

Abstract
For the analysis of cancer, there is great interest in rapid and accurate detection of cancer genome amplifications containing oncogenes that are potential therapeutic targets. The vast majority of cancer tissue samples are formalin fixed and paraffin embedded (FFPE) which enables histopathological examination and long term archiving. However, FFPE cancer genomic DNA is oftentimes degraded and generally a poor substrate for many molecular biology assays. To overcome the issues of poor DNA quality from FFPE samples and detect oncogenic copy number amplifications with high accuracy and sensitivity, we developed a novel approach. Our assay requires nanogram amounts of genomic DNA, thus facilitating study of small amounts of clinical samples. Using droplet digital PCR (ddPCR), we can determine the relative copy number of specific genomic loci even in the presence of intermingled normal tissue. We used a control dilution series to determine the limits of detection for the ddPCR assay and report its improved sensitivity on minimal amounts of DNA compared to standard real-time PCR. To develop this approach, we designed an assay for the fibroblast growth factor receptor 2 gene (FGFR2) that is amplified in a gastric and breast cancers as well as others. We successfully utilized ddPCR to ascertain FGFR2 amplifications from FFPE-preserved gastrointestinal adenocarcinomas.
Keywords: cancer, gene amplifications, copy number variations, archival cancer samples
Introduction
Genomic amplifications, a type of DNA copy number variation (CNV), are common features of cancer genomes. These amplifications often lead to overexpression of specific cancer oncogenes that act as “drivers” in cancer development. As a result, these amplified oncogenes provide pro-growth signals that lead to tumor proliferation (1). Inhibition of these amplified oncogenes and their effector signaling cascades often leads to arrest of tumor growth and cell death. Thus, oncogenes residing within cancer genome amplifications provide potential therapeutic targets that are specific to a given tumor (2). For example, a significant proportion of breast and gastric cancers have amplifications of the gene ERBB2, otherwise referred to as HER2, which is an oncogenic growth factor receptor. The ERBB2 gene product is a target for the therapeutic antibody, trastuzumab and other small molecule inhibitors in cancer (3). Given the existence of therapeutics to treat specific gene amplifications in a “personalized medicine” approach, there is great interest in the accurate and timely identification of genomic amplifications of specific oncogenes. Current detection methods include real-time PCR, highdensity array comparative genomic hybridization (CGH) methods, single nucleotide polymorphism (SNP) microarrays and fluorescent in situ hybridization (FISH). While these approaches have generally performed well, they are handicapped by the issues of sensitivity of detection. For example, tumors are oftentimes associated with normal stroma that effectively dilutes the presence of genomic amplifications and thus makes detection of amplifications substantially more difficult.
Another challenge for cancer genome amplification analysis is that the vast majority of clinical cancer samples are processed as formalin fixed paraffin embedded (FFPE) tissues. For clinical pathology laboratories, this is a universal preservation method because (1) it maintains morphological features of the tumor, (2) it enables histopathologic examination with a number of immunohistological staining processes and (3) it can be stored indefinitely at room temperature. However, the FFPE fixation process causes irreversible damage to the sample genomic DNA via cross linkages and increased fragmentation. As a result, genomic DNA extracted from FFPE material is often of poor quality. Therefore, analysis of FFPE-derived genomic DNA using PCR-based or microarray-based assays for genomic amplification can be technically challenging.
We developed a robust solution for measuring genomic amplifications in FFPE tumor samples using droplet digital PCR (ddPCR) that is sensitive enough to detect abnormal genomic amplifications even if the sample contains only a small fraction of tumor cells. The ddPCR method requires nanogram amounts of genomic DNA, thus facilitating the study of rare samples. As described by Hindson et al., the ddPCR method involves emulsifying the sample which provides specific advantages for highly sensitive and specific detection of certain genomic events including CNVs such as amplifications (5). In the case of our copy number assay in the post-amplification reaction, emulsion droplets are streamed single-file into a capillary that leads past a two-color detector; where the positive droplets for the target and reference genes are “counted” for quantitation as generally shown in Figure 1. We demonstrate here the robustness of ddPCR for highly sensitive and specific detection of a cancer gene amplification specific for the FGFR2 gene from minute amounts of genomic DNA derived from clinical cancer samples (6). This gene encodes the fibroblast growth factor receptor 2 and is a potential therapeutic target for cancer in clinical trials (7).
Figure 1.
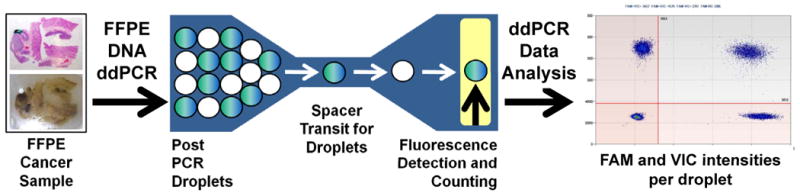
General workflow of droplet digital PCR in amplification analysis of archival cancer samples.
DNA is extracted from an archival cancer sample. An example of a formalin-fixed paraffin block of gastric adencarcinoma with an accompanying stained section is shown. After DNA extraction, droplet PCR is conducted with a specific set of PCR primers and fluorescent probe. Post-PCR emulsion droplets are streamed single-file into a capillary that leads past a two-color detector; where the positive droplets for the target and reference genes are counted for copy number quantitation with two different dyes such as 6-FAM and VIC. One dye is specific to the control loci and the other to the loci being measured.
Materials and Methods
Sample DNA preparation
This study was approved by the institutional review board (IRB) at Stanford University School of Medicine. Samples were obtained from the Stanford Cancer Institute Tissue Bank. Frozen tissue sections were prepared and hematoxylin-eosin (HE) staining was performed on a single section from each tumor. On a subset of samples, we conducted our analysis on matched gastric cancer and normal tissue from the same individual. Tumor samples were macro-dissected from areas where tumor cellular composition was estimated to be 60% or higher. For a number of cases, we also used matched tissue that was confirmed to be normal using pathological examination. For FFPE cancer samples, genomic DNA was extracted using the E.Z.N.A. SQ DNA/RNA Protein Kit (Omega Bio-Tek). For the gastric cancer cell line KatoIII, we extracted genomic DNA with the DNAeasy Tissue Kit (Qiagen) following the manufacturer's protocol. Concentrations of genomic DNA were determined with a Nanodrop instrument (Thermo Scientific).
Standard real-time PCR for copy number analysis
We conducted traditional real-time PCR to detect copy number changes in FGFR2. Briefly, a 20-μl reaction mixture was made up of 2× Quantitech SYBR Green PCR master mix (Qiagen), 1uM each forward and reverse primers, and 50 ng of template DNA. All real-time PCR assays were performed in triplicate with an iCycler thermocycler (BioRad). Thermal cycling conditions were as follows: 95°C at 15 min (1 cycle); 94°C for 15 s, 60°C for 30 s and 72°C for 15 s (40 cycles). Melting curves and product validation proceeded per manufacturer's guidelines. Primers used in this study were ordered from Integrated DNA Technologies and had the sequences 5′-ACTTGGGCTGGAGTGATTTG-3′ (forward primer), and 5′-AATCCCATCTGCACACTTCC-3′, (reverse primer). Results were normalized using normal genomic DNA.
SNP array analysis
Standard protocols for DNA preparation, array hybridization and scanning were used to analyze the normal, primary tumor and metastatic tumor samples using SNP 6.0 arrays (Affymetrix, Santa Clara, CA). We used 1 μg of genomic DNA from our samples for array hybridization. We used the Matlab programming language (Mathworks) for quantile probe normalization, probe level summarization and estimation of the copy number between a tumor and normal samples using log2 ratio of the normalized signals (http://www.mathworks.com/products/bioinfo/examples.html?file=/products/demos/shipping/bioinfo/affysnpcnvdemo.html).
Droplet digital PCR workflow
Digital PCR was performed as previously described on the Q×100 droplet digital PCR system (Bio-Rad) (5). Briefly, Taq polymerase PCR reaction mixtures were assembled using 2× ddPCR Supermix for Probes, 20× assays (18 μM primers and 5 μM probe) and restriction digested FFPE sample. DG8 cartridges were loaded with 20 μL PCR reaction mixtures and 70 μL of droplet generation oil for each sample. The cartridges were placed into a droplet generator for emulsification and the emulsified samples were transferred onto 96-well plate as per manufacturer's instructions. Plates were heat-sealed and underwent 40 cycles in a C-1000 thermal cycler (Bio-Rad). After PCR, the 96-well droplet PCR plates were loaded into a droplet reader, which sequentially reads the droplets from each well of the plate. Analysis of the ddPCR data was performed using the CNV mode of the Q×100 analysis software (version 1.2.9.0), relying on two probes, one specific to the target with a FAM fluorescent signal and the second representing a control with a VIC fluorescent signal. We had replicates for all of the ddPCR wells for any given sample. A dilution series with the genomic DNA from Kato III and NA18507 involved seven replicates.
Determination of cancer genomic amplifications in FFPE tumor samples
For the assessment of copy number on high quality genomic DNA samples, we digested the samples with a restriction enzyme in order to unlink tandem gene copies for random distribution into droplets for precise DNA quantitation using Poisson statistics. The digestion step was not required for FFPE samples as the DNA tends to be highly fragmented, but was performed here to maintain experimental consistency. To assess FGFR2 copy number, 125 ng of each FFPE sample was digested with 1.25 units of BsaJI (New England BioLabs) in 15 μL for 1 h at 60°C. The restriction enzyme reactions were diluted 1.67-fold to 25 μL with nuclease free water then 25 ng (5 μL) was assayed per 20 μL ddPCR reaction. FGFR2 assay sequences were (forward primer) 5′-GGCTGGCTGCTGAAGTCT-3′, (reverse primer) 5′-CTTAATCGCCTGTATGGTGGTAACA-3′, and (probe) 5′-FAM-TCTTGGTCGTGTTCTTCATTCGGCACAG-BHQ1-3′ (Biosearch Technologies). This sequence localizes around genome location chr10:123274680-123274760 based on the NCBI build 37. The FGFR2 assay was duplexed with a reference assay targeting highly conserved sequence on Chromosome 1 that was used for comparison during the counting assay. This reference assay used the following primers: (forward primer) 5′-TGAGGGATTCGGCAGATGTTG-3′, (reverse primer) 5′-CTGAAAGGCTGGACTTGACAGA-3′, and (probe) 5′-VIC-ACTGTGTGCTGGACCT-MGB-3′ (Life Technologies). All assay primers were ordered from Integrated DNA Technologies. Thermal cycling conditions were 95 °C 10 min (1 cycle); 94°C 30s and 60°C 60s (40 cycles); 98°C 10 min (1 cycle); a 12°C hold. FGFR2 copy number per cell was estimated as the ratio of the FGFR2 and chromosome 1 marker concentrations multiplied by two to account for the two copies that are expected per a normal diploid genome.
Results
As previously described, a major issue in the detection of gene amplifications is the heterogeneous nature of tumor samples. Tumor and normal cells are admixed and this can reduce the sensitivity of detection when determining the presence or extent of genomic amplification. To address this challenge of mixed sample copy number measurement, we determined the limits of detection of the ddPCR assay for FGFR2 amplification. We identified a gastric cancer cell line, KatoIII, containing a well-described amplification of the FGFR2 gene (6). KatoIII genomic DNA was diluted in decreasing ratios with DNA from a well-described normal diploid genome sample (NA18507, Coriell Institute). After we verified the FGFR2 amplification in KatoIII with ddPCR, we calculated the predicted FGFR2 amplification copy number when diluted with a normal diploid genome in various ratios. The equation for the predicted copy number for the dilutions series is expressed as follows: CN = X * mutF+2 * (1 - mutF) where CN is the predicted copy number of the mixed sample, X is the copy number of the genomic loci from a pure “mutant” cell line and mutF is the mutant fraction of the mixed sample. The assumption is that the normal cells in the sample have 2 copies per cell for the gene or loci of interest and the copy number estimate is a weighted average of mutant and normal cells.
Using the equation described above, we determined the predicted FGFR2 copy number for various dilutions of the KatoIII:NA18507 dilution as indicated in Table 1. For example, in the case of 90% wildtype genome (NA18507) admixed with 10% mutant genome (KatoIII), X is equal to 310 (the FGFR2 copy number), mutF is equal to 0.1, and CN is the predicted FGFR2 copy number. Solving for CN confirms that the FGFR2 CN is predicted to be 32.8. This calculation was repeated for each dilution to determine the predicted copy number.
Table 1.
Predicted versus measured FGFR2 copy number across a dilution series with KatoIII and normal diploid genomic DNA.
| % Kato III (tumor DNA) | % Wildtype (normal DNA) | Predicted FGFR2 Copy Number | Measured average FGFR2 Copy Number | Coefficient of variation for measured FGFR2 CN |
|---|---|---|---|---|
| 0.00 | 100.00 | 2.00 | 1.99 | 0.02 |
| 0.10 | 99.90 | 2.30 | 2.16 | 0.03 |
| 1.00 | 99.00 | 4.80 | 3.88 | 0.02 |
| 10.00 | 90.00 | 29.80 | 38.66 | 0.02 |
| 20.00 | 80.00 | 57.60 | 91.30 | 0.03 |
| 50.00 | 50.00 | 141.00 | 196.57 | 0.05 |
Seven replicates were run for each dilution point to empirically determine the actual FGFR2 copy number values. The reproducibility between replicates was tight, as evidenced by the low coefficient-of-variance. This analysis confirmed the accuracy and sensitivity of ddPCR in quantifying the FGFR2-amplified locus even when there was 1,000-fold dilution with normal diploid genomic DNA (Figure 2). Using this cell line with an amplified locus, the 0.1% dilution was found to be statistically distinguishable from a pure normal DNA source. The empirically measured copy number versus the extent of dilution with normal genomic DNA demonstrated a linear relationship with a linear regression R coefficient of 0.99. Based on our analysis, the level of detection based on dilution is thus related to the level of amplification and the ratio of tumor to normal DNA.
Figure 2.
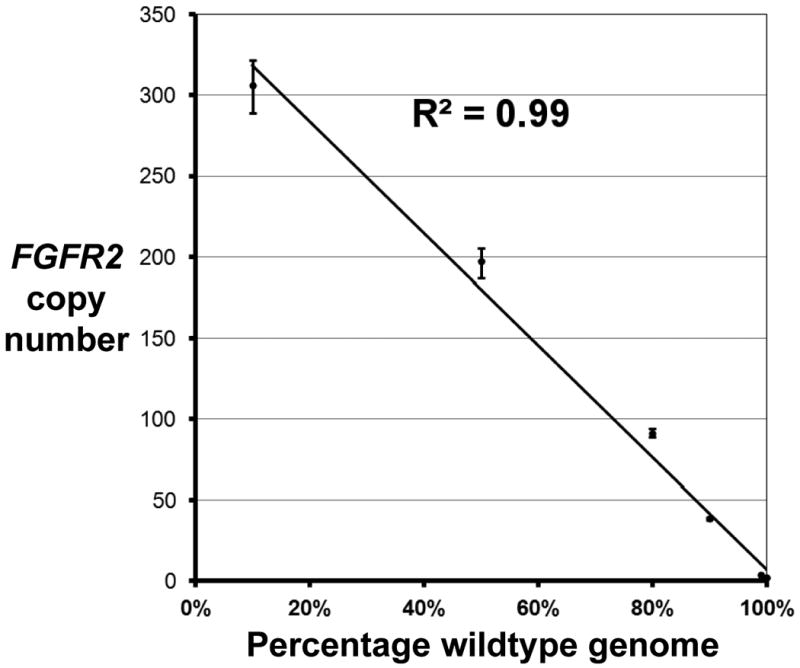
Measurement of FGFR2 copy number from a dilution series of tumor versus normal genomic DNA using ddPCR.
To ascertain the limits of quantifying amplifications, KatoIII genomic DNA, known to harbor FGFR2 amplification, was diluted with a normal diploid genomic DNA in decreasing ratios. Data points with error bars represent the drop in FGFR2 copy number as the KatoIII genomic DNA is diluted with normal genomic DNA.
Amplification analysis comparison between a matched FFPE and flash frozen tumor sample
In contrast to the high quality genomic DNA obtained from cell lines, genomic DNA obtained from FFPE samples is highly degraded, making standard molecular biology techniques, such as PCR, more difficult. We sought to determine the ability of ddPCR to detect amplification in FFPE-derived cancer and normal tissue samples. As a test case, we analyzed matched FFPE and flash frozen gastric cancer samples from one individual (ID 525). Using high quality genomic DNA from the flash frozen gastric tumor, we identified a 6-fold amplification of the FGFR2 locus using an Affymetrix 6.0 microarray. From the same tumor which had a portion that was FFPE processed, we used ddPCR to measure the FGFR2 amplification and confirmed the presence of an approximate 7-fold amplification of the FGFR2 locus (Fig. 3A). As a performance comparison, we used a standard real-time PCR assay on the same FFPE tumor sample and measured a copy of number estimate of 35 (Fig. 3B). These results suggest ddPCR is more accurate than real-time PCR for determining copy number variants in FFPE-derived samples. As a control, we also analyzed a normal gastric tissue sample (ID 2502) from an FFPE block with ddPCR (Fig. 3A) and real-time PCR (Fig. 3B). In both cases, there was no evidence of the FGFR2 amplification, supporting the specificity of our method.
Figure 3.
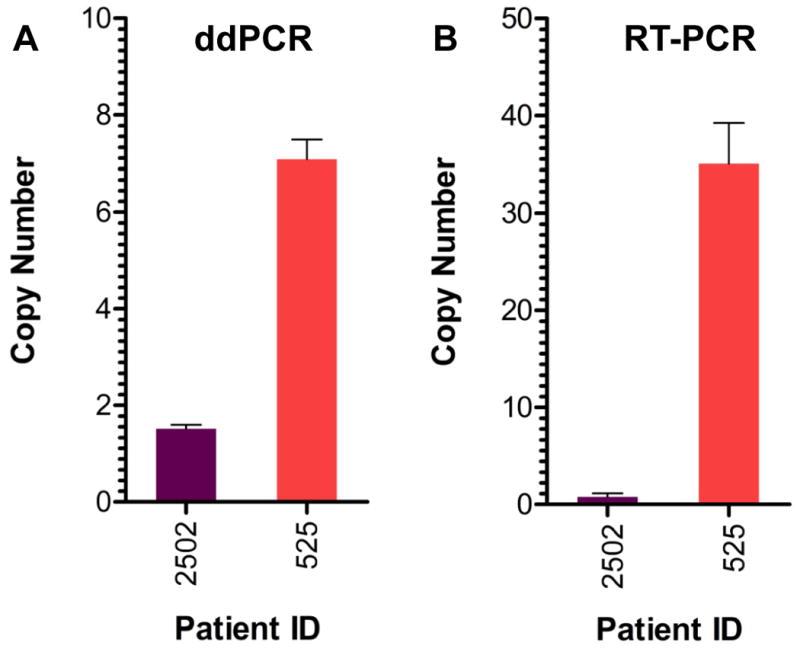
Comparison of ddPCR versus real-time PCR for measuring FGFR2 copy number in FFPE tissue.
FGFR2 copy number was measured in two different patient samples by ddPCR (panel A) and real-time PCR (panel B). The FFPE sample from Patient 2502 (purple bars) represents normal gastric tissue. The FFPE sample from Patient 525 (red bars) is a gastric adenocarcinoma sample. Data are plotted as copy number per diploid genome.
Genomic amplification analysis in FFPE gastrointestinal cancer samples
Given that ddPCR accurately detects genomic amplifications from archival FFPE tumors, we analyzed FFPE colorectal and gastric adenocarcinomas for the presence of FGFR2 amplification. This included two different subtypes of gastric cancer referred to as intestinal versus diffuse. We also analyzed diffuse gastric cancer samples that were either sporadic or originated from individuals with Hereditary Diffuse Gastric Cancer, an inherited cancer syndrome. Germline carriers of a mutation in the CDH1 gene are at increased risk for the diffuse subtype.
Analysis of four separate colorectal carcinoma samples (IDs 602, 1428, 1427, 1585) did not identify any instances of FGFR2 amplifications, an expected result given that this amplification has not been described in colon cancer. We assessed the presence of FGFR2 amplifications in six sporadic gastric carcinomas (IDs 2521, 2512, 709, 2505, 2502, 2504) (Fig. 4). FGFR2 amplifications were detected in two (IDs 2521, 2512) of the six. For five of the diffuse gastric cancer samples, we had matched normal FFPE tissue from the same individual (IDs 2521, 2512, 2505, 2502, 2504) with the exception of one sample (ID 709). Every tested matched normal gastric tissue contained no FGFR2 amplification, thus confirming that the amplifications in 2521 and 2512 were somatic in nature and related to the tumor (Fig. 4). We also evaluated seven hereditary diffuse gastric carcinomas (IDs 525, 2618, 379, 2619, 561, 778 and 619) of the diffuse subtype and identified a single FGFR2 amplification (Fig. 4). All six of these individuals inherited a germline CDH1 mutation and thus are at risk for developing gastric cancer. With the exception of 525 as previously described, none of these samples had FGFR2 amplifications. From the intestinal subtype (IDs 15314, 15290, 15313, 15289) of the gastric cancer samples, none had FGFR2 amplifications (Fig. 4).
Figure 4.
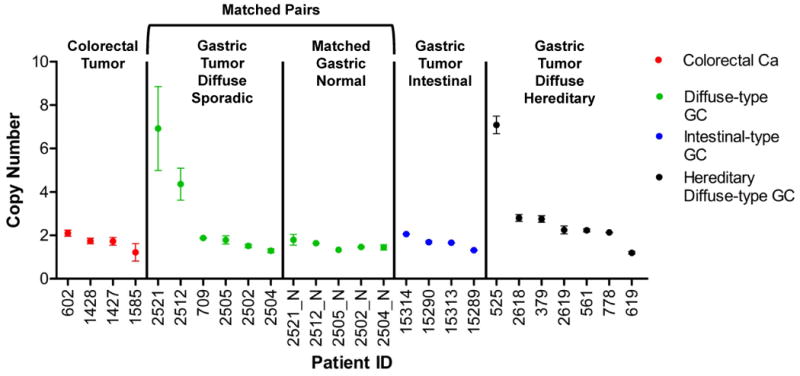
FGFR2 copy number measured by ddPCR in archival FFPE cancer samples.
Copy number of the FGFR2 gene were measured in human colon carcinomas (red dots, n=4), diffuse-type gastric carcinomas (green dots, n=6), intestinal-type gastric carcinomas (blue dots, n=4), and hereditary diffuse gastric cancers (black dots, n=7). Patient 525 is a positive control for the FGFR2 amplification. The X-axis numbers indicate patient identification numbers; “N” indicates a matched normal tissue control for that patient identification number. Error bars represent standard deviation of a minimum of 3 experimental replicates.
Discussion
We developed a droplet digital PCR assay method of determining genomic amplifications from archival FFPE cancer samples. We demonstrated ddPCR offers improved accuracy and precision in measuring genome amplification from FFPE samples over traditional real-time PCR and other techniques that are adversely affected by PCR amplification inequalities, sample quality, sample quantity, and sample purity. Clinical cancer specimens from surgical resections and biopsy are frequently comprised of both malignant and adjacent normal tissue. This admixture of tissues with inherently different genetic compositions can subsequently confound copy number estimations. The digital PCR method employed here is highly sensitive and capable of accurately detecting amplified loci present at ratios less than 1:100. Thus, biopsy specimens of tumor admixed with significant amounts of surrounding normal tissue can be accurately evaluated for the presence of an amplified locus. However, the detection limit of ddPCR is dependent on the extent of target locus amplification. Highly amplified genomic loci are more readily detected in highly heterogeneous populations than low-level amplification events which require a more pure sample for accurate detection.
CNVs, such as genomic amplifications, represent critical genetic events that contribute to the development and progression of human malignancy. In this regard, accurately identifying and characterizing copy number alterations is important for understanding the genetic, genomic and underlying biology of human tumors. Of significant clinical relevance is the detection of genomic amplifications containing oncogenes that can be inhibited with targeted therapies. This represents an increasingly important approach for identifying personalized targets for pharmaceutical intervention. This principle is exemplified by the detection of ERBB2 (e.g. Her2) amplification in gastric and breast tumors with subsequent targeting through small molecules (e.g. lapatinib) and biologics (e.g. trastuzumab), which result in improved patient outcomes and survival. As a demonstration of the translational utility of the ddPCR analysis in cancer studies, we demonstrated reliable detection of amplification of the FGFR2 gene. Several small molecule FGFR2 inhibitors have been recently developed for clinical use in cancer patients whose tumors harbor amplification or activating mutations of the FGFR2 gene. The reliable detection of an FGFR2 amplification event will be of critical importance in appropriately selecting individuals with cancer that might benefit from pharmacological blockade of the FGF receptor signaling cascade. Overall, the ddPCR approach is another tool for evaluating and measuring the diverse genomic events that drive human malignancies such as FGFR2 amplifications among others.
During the course of our study, we analyzed gastric adenocarcinoma samples. Based on specific histopathological and clinical features, gastric carcinoma is generally categorized into diffuse and intestinal subtypes (9). Compared to the intestinal subtype, diffuse gastric cancer has a higher incidence of metastatic disease and a generally worse prognosis (10,11). The amplification events reported in this study were restricted to diffuse-type gastric tumors, but not the intestinal type. Although the sample size in this study was too small to extrapolate a significant difference in amplification frequency or elevation in copy number between these types of gastric cancer, the trend of this small data set is intriguing and warrants additional study. The presence of the amplification events in gastrointestinal cancers reported here and elsewhere suggests that targeted treatments of these solid tumors is feasible and represents an alternative to cytotoxic chemotherapies. Several early-phase trials evaluating the role of oral tyrosine-kinase inhibitors, including FGFR2 inhibitors, in gastrointestinal malignancies are underway.
Acknowledgments
This work was supported by grants awarded from the Gastric Cancer Foundation to HPJ and JMF. Additional support for HPJ came from the Doris Duke Clinical Foundation and the Howard Hughes Medical Foundation. We thank Patrick Flaherty for assistant with the Affymetrix array data analysis.
References
- 1.Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68:3077–3080. doi: 10.1158/0008-5472.CAN-07-3293. discussion 3080. [DOI] [PubMed] [Google Scholar]
- 2.Settleman J. Oncogene addiction. Curr Biol. 2012;22:R43–44. doi: 10.1016/j.cub.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Kallioniemi OP, Kallioniemi A, Kurisu W, Thor A, Chen LC, Smith HS, Waldman FM, Pinkel D, Gray JW. ERBB2 amplification in breast cancer analyzed by fluorescence in situ hybridization. Proc Natl Acad Sci U S A. 1992;89:5321–5325. doi: 10.1073/pnas.89.12.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8817–8822. doi: 10.1073/pnas.1133470100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kunii K, Davis L, Gorenstein J, Hatch H, Yashiro M, Di Bacco A, Elbi C, Lutterbach B. FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer research. 2008;68:2340–2348. doi: 10.1158/0008-5472.CAN-07-5229. [DOI] [PubMed] [Google Scholar]
- 7.Katoh Y, Katoh M. FGFR2-related pathogenesis and FGFR2-targeted therapeutics (Review) Int J Mol Med. 2009;23:307–311. doi: 10.3892/ijmm_00000132. [DOI] [PubMed] [Google Scholar]
- 8.Nakatani H, Sakamoto H, Yoshida T, Yokota J, Tahara E, Sugimura T, Terada M. Isolation of an amplified DNA sequence in stomach cancer. Japanese journal of cancer research: Gann. 1990;81:707–710. doi: 10.1111/j.1349-7006.1990.tb02631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lauren P. Histogenesis of intestinal and diffuse types of gastric carcinoma. Scand J Gastroenterol Suppl. 1991;180:160–164. [PubMed] [Google Scholar]
- 10.Cunningham D, Allum WH, Stenning SP, Thompson JN, Van de Velde CJ, Nicolson M, Scarffe JH, Lofts FJ, Falk SJ, Iveson TJ, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med. 2006;355:11–20. doi: 10.1056/NEJMoa055531. [DOI] [PubMed] [Google Scholar]
- 11.Lim L, Michael M, Mann GB, Leong T. Adjuvant therapy in gastric cancer. J Clin Oncol. 2005;23:6220–6232. doi: 10.1200/JCO.2005.11.593. [DOI] [PubMed] [Google Scholar]