-AG and endocannabinoid-mediated antinociception in vivo, consistent with a role for 2-AG as a retrograde endocannabinoid messenger.
Discussion
Our results indicate that acute footshock stress suppresses pain sensitivity in vivo by engaging the mGlu5–DGL-α signalosome and initiating 2-AG-mediated retrograde signaling in the dlPAG. Several lines of evidence support this conclusion. First, blockade of mGlu5 depressed endocannabinoid-mediated stress-induced antinociception. Second, activation of mGlu5 enhanced stress-induced antinociception while concomitantly stimulating 2-AG production. Third, blockade of DGL activity, established using structurally distinct DGL inhibitors that do not discriminate between the known DGL isoforms, suppressed both stress-induced antinociception and 2-AG formation. Fourth, mGlu5 and the putative 2-AG-synthesizing enzyme DGL-α were colocalized in dendritic spines of the dlPAG, whereas CB1 receptors were confined to axon terminals, consistent with a role for 2-AG as a retrograde signaling messenger. Finally, virally mediated RNA silencing of DGL-α, but not DGL-β, mimicked the effects of pharmacological inhibition of DGL on both stress-induced antinociception and 2-AG accumulation. These studies thus provide direct evidence that 2-AG is synthesized in vivo through activation of the mGlu5–DGL-α signalosome and represent, to our knowledge, the first direct demonstration of a functional role for DGL-α in a behavioral paradigm. The implications of this work likely extend beyond endocannabinoid-mediated stress-induced analgesia to include other forms of 2-AG-mediated control of behavior.
Exogenous cannabinoids are known to produce CB1-mediated suppression of both GABAergic and glutamatergic synaptic transmission in the rat PAG in vitro through presynaptic mechanisms, but postsynaptic effects on PAG neurons are strikingly absent (Vaughan et al., 2000). These electrophysiological findings are consistent with the present results localizing CB1 receptors to both excitatory and inhibitory axon terminals in the PAG, and with the well established primarily presynaptic localization of CB1 throughout the brain (Katona and Freund, 2008). Although a substantial portion of CB1 immunoreactivity is also present on the preterminal segment of the axons, receptors in the axonal plasma membrane do not influence action potential propagation or modulate transmitter release, indicating that the functional location of CB1 receptors was the axon terminal (Mátyás et al., 2006). In PAG slices, neuronal inhibition of glutamate transporters has been shown to enhance mGlu5-mediated control of GABAergic synaptic transmission through a CB1-dependent mechanism (Drew et al., 2008). These in vitro studies suggest that, in the PAG, activation of mGlu5 modulates neuronal excitability by mobilizing an unidentified endocannabinoid mediator. However, a major gap has remained in the present knowledge base to identify this mediator and the biosynthetic pathway responsible for its formation in vivo. Our studies have closed this gap by identifying 2-AG as the endocannabinoid mediator responsible for retrograde signaling in the PAG.
The present work validates a key role for the mGlu5–DGL-α pathway in triggering both 2-AG formation and endocannabinoid-mediated antinociception. Our studies provide direct evidence implicating a role for on-demand 2-AG formation in behavior. We found that mGlu5 immunoreactivity, localized perisynaptically on dendrites and dendritic spines (Uchigashima et al., 2007), was concentrated in the dorsolateral columns at all levels of the PAG. The dlPAG is implicated in fear-conditioned analgesia (Olango et al., 2012) as well as opioid-independent forms of both stimulation-produced (Walker et al., 1999) and stress-induced (Hohmann et al., 2005) analgesia. Our studies suggest that the role of mGlu5 activation in initiating 2-AG formation may account for the previously unexplained pivotal role of the dlPAG in endocannabinoid-mediated stress-induced analgesia (Hohmann et al., 2005; Hohmann and Suplita, 2006). In the dlPAG, mGlu5 colocalized with the 2-AG synthesizing enzyme DGL-α at postsynaptic sites and stimulation of this pathway triggered both endocannabinoid-mediated antinociception and 2-AG formation. 2-AG was likely to be produced on demand because pharmacological and RNA silencing manipulations did not alter the basal nociceptive threshold in the absence of the stressor. 2-AG, formed via activation of the mGlu5–DGL-α signalosome, may thus underlie other nonopioid forms of analgesia (Akil et al., 1976; Lewis et al., 1980; Cannon et al., 1982; Terman et al., 1986) that are mediated specifically by the PAG (Ho et al., 2011; Olango et al., 2012).
An important finding in our studies was that virally mediated RNA silencing of DGL-α, but not DGL-β, suppressed both endocannabinoid-mediated stress antinociception and 2-AG formation. Of note, silencing of DGL-α transcription produced profound deficits in stress antinociception under conditions in which the basal nociceptive threshold was unchanged. These observations further highlight the functional significance of DGL-α in controlling both stimulation-contingent 2-AG production and endocannabinoid-mediated antinociception. These effects were specific for 2-AG because virally mediated RNA silencing of DGL-α in the dlPAG did not alter anandamide levels. By contrast, prior experiments have shown that DGL-α−/− and DGL-β−/− mice show marked, parallel reductions in brain 2-AG and arachidonic acid levels, but also substantial decreases in brain anandamide (Gao et al., 2010). In our studies, virally mediated RNA silencing of DGL-α, but not DGL-β, reduced 2-AG formation and markedly inhibited endocannabinoid-mediated stress antinociception. Sampling methods may have diluted the magnitude of changes in mRNA and/or endocannabinoid content produced by RNA silencing. Nonetheless, it is important to note that both viruses produced comparable reductions in DGL activity in the PAG. It is reasonable to conclude that mobilization of the signaling-competent pool of 2-AG that engages presynaptic CB1 receptors and mediates stress-induced analgesia is under the control of DGL-α rather than DGL-β. Our studies validate the use of in vivo RNA silencing as a tool for studying DGL-α function that circumvents limitations associated with constitutive, global DGL-α deletion. To our knowledge, the present studies are the first to use and validate in vivo virally mediated RNA silencing with targeted lipidomic analyses.
2-AG and anandamide are mobilized with strikingly dissimilar time courses in dorsal midbrain (Hohmann et al., 2005). This observation suggests that the physiological roles of 2-AG and anandamide in the PAG are not redundant. Because the time course of endocannabinoid-dependent stress antinociception closely correlates with the mobilization of 2-AG, but not anandamide (Hohmann and Suplita, 2006), the present studies focused on uncovering mechanisms responsible for 2-AG formation under physiological conditions. The role of anandamide in the PAG, and the mechanism responsible for its formation in vivo, remain to be elucidated. In the PAG, inhibition of the anandamide-hydrolyzing enzyme FAAH (fatty-acid amide hydrolase) produces antinociceptive (de Novellis et al., 2008) and anxiolytic-like (Lisboa et al., 2008) effects and enhances endocannabinoid-mediated stress antinociception (Suplita et al., 2005). Inhibitors of endocannabinoid transport also enhance the anxiolytic effects of anandamide (Moreira et al., 2007) and reduce expression of contextual fear conditioning (Resstel et al., 2008) following microinjection into the dlPAG. A role for anandamide in the regulation of anxiety (Moreira et al., 2008) may, therefore, contribute to the distinct time courses of 2-AG and anandamide mobilization in the PAG observed during stress-induced analgesia. Electrical stimulation of the dlPAG also produces both CB1-dependent antinociception and anandamide release in the PAG (Walker et al., 1999). Nonetheless, because 2-AG release was not measured in this latter study (Walker et al., 1999), a role for 2-AG mobilization in contributing to CB1-dependent stimulation-produced analgesia cannot be discounted. More work is necessary to elucidate possible functional interactions between 2-AG and anandamide signaling (Maccarrone et al., 2008) in the PAG. In conclusion, our studies provide the first definitive evidence that activation of the mGlu5–DGL-α pathway triggers 2-AG formation in vivo, consistent with a retrograde 2-AG signaling mechanism. A key behavioral consequence of activation of this pathway, antinociception, may be directly attributed to stimulation-contingent formation of 2-AG.
Footnotes
This work was supported by NIH Grants DA021644 and DA022478 (A.G.H., D.P.), and DA011322 and DA021696 (K.M.), and by a grant from Switzerland through the Swiss Contribution (SH7/2/18), the Hungarian Scientific Research Fund–Norwegian Financial Mechanism Joint Program (NNF 78918), and the European Research Council Grant 243153 (R.N., I.K.). We are grateful to Nathan Bolton, Erika Tischler, and Balázs Pintér for technical assistance. We also thank László Barna, the Nikon Microscopy Center at Institute of Experimental Medicine, Nikon Austria GmbH, and Auro-Science Consulting, Ltd., for kindly providing microscopy support.
References
- Akil H, Mayer DJ, Liebeskind JC. Antagonism of stimulation-produced analgesia by naloxone, a narcotic antagonist. Science. 1976;191:961–962. doi: 10.1126/science.1251210. [DOI] [PubMed] [Google Scholar]
- Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68:247–286. doi: 10.1016/s0301-0082(02)00080-1. [DOI] [PubMed] [Google Scholar]
- Alvarez FJ, Villalba RM, Carr PA, Grandes P, Somohano PM. Differential distribution of metabotropic glutamate receptors 1a, 1b, and 5 in the rat spinal cord. J Comp Neurol. 2000;422:464–487. doi: 10.1002/1096-9861(20000703)422:3<464::aid-cne11>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Azkue JJ, Knöpfel T, Kuhn R, Mateos JM, Grandes P. Distribution of the metabotropic glutamate receptor subtype mGluR5 in rat midbrain periaqueductal grey and relationship with ascending spinofugal afferents. Neurosci Lett. 1997;228:1–4. doi: 10.1016/s0304-3940(97)00357-1. [DOI] [PubMed] [Google Scholar]
- Bandler R, Keay KA, Floyd N, Price J. Central circuits mediating patterned autonomic activity during active vs. passive emotional coping. Brain Res Bull. 2000;53:95–104. doi: 10.1016/s0361-9230(00)00313-0. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon JT, Prieto GJ, Lee A, Liebeskind JC. Evidence for opioid and non-opioid forms of stimulation-produced analgesia in the rat. Brain Res. 1982;243:315–321. doi: 10.1016/0006-8993(82)90255-4. [DOI] [PubMed] [Google Scholar]
- Carey MR, Myoga MH, McDaniels KR, Marsicano G, Lutz B, Mackie K, Regehr WG. Presynaptic CB1 receptors regulate synaptic plasticity at cerebellar parallel fiber synapses. J Neurophysiol. 2011;105:958–963. doi: 10.1152/jn.00980.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Endocannabinoid Research Group. de Novellis V, Palazzo E, Rossi F, De Petrocellis L, Petrosino S, Guida F, Luongo L, Migliozzi A, Cristino L, Marabese I, Starowicz K, Di Marzo V, Maione S. The analgesic effect of N-arachidonoyl-serotonin, a FAAH inhibitor and TRPV1 receptor antagonist, associated with changes in rostral ventromedial medulla and locus coeruleus cell activity in rats. Neuropharmacology. 2008;55:1105–1113. doi: 10.1016/j.neuropharm.2008.06.023. [DOI] [PubMed] [Google Scholar]
- Drew GM, Mitchell VA, Vaughan CW. Glutamate spillover modulates GABAergic synaptic transmission in the rat midbrain periaqueductal grey via metabotropic glutamate receptors and endocannabinoid signaling. J Neurosci. 2008;28:808–815. doi: 10.1523/JNEUROSCI.4876-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukudome Y, Ohno-Shosaku T, Matsui M, Omori Y, Fukaya M, Tsubokawa H, Taketo MM, Watanabe M, Manabe T, Kano M. Two distinct classes of muscarinic action on hippocampal inhibitory synapses: M2-mediated direct suppression and M1/M3-mediated indirect suppression through endocannabinoid signalling. Eur J Neurosci. 2004;19:2682–2692. doi: 10.1111/j.0953-816X.2004.03384.x. [DOI] [PubMed] [Google Scholar]
- Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, Mark L, Piesla MJ, Deng K, Kouranova EV, Ring RH, Whiteside GT, Bates B, Walsh FS, Williams G, Pangalos MN, et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci. 2010;30:2017–2024. doi: 10.1523/JNEUROSCI.5693-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Rodríguez de Fonseca F, Piomelli D. Quantification of bioactive acylethanolamides in rat plasma by electrospray mass spectrometry. Anal Biochem. 2000;280:87–93. doi: 10.1006/abio.2000.4509. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi K, Shin HS, Kano M. Phospholipase Cbeta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron. 2005;45:257–268. doi: 10.1016/j.neuron.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Maejima T, Fukami K, Kano M. Pharmacological evidence for the involvement of diacylglycerol lipase in depolarization-induced endocanabinoid release. Neuropharmacology. 2008;54:58–67. doi: 10.1016/j.neuropharm.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Yamazaki M, Sakimura K, Kano M. Neuronal protease-activated receptor 1 drives synaptic retrograde signaling mediated by the endocannabinoid 2-arachidonoylglycerol. J Neurosci. 2011;31:3104–3109. doi: 10.1523/JNEUROSCI.6000-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YC, Lee HJ, Tung LW, Liao YY, Fu SY, Teng SF, Liao HT, Mackie K, Chiou LC. Activation of orexin 1 receptors in the periaqueductal gray of male rats leads to antinociception via retrograde endocannabinoid (2-arachidonoylglycerol)-induced disinhibition. J Neurosci. 2011;31:14600–14610. doi: 10.1523/JNEUROSCI.2671-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL., 2nd Endocannabinoid mechanisms of pain modulation. AAPS J. 2006;8:E693–E708. doi: 10.1208/aapsj080479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Jung KM, Mangieri R, Stapleton C, Kim J, Fegley D, Wallace M, Mackie K, Piomelli D. Stimulation of endocannabinoid formation in brain slice cultures through activation of group I metabotropic glutamate receptors. Mol Pharmacol. 2005;68:1196–1202. doi: 10.1124/mol.105.013961. [DOI] [PubMed] [Google Scholar]
- Jung KM, Astarita G, Zhu C, Wallace M, Mackie K, Piomelli D. A key role for diacylglycerol lipase-alpha in metabotropic glutamate receptor-dependent endocannabinoid mobilization. Mol Pharmacol. 2007;72:612–621. doi: 10.1124/mol.107.037796. [DOI] [PubMed] [Google Scholar]
- Katona I, Freund TF. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat Med. 2008;14:923–930. doi: 10.1038/nm.f.1869. [DOI] [PubMed] [Google Scholar]
- Katona I, Urbán GM, Wallace M, Ledent C, Jung KM, Piomelli D, Mackie K, Freund TF. Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci. 2006;26:5628–5637. doi: 10.1523/JNEUROSCI.0309-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafourcade M, Elezgarai I, Mato S, Bakiri Y, Grandes P, Manzoni OJ. Molecular components and functions of the endocannabinoid system in mouse prefrontal cortex. PLoS One. 2007;2:e709. doi: 10.1371/journal.pone.0000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JW, Cannon JT, Liebeskind JC. Opioid and nonopioid mechanisms of stress analgesia. Science. 1980;208:623–625. doi: 10.1126/science.7367889. [DOI] [PubMed] [Google Scholar]
- Lisboa SF, Resstel LB, Aguiar DC, Guimarães FS. Activation of cannabinoid CB1 receptors in the dorsolateral periaqueductal gray induces anxiolytic effects in rats submitted to the Vogel conflict test. Eur J Pharmacol. 2008;593:73–78. doi: 10.1016/j.ejphar.2008.07.032. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Rossi S, Bari M, De Chiara V, Fezza F, Musella A, Gasperi V, Prosperetti C, Bernardi G, Finazzi-Agrò A, Cravatt BF, Centonze D. Anandamide inhibits metabolism and physiological actions of 2-arachidonoylglycerol in the striatum. Nat Neurosci. 2008;11:152–159. doi: 10.1038/nn2042. [DOI] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–475. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- Mátyás F, Yanovsky Y, Mackie K, Kelsch W, Misgeld U, Freund TF. Subcellular localization of type 1 cannabinoid receptors in the rat basal ganglia. Neuroscience. 2006;137:337–361. doi: 10.1016/j.neuroscience.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- Moreira FA, Aguiar DC, Guimarães FS. Anxiolytic-like effect of cannabinoids injected into the rat dorsolateral periaqueductal gray. Neuropharmacology. 2007;52:958–965. doi: 10.1016/j.neuropharm.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Moreira FA, Kaiser N, Monory K, Lutz B. Reduced anxiety-like behaviour induced by genetic and pharmacological inhibition of the endocannabinoid-degrading enzyme fatty acid amide hydrolase (FAAH) is mediated by CB1 receptors. Neuropharmacology. 2008;54:141–150. doi: 10.1016/j.neuropharm.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Nyilas R, Gregg LC, Mackie K, Watanabe M, Zimmer A, Hohmann AG, Katona I. Molecular architecture of endocannabinoid signaling at nociceptive synapses mediating analgesia. Eur J Neurosci. 2009;29:1964–1978. doi: 10.1111/j.1460-9568.2009.06751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olango WM, Roche M, Ford GK, Harhen B, Finn DP. The endocannabinoid system in the rat dorsolateral periaqueductal grey mediates fear-conditioned analgesia and controls fear expression in the presence of nociceptive tone. Br J Pharmacol. 2012;165:2549–2560. doi: 10.1111/j.1476-5381.2011.01478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 4. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Resstel LB, Lisboa SF, Aguiar DC, Corrêa FM, Guimarães FS. Activation of CB1 cannabinoid receptors in the dorsolateral periaqueductal gray reduces the expression of contextual fear conditioning in rats. Psychopharmacology (Berl) 2008;198:405–411. doi: 10.1007/s00213-008-1156-1. [DOI] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- Suplita RL, 2nd, Farthing JN, Gutierrez T, Hohmann AG. Inhibition of fatty-acid amide hydrolase enhances cannabinoid stress-induced analgesia: sites of action in the dorsolateral periaqueductal gray and rostral ventromedial medulla. Neuropharmacology. 2005;49:1201–1209. doi: 10.1016/j.neuropharm.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Suplita RL, 2nd, Gutierrez T, Fegley D, Piomelli D, Hohmann AG. Endocannabinoids at the spinal level regulate, but do not mediate, nonopioid stress-induced analgesia. Neuropharmacology. 2006;50:372–379. doi: 10.1016/j.neuropharm.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, Sakimura K, Kano M. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron. 2010;65:320–327. doi: 10.1016/j.neuron.2010.01.021. [DOI] [PubMed] [Google Scholar]
- Terman GW, Morgan MJ, Liebeskind JC. Opioid and non-opioid stress analgesia from cold water swim: importance of stress severity. Brain Res. 1986;372:167–171. doi: 10.1016/0006-8993(86)91472-1. [DOI] [PubMed] [Google Scholar]
- Uchigashima M, Narushima M, Fukaya M, Katona I, Kano M, Watanabe M. Subcellular arrangement of molecules for 2-arachidonoyl-glycerol-mediated retrograde signaling and its physiological contribution to synaptic modulation in the striatum. J Neurosci. 2007;27:3663–3676. doi: 10.1523/JNEUROSCI.0448-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Connor M, Bagley EE, Christie MJ. Actions of cannabinoids on membrane properties and synaptic transmission in rat periaqueductal gray neurons in vitro. Mol Pharmacol. 2000;57:288–295. [PubMed] [Google Scholar]
- Walker JM, Huang SM, Strangman NM, Tsou K, Sañudo-Peña MC. Pain modulation by release of the endogenous cannabinoid anandamide. Proc Natl Acad Sci U S A. 1999;96:12198–12203. doi: 10.1073/pnas.96.21.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]