

Abstract
Biomarker discovery efforts in serum and plasma are greatly hindered by the presence of high abundance proteins that prevent the detection and quantification of less abundant, yet biologically significant proteins. The most common method for addressing this problem is to specifically remove the few abundant proteins through immunoaffinity depletion/subtraction. Herein, we improved upon this method by utilizing multiple depletion columns in series, so as to increase the efficiency of the abundant protein removal and augment the detection/identification of less abundant plasma proteins. Spectral counting was utilized to make quantitative comparisons between un-depleted plasma, plasma depleted with a single depletion column, and plasma depleted using two or three depletion columns in tandem. In the un-depleted plasma only 29 lower abundance protein groups were identified with the top-scoring protein from each group having a median spectral count of 3, while in the plasma processed using a single HSA depletion column 61 such protein groups were identified with a median spectral count of 8. In comparison, 76 lesser abundant protein groups were identified with a median spectral count of 11.5 in the two column setup (i.e., HSA followed by MARS Hu14). However, in the ultimate depleted plasma sample, which was created using three depletion columns in tandem, the number of less abundant protein groups identified increased to 81 and the median, average spectral count for the top-scoring proteins from each group increased to 15 counts per protein. Moreover, exogenous B-type Natriuretic Peptide-32, which was added to the plasma as a detection benchmark at 12 μg/mL, was only detected in the plasma sample depleted using three depletion columns in tandem. Collectively, these data demonstrate this method, Tandem Removal of Abundant Proteins or TRAP, provides superior removal efficiency compared to traditional applications and improves the depth of proteome coverage in plasma.
Introduction
The potential utility of plasma as a source for diagnostic and prognostic markers of human disease is unparalleled by any other clinical specimen given its ease of access and the diversity of its constituents. In particular, the use of plasma proteins, such as Prostate Specific Antigen1, CA-1252 or B-type Natriuretic Peptide-32 (BNP-32)3, as biomarkers of disease progression is particularly useful to clinicians. However, the identification of valid protein biomarkers within plasma or elsewhere is exceedingly difficult and, as such, the rate at which protein biomarkers are being introduced into clinical practice is prohibitively slow.4 The challenge with identifying biomarkers in plasma is due in part to the complexity of the plasma proteome and the limitations it places on traditional protein fractionation methods and global-scale quantitative strategies. For instance, the majority of protein mass in plasma is comprised from a relatively small number of highly abundant proteins though the dynamic range of all plasma proteins spans over 10 orders of magnitude.4 Consequently, low abundance proteins of biological importance inevitably go undetected as a result of their low absolute concentration and the suppressive effect of these highly abundant proteins.
Of all the pre-fractionation methods, perhaps the most effective technology for combating interference from abundant plasma proteins is affinity depletion.5 In this method serum or plasma is passed through an affinity column, which specifically retains (i.e., depletes) one or more of the most abundant plasma proteins, thereby decreasing the dynamic range of the unbound proteins. One of the first examples of this was the utilization of immobilized Cibracon Blue 3G dye to remove human serum albumin (HSA) from plasma in a chromatography platform;6 however, the poor specificity of this reagent makes its use unfavorable for quantitative studies.7 To improve upon this technique, Lollo et al. developed an affinity column comprised of proprietary “polypeptides” that specifically bound both HSA and immunoglobulin G (IgG).8 Using this method, they were able to reproducibly obtain greater than 96% removal efficiency of the two target proteins and obtain better visualization of the remaining serum proteins in the subsequent 2D-SDS-PAGE analysis. Since this report, several similar methods have been developed using a mix of polyclonal antibodies to specifically bind and remove multiple abundant plasma proteins as well as their associated fragments and, currently, there are a variety of immunoaffinity depletion/subtraction platforms commercially available that target between 6 and 20 abundant proteins.9-11 Although these antibody-based techniques can reportedly obtain greater than 95% removal efficiency, there is still significant room for improvement since the portion of the abundant target proteins remaining after depletion still have comparable concentrations to the other protein constituents and, as a result, can still interfere during subsequent analyses. For example, if each of the 14 most abundant plasma proteins, which comprise approximately 94% of the total protein mass in plasma, is depleted at an efficiency of 98%, those same 14 proteins would still account for nearly one fourth of the total protein mass following depletion. In contrast, if two of the same depletion columns were used in series (each with 98% efficiency), the overall depletion efficiency in theory would be 99.97% and less than one percent of the remaining protein mass would be from the 14 abundant proteins. To demonstrate this principle and determine if it results in improved detection and identification of less abundant proteins, herein we have performed shotgun analyses on plasma depleted with multiple columns connected in series. In order to validate this method, termed Tandem Removal of Abundant Proteins (TRAP), spectral counting was used to make quantitative comparisons between the tandem-depleted plasma sample, un-depleted plasma, as well as plasma depleted using a single removal column.
Spectral counting, which equates a protein’s abundance to the number of MS/MS spectra identifying that protein,12 has seen considerable use as a tool for biomarker discovery in cell lines13-15 and tissue specimens16-18; however, it has seen limited applications in bodily fluids such as urine19, saliva20 or blood.21, 22 It has been reported by Old et al.23 and recently by our group24 that multiple spectral counts are required to accurately quantify relative proteins abundances; thus, to practically apply spectral counting in plasma or serum it is essential to remove the highly abundant proteins that would otherwise dominate the spectral data. Indeed, the two reports we discovered in the literature both employed ethanol precipitation to remove HSA and improve identification and detection of the less abundant proteins.21, 22 However, it has not been demonstrated to what extent immunoaffinity depletion of multiple abundant proteins will improve spectral counting in plasma. Thus, another aim of the present work was to systematically characterize the effects of abundant protein depletion on the application of spectral counting in plasma, on which there is few reports, in an effort to detect low abundant proteins (e.g., BNP-32) with significant clinical utility.
Experimental
Materials
All solvents used in these studies were of HPLC grade and were purchased from Burdick and Jackson (Muskegon, MI) unless otherwise noted. The chemical reagents iodoacetamide, urea, cysteine, formic acid, and acetic acid were obtained from Sigma-Aldrich (St. Louis, MO), as was the TPCK-treated trypsin. The ammonium bicarbonate used to produce the digestion buffer was from Fisher Scientific (Fair Lawn, NJ), while the dithiothreitol (DTT) was from Bio-Rad (Hercules, CA). Lastly, synthetic B-type Natriuretic Peptide-32 was purchased from the American Peptide Company (Sunnyvale, CA).
Protein Depletions
Three distinct depleted plasma samples were created using a combination of Multiple Affinity Removal System (MARS) LC columns (Agilent, Santa Clara, CA). The depletion columns consisted of one 4.6 × 50 mm serum albumin (HSA) removal column and two identical 4.6 × 100 mm Human 14 (Hu14) removal columns. The latter columns target the 14 most abundant plasma proteins for removal, which include: HSA, Immunoglobulin G (IgG), Immunoglobulin A (IgA), Immunoglobulin M (IgM), Fibrinogen, Apolipoprotein A1, Apolipoprotein A2, α2-Macroglobulin, Transferrin, Transthyretin, α1-Antitrypsin, α1-Acid Glycoprotein, Complement C3, and Haptoglobin. The first depleted plasma sample, termed sample A, was created by removing only the HSA using the corresponding MARS column according to the manufacturer’s protocol. The two tandem-depleted samples (TRAP samples), termed sample B and C were created by connecting the HSA and Hu14 depletion column(s) in series using PEEK tubing (0.01” I.D. × 5 cm). The former sample was produced using only the HSA removal column and a single Hu14 removal column in tandem, while the latter sample was created using an additional Hu14 removal column.
For all depletions, 50 μL of normal pooled human plasma (Innovative Research Inc., Novi, MI) was spiked with 12 μg/mL of BNP-32 to serve as a benchmark for these studies (vide infra). After diluting the spiked plasma 4-fold with MARS buffer A, the diluted plasma was passed through a 0.22 μm centrifugal filter (Agilent, Santa Clara, CA), 160 μL of filtrate (equivalent to 40 μL of undiluted plasma) and loaded into the sample loop for depletion. All depletions were performed on an LC-20AD HPLC system equipped with an SPD-20A UV/Vis detector (Shimadzu, Columbia, MD) and an FC 203B fraction collector (Gilson, Middleton, WI). In producing the tandem depleted plasma samples (Samples B and C), the flow-rate of 0.125 mL/min was used in order to maintain the optimum efficiency for the Hu14 removal columns and the binding portion of the gradient was extended to account for the increased bed volume (see Table S-1). Immediately following their elution the unbound proteins were immediately concentrated to < 100 μL using a Vivaspin 2, 2000 MWCO filter (Vivaproducts Inc., Littleton, MA), buffer exchanged 3 times with 2 mL of 100 mM ammonium bicarbonate buffer (pH 8) and, finally, concentrated to dryness, in vacuo, prior to storage at -20 °C until needed. For subsequent analyses by 1D-SDS-PAGE and nanoLC-MS, the dried depleted plasma samples were initially reconstituted in 6M urea and then an aliquot of each was diluted 3-fold in water in order to measure the total protein concentration using a Coomassie Plus Bradford Assay Kit (Thermo-Fisher Scientific). Afterwards, the samples were diluted with 6M urea to create final working samples with a total protein concentration of 1 mg/mL. The un-depleted plasma sample used in these studies was created simply by spiking 12 μg/mL BNP-32 and diluting with 6 M urea to a final total protein concentration of 1 mg/mL.
In-Solution Digestion
For each depletion sample, 35 μL of the 1 mg/mL protein solution was reduced with 2 μL of 50 mM DTT for 1 hour at 37 °C and then alkylated with 6 μL of 200 mM iodoacetamide for 1 hour at 37 °C. After allowing the sample to cool, the alkylation reaction was quenched by adding 100 μL of 120 mM cysteine and allowing the mixture to sit for an additional 30 minutes at room temperature. This step also served to dilute the concentration of urea to less than 2 M. The sample was then subjected to tryptic digestion at a 1-to-50 substrate-to-enzyme ratio by adding 700 ng of enzyme. After incubating at 37 °C for 4 hours, a fresh 700 ng aliquot of enzyme was added, bringing the total substrate-to-enzyme ratio to 1-to-25. The digestion was then allowed to proceed for an additional 12 hours at 37 °C prior to quenching with 1% formic acid. Finally, all samples were diluted to a final concentration 100 ng/μL and stored at -20 °C until nanoLC-MS/MS analysis. All reagents were made in 100 mM ammonium bicarbonate buffer (pH 8), while the trypsin was initially reconstituted in 50 mM acetic acid and then diluted 50-fold in the ammonium bicarbonate buffer prior to its use.
NanoLC-MS/MS
Tryptic digestions from each sample were thawed and analyzed in triplicate, in a randomized order, using an LTQ-Orbitrap mass spectrometer (Thermo Scientific, San Jose, CA). Online desalting and reversed phase chromatography was performed using a vented column configuration as previously described25 with a Nano2D-LC system equipped with an AS1 autosampler (Eksigent, Dublin, CA). Mobile phases A and B for these analyses were 98/2/0.2 and 2/98/0.2 (water/acetonitrile/formic acid), respectively, and the trap and analytical columns were packed with 4 μm Jupiter Proteo C12 90 Å stationary phase (Phenomenex, Torrance, CA). For each run 10 μL of a 100 ng/μL tryptic digestion was aspirated and injected onto the trap/desalting column at 3 μL/min in 2% mobile phase B by way of a 13.5 μL metered injection. The trap column was packed in-house to 3 cm using a 100 μm ID IntegraFrit column (New Objective, Woburn, MA). After desalting, the 350 nL/min gradient was directed across the trap and onto the analytical column, which was packed in-house to 10 cm using a 75 μm ID PicoFrit column (New Objective, Woburn, MA). The gradient was initially held at 2% mobile phase B for 5 minutes and then ramped to 45% mobile phase B over the next 58 minutes. The column was then flushed with 90% mobile phase B for 3 minutes prior to re-equilibrating the column at initial conditions for 7 minutes. Precursor mass spectra were acquired at a resolving power of 60,000FWHM at m/z=400 using a lock mass injection of 445.120025 m/z and subsequent MS/MS spectra were acquired on the 6 most abundant precursor ions using the data dependent facilities of the instrument with an isolation window of 2 m/z units. Dynamic exclusion was utilized allowing for 1 repeat, a 30 second repeat duration, a 90 second exclusion duration, and an exclusion list size of 100. The AGC for the Orbitrap and ion trap were set to 1 × 106 and 1 × 104, respectively. Other instrumental parameters included an ESI potential of 2000 V, a capillary offset of 35 V, and a capillary temperature of 250 °C.
Data Processing
The .RAW files produced in these experiments were initially processed and then searched using Mascot Daemon (Matrix Science, Boston, MA), which created the .dat files (i.e., the search results files) used for spectral counting analysis within ProteoIQ 1.5.01 (BioInquire, Athens, GA). The data was searched against the IPI Human Database v.3.68 that was modified to produce a concatenated forward and reverse database for estimating the false discovery rate (FDR). Within Mascot Daemon, trypsin was selected as the enzyme and two missed cleavages were allowed per peptide. Carbamidomethyl groups were selected as a fixed modification on all cysteine residues, while variable modifications included oxidation of methionine residues and deamidation of asparagine and glutamine residues. Other search parameters included a 5 ppm peptide tolerance and a 0.6 Da MS/MS tolerance. In ProteoIQ, each of the 4 samples was treated as a different biological sample with 3 replicates, and all identifications were made using a minimum protein probability of 0.5 and maximum protein FDR of 1%. Since the complexity and dynamic range of protein concentrations in each sample were drastically different, each of the four samples were not expected to yield the same total number of spectral counts; thus, the spectral counts reported here were not normalized by the total number of spectral counts in each replicate or sample and were only normalized according to protein length where noted in the text. Additionally, all spectral count values reported are the total obtained across the three replicate injections and only to top scoring proteins from each protein group identified were included in the spectral counting analyses.
Results and Discussion
Three MARS LC columns (one HSA removal and two Hu14 removal) were systematically evaluated to determine if using the three columns in tandem could benefit a shotgun proteomics analysis of plasma and, in particular, the application of spectral counting and detection of BNP-32. The premise behind the TRAP method was that connecting multiple columns in series would inherently increase the depletion efficiency of the protein removal relative to using a single column, thereby improving the enrichment of lower abundance proteins, and improving their detection and identification. The total protein concentration of the depleted plasma (i.e., the collected unbound fractions) was measured for each sample using a Bradford assay in order to gauge the efficiency of the depletion setups – these data are summarized in Table 1. It was determined the un-depleted plasma had a total protein concentration of approximately 54 mg/mL, which corresponds to a total of 2.15 mg of protein in each 40 μL aliquot of plasma used for depletion. In comparison, the depleted plasma samples A, B and C had 64, 93 and 94% less protein after depletion, respectively. For sample A these data correlate well with the reported mass contribution of HSA in plasma.8, 26 In samples B and C, the amount of protein removed was also consistent with the estimated mass contribution of the 14 targeted abundant plasma proteins26, 27 and the 1% increase in depletion efficiency from sample B to C was on par with an expected depletion efficiency of 98-99% per Hu14 removal column. Even though this was only a marginal increase in the overall amount of protein removed, the unbound proteins in sample C were enriched 15.8-fold with respect to the un-depleted plasma, which is 15% greater than the unbound proteins in sample B were enriched. These results were also noticeable during analysis of the various samples by 1D-SDS-PAGE (see Supporting Information and Figure S-2).
Table 1.
Bradford Assay Results
| Sample | Protein Mass, mg (±SD) | % Depletion | Enrichment Factor, -fold* |
|---|---|---|---|
| Raw (Un-depleted) | 2150 (±70) | - | - |
| A - HSA Depleted | 767 (±127) | 64.3 | 2.8 |
| B - HSA/Hu14 TRAP | 157 (±5) | 92.7 | 13.7 |
| C - HSA/Hu14/Hu14 TRAP | 136 (±6) | 93.7 | 15.8 |
Relative to the raw plasma
Proteomic Analysis
In total, 268 proteins were identified between the un-depleted and depleted plasma samples. The number of proteins identified in each sample is shown in Figure 1 and are broken-down into the number of Hu14 proteins (those 14 abundant proteins targeted for depletion) and non-Hu14 proteins identified. Of the four samples the greatest number of protein identifications was obtained in sample A; however, over one third of these protein identifications (77 out of 201) were attributed to various annotated forms of the targeted Hu14 proteins. The most abundant proteins, likewise, dominated the protein identifications in the raw, un-depleted plasma with over half of the identifications coming from different annotated forms of the Hu14 proteins. In comparison, the number of Hu14 proteins identified was substantially lower in the TRAP samples, which resulted in an increase in the number of non-Hu14 identified. In samples B and C, respectively, approximately 91% and 94% of the proteins identified were lower abundance proteins. In comparing the two TRAP setups, 11 additional non-Hu14 proteins were identified in sample C relative to sample B. These results directly indicate the use of multiple depletion columns (even identical ones) in tandem can improve the efficiency of the abundant protein removal and can improve the detection/identification of lesser abundant proteins. Indeed, even greater improvements would likely be observed using additional protein or peptide level pre-fractionation methods such as in a MuDPIT or GeLC-MS workflow.
Figure 1.
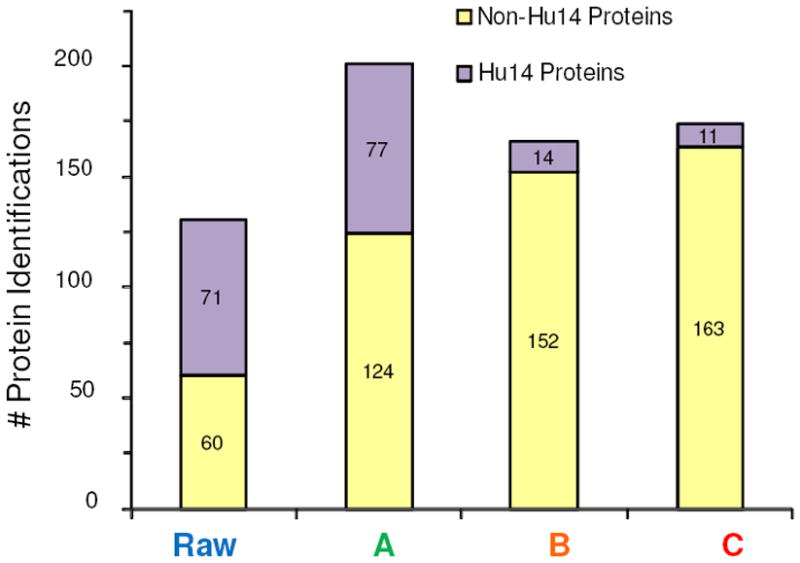
Bar graph showing the number of abundant proteins (Hu14) and lesser abundant proteins (non-Hu14) identified in the raw, un-depleted plasma and various depleted plasma samples. Please note these numbers correspond to all proteins identified in these studies and not the number of protein groups identified.
Spectral Counting
Compiled in Table 2 is a list of proteins identified in these studies whose physiological concentrations have been previously reported and their total number of spectral counts they obtained within each sample. For proteins with multiple annotated forms identified, only the top scoring form was considered when creating this table. The spectral counts for protein subunits were summed in order to obtain the total number of spectral counts for any multimeric proteins and, similarly, the spectral counts for all immunoglobulins were summed due to the inability to determine the origin of each identified subunit. Unsurprisingly, the spectral counts for HSA and the other targeted Hu14 proteins decreased dramatically in the depleted plasma samples. Several of these proteins were undetected in the depleted plasma samples, which indicate they were depleted sufficiently to fall below the detection threshold of the instrumentation. However, three of the targeted Hu14 proteins (Fibrinogen, Complement C3, and Apolipoprotein AII) were still detected following depletion, two which are known to circulate as fragments or subunits of the parent protein.
Table 2.
Protein Spectral Counts
| Protein | Total Spectral Counts | Physiological Concentration (μg/mL) | |||
|---|---|---|---|---|---|
| Raw | A | B | C | ||
| Serum Albumin | 319 | - | - | - | 35000 - 5000039 |
| Immunoglobulins (G, M, A) | 220 | 639 | - | - | 8230 - 2352039, a |
| Serotransferrin | 45 | 212 | - | - | 2000 - 400039 |
| Haptoglobin | 38 | 121 | - | - | 200 - 190039 |
| Apolipoprotein A1 | 37 | 86 | - | - | 1280 - 276039 |
| Fibrinogen | 30 | 226 | 56 | 29 | 1850 - 434039 |
| Complement C3 | 30 | 120 | 12 | 11 | 900 - 180039 |
| Alpha-2-Macroglobulin | 10 | 47 | - | - | 1060 - 279039 |
| Apolipoprotein A2 | 8 | 21 | 2 | 1 | 52040 |
| Alpha-1-Antitrypsin | 7 | 65 | - | - | 900 - 220039 |
| Complement C4 | 7 | 47 | 143 | 179 | 160 - 47039 |
| Vitamin D-binding Protein | 6 | 37 | 119 | 138 | 320 - 46041 |
| Vitronectin | 6 | 8 | 30 | 35 | 240 - 53042 |
| Complement Factor H | 5 | 45 | 100 | 146 | 160 - 41239 |
| Alpha-1-Acid Glycoproteina | 2 | 27 | - | - | 390 - 115039 |
| Clusterin | 1 | 10 | 34 | 33 | 250 - 42042 |
| Gelsolin | 1 | 3 | 22 | 42 | 190 - 30043 |
| Hemopexin | - | 22 | 55 | 70 | 400 - 150044 |
| Ceruloplasmin | - | 15 | 52 | 81 | 170 - 48039 |
| Plasminogen | - | 16 | 38 | 53 | 80 - 14039 |
| Apolipoprotein B | - | 12 | 40 | 36 | 40 - 13039 |
| Hemoglobin | - | 4 | - | - | 5 - 8039 |
| Apolipoprotein M | - | 3 | - | - | 12.3 - 27.645 |
| Heparin Cofactor II | - | 2 | 9 | 13 | 9046 |
| Angiotensinogen | - | 2 | 4 | 11 | 6947 |
| Complement Factor I | - | 1 | 14 | 32 | 29.3 - 58.539 |
| Complement C5 | - | 1 | 12 | 21 | 60 - 20039 |
| Complement C1q | - | 1 | 11 | 13 | 70 - 48039 |
| Complement C6 | - | 1 | 7 | 11 | 71 - 12839 |
| Afamin | - | - | 11 | 43 | 34.6 - 116.148 |
| Complement C9 | - | - | 11 | 16 | 60 - 29039 |
| Coagulation Factor XII | - | - | 11 | 15 | 12 - 4749 |
| Leucine-rich Alpha-2-Glycoprotein | - | - | 11 | 4 | 5050 |
| Complement C8 | - | - | 10 | 31 | 107 - 24939 |
| Pigment Epithelium-derived Factor | - | - | 9 | 2 | 3.1 - 6.251 |
| Serum paraoxonase/arylesterase 1 | - | - | 7 | 8 | 16.8 - 527.452 |
| Tetranectrin | - | - | 5 | 7 | 7.8 - 1753 |
| Properdin | - | - | 5 | 3 | 22.3 - 67.639 |
| Alpha-2-Antiplasmin | - | - | 3 | 4 | 6954 |
| Zinc Alpha-2-Glycoprotein | - | - | 3 | - | 27.455 |
| Complement C7 | - | - | 2 | 9 | 40 - 11039 |
| Serum Amyloid P Component | - | - | 1 | 3 | 2756 |
| B-type Natriuretic Petide-32 | - | - | - | 7 | 12b |
| Fibulin 1 | - | - | - | 7 | 3357 |
| Insulin-like Growth Factor Binding Protein-3 | - | - | - | 7 | 3.2 - 8.739 |
| Phosphatidylinositol-Glycan Specific Phospholipase D | - | - | - | 2 | 18.3 - 106.758 |
| Apolipoprotein(a) | - | - | - | 1 | < 3039 |
| Cysteine-rich Secretory Protein-3 | - | - | - | 1 | 6.359 |
| Protein Z-dependent Protease Inhibitor | - | - | - | 1 | 1 - 1.660 |
Range was obtained by summing reference values for the three immunoglobulins
Concentration of exogenous protein spiked into plasma prior to depletion
With respect to the non-Hu14 proteins, the spectral counts for the majority of these proteins demonstrate the expected increase across the depleted plasma samples (see Table 3 for those non-Hu14 proteins whose spectral counts did not increased). Based on the known physiological concentration of the un-depleted proteins and their observed spectral counts (Table 2), it was apparent that lower concentration proteins were detected as a greater fraction of abundant proteins were depleted. In the un-depleted plasma, for instance, the two proteins detected with the lowest reported concentrations were Complement C4 and Complement Factor H. These proteins, whose reported concentrations go as low as 160 μg/mL, had a total of 7 and 5 spectral counts, respectively. However, the removal of HSA resulted in obvious improvements to the depth of coverage as a number of new proteins were identified in sample A having reported concentration in the tens of microgram per milliliter range, such as Complement Factor I and Angiotensinogen. In comparison, the majority of new proteins detected in sample B had reported concentrations in the tens of microgram per milliliter range and even a few were detected with reported concentrations in the low microgram per milliliter range (e.g., Pigment Epethelium-derived Factor and Tetranectrin). Finally, several new proteins with reported concentrations in the low micrograms per milliliter range were identified in sample C. Most notably, Protein Z-dependent Protease Inhibitor (PZI) was detected with a reported circulating concentration range of only 1 – 1.6 μg/mL. Although these observations are inherently qualitative as a result of the uncertainty in the true protein concentrations, it is apparent that lower concentrations of protein are detectable in the TRAP samples compared to those detected in the un-depleted and HSA depleted plasma.
Table 3.
Spectral Counts for Depleted Proteins
| Protein | Total Spectral Counts | |||
|---|---|---|---|---|
| Raw | A | B | C | |
| Apolipoprotein C-II | - | 1 | - | - |
| Apolipoprotein C-III | 4 | 9 | 6 | 4 |
| Apolipoprotein D | - | 3 | - | - |
| Apolipoprotein E | 1 | 13 | 3 | 6 |
| Apolipoprotein L1 | - | 5 | - | - |
| Apolipoprotein M | - | 3 | - | - |
| Serum Amyloid A-4 Protein | 1 | 6 | - | - |
| Apolipoprotein B-100 | - | 12 | 40 | 36 |
| C4 Binding Protein | - | 7 | 4 | - |
| CD5 Antigen-like Protein | 1 | 6 | - | - |
| Hemoglobin | - | 3 | - | - |
| Histidine-rich Glycoprotein | - | 10 | 14 | - |
| C1 Inhibitor | 1 | 2 | 1 | 1 |
| Reumatoid Factor D5 | - | 3 | - | - |
| Protein S | - | 4 | 2 | 2 |
| Zinc-Alpha-2-Glycoprotein | - | - | 3 | - |
| Leucine-rich Alpha-2-Glycoprotein | - | - | 11 | 4 |
| Clusterin | 1 | 10 | 34 | 33 |
| Properdin | - | - | 7 | 5 |
In order to validate this claim, exogenous BNP-32 was spiked into the plasma prior to any depletion at a concentration of 12 μg/mL to provide a known benchmark within each sample. We were confident in using this exogenous protein to gauge the level of detection in these studies given its low endogenous concentration (pg/mL)28, 29 and the fact it has been shown previously to be unaffected during depletion of the top 6 abundant plasma proteins using a similar MARS column platform.30 As expected, the exogenous protein standard went undetected in the un-depleted plasma as well as in sample A. The BNP-32 was also undetected in sample B, which was created using an HSA removal column and a single Hu14 removal column in tandem. However, the same concentration of BNP-32 was detected with a total of 7 spectral counts in sample C, which was created using an additional Hu14 removal in the tandem depletion setup. This detection of the exogenous BNP-32 only in sample C unequivocally demonstrates the intended advantage of the TRAP strategy to increase the dynamic range of protein concentrations detectable within human plasma.
In Figure 2 are histograms showing the distribution of normalized spectral counts for the proteins identified in each sample. As can be seen from the first histogram, the majority of spectral counts in the un-depleted plasma were attributed to HSA while all other proteins identified had significantly fewer spectral counts, with the median, normalized, spectral count for the non-Hu14 proteins being only 0.45×10-2. After depletion of HSA the spectral counts for the remaining proteins increased significantly, as demonstrated by Serotransferrin’s increase from 45 spectral counts in the un-depleted plasma to 212 in sample A. Moreover, the median, normalized spectral count obtained in sample A for the non-Hu14 proteins was increased to 1.48×10-2 – more than triple what was obtained in the un-depleted sample. Utilizing the Mann-Whitney U-test, the median normalized spectral counts for the non-Hu14 proteins were determined to be statistically different (p-value = 0.0038) between the un-depleted plasma and sample A. The improvement continued in the TRAP samples with the median, normalized spectral counts for the non-Hu14 proteins increasing to 2.33×10-2 and 2.86×10-2 in samples B and C, respectively. By the Mann-Whitney U-test the increase between samples A and B was determined to be statistically significant (p-value = 0.0272), while the increase between samples B and C was insignificant (p-value = 0.2731); however, when comparing common proteins identified between the respective samples, the increase in the normalized spectral counts was readily apparent as well as statistically significant (see Supporting Information and Figure S-3). When considering non-normalized spectral counts, the median value for the non-Hu14 proteins was 3 in the un-depleted plasma, 8 in sample A, 11.5 in sample B, and 15 in sample C. It is also important to note the total number proteins identified with greater than 4 total spectral counts increased across theses samples, given it has been reported this is the threshold required to obtain accurate quantification.23, 24 In the un-depleted plasma, for example, only 21 of the 60 (~35%) non-Hu14 proteins had 4 or more spectral counts while in sample A 80 of the 124 (~65%) non-Hu14 proteins met the same criteria. In comparison, 117 of the 152 (~77%) and 139 of the 163 (~85%) of the all the non-Hu14 proteins identified in samples B and C, respectively, met or exceed the 4 spectral counts threshold.
Figure 2.
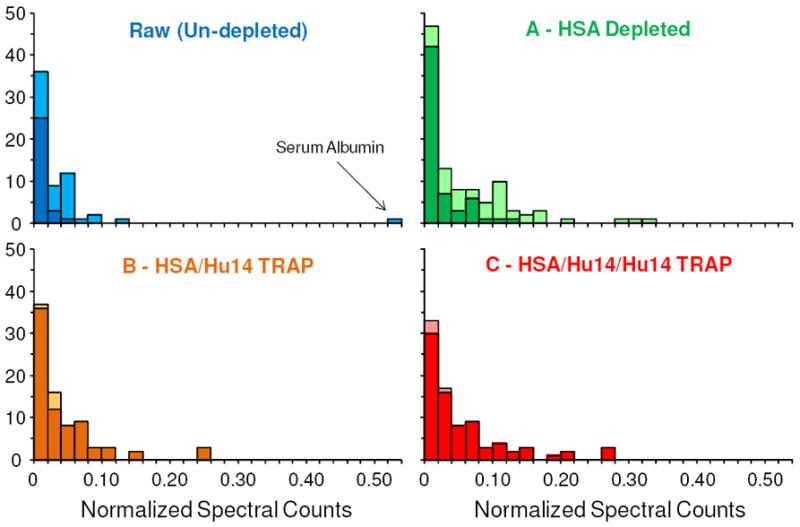
Shown are histograms of the normalized spectral counts (total spectral counts divided by protein length), for the raw, un-depleted plasma and various depleted plasma samples. The darker colored boxes highlight the normalized spectral counts for only the lesser abundant (non-Hu14) proteins identified. Note, only the top-scoring proteins from each protein group were utilized in these graphs.
Non-specific Protein Depletion
In these studies the spectral counts for the majority of non-Hu14 proteins identified trended upwards as the removal of the targeted abundant proteins progressed; however, a select few non-Hu14 proteins demonstrated trends consistent with a depleted protein. More specifically, a decrease in spectral counts from sample A to B or from sample C to D indicated that the protein was depleted by the additional Hu14 removal column. A list of these un-targeted, yet depleted proteins and their spectral counts are compiled in Table 3. Although non-specific adsorption to the column resin has been proposed, it is an unlikely explanation in this case since no decrease in spectral counts was observed between the un-depleted and HSA depleted plasma sample for any protein. Rather, several of the proteins exhibiting the downward progression were apolipoproteins, which would suggest cross reactivity between the anti-Apolipoprotein A1 or anti-Apolipoprotein A2 polyclonal antibodies found in the Hu14 removal columns. Co-depletion of a Hu14 protein and its binding partner(s) is also a likely explanation for the observed depletion of the un-targeted proteins; as in the case of Apolipoprotein D, which it is known to form a heterodimer with the targeted Hu14 protein, Apolipoprotein A2 via a disulfide bridge.31 In fact, several of the identified proteins that demonstrated the decrease in spectral counts are known to bind one or more of the Hu14 proteins targeted for depletion. For instance, CD5 Antigen-like Protein has been found in immunopurified fractions of both monoclonal and polyclonal IgM originating from plasma32 and it is well known Haptoglobin binds free Hemoglobin in circulation in order to prevent oxidative stress.33 Clusterin immobilized on an anti-clusterin immunoaffinity column has been shown to selectively retain Apolipoprotein A1 in plasma, and both proteins are believed to form the base of high density lipoproteins complexes.34 Additionally, Histidine Rich Glycoprotein35 and C1 Inhibitor36 are known to bind Fibrinogen and C1 Inhibitor36, along with Properdin37, is also known to participate in the alternative pathway of the compliment system by binding C3b, a fragment of Complement C3. Moreover, application notes released by the manufacturer of these MARS columns have reported non-specific depletion of proteins.27, 38 Most notably, C1 Inhibitor, Zinc Alpha-2-Glycoprotein (ZAG), and Apolipoproteins B100 and D have been identified in the bound fraction obtained from depletion of the plasma, which corroborates the findings obtained herein using spectral counting.
Conclusions
Multiple abundant protein depletion columns were connected in series to determine if the overall depletion efficiency of the top 14 most abundant proteins from plasma could be increased and to evaluate how this would affect spectral counting for lesser abundant proteins. The TRAP method was systematically evaluated and compared in parallel to un-depleted plasma as well as plasma depleted of only HSA. Both Bradford assays and spectral counting were employed to quantitatively compare the enrichment of proteins between the respective depleted plasma samples as well as the un-depleted plasma. The data collected here consistently demonstrated TRAP technique provides improved depletion of the targeted abundant proteins and enhanced the detection of lesser abundant proteins and increased their spectral counts compared to traditional abundant protein depletion. Although the gains may seem marginal compared to the cost associate with using multiple columns, a more cost effective method of performing TRAP could possibly be achieved using a newly purchased column and an older column in tandem. It should also be noted the problem of non-specific depletion was observed and compounded by TRAP. By observing a systematic decrease in spectral counts between the un-depleted and tandem-depleted samples, 19 proteins were identified as being non-specifically depleted – most likely as a result of antibody cross-reactivity or co-depletion. Overall though, the vast majority of the less abundant proteins identified were positively affected by abundant protein depletion and even more so by the TRAP stratagem.
Supplementary Material
Acknowledgments
The authors are appreciative of the financial support from the National Institutes of Health (Grant 5RO1HL036634) and the NIH/NCSU Molecular Biotechnology Training Program (Grant 5T32GM00-8776-08), which supported CMS in this work.
Footnotes
Supporting Information Available The supporting information referenced in the text is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hernandez J, Thompson IM. Cancer. 2004;101(5):894–904. doi: 10.1002/cncr.20480. [DOI] [PubMed] [Google Scholar]
- 2.Bast RC, Xu FJ, Yu YH, Barnhill S, Zhang Z, Mills GB. Int J Biol Marker. 1998;13(4):179–187. doi: 10.1177/172460089801300402. [DOI] [PubMed] [Google Scholar]
- 3.Hawkridge AM, Heublein DM, Bergen HR, Cataliotti A, Burnett JC, Muddiman DC. P Natl Acad Sci USA. 2005;102(48):17442–17447. doi: 10.1073/pnas.0508782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson NL, Anderson NG. Mol Cell Proteomics. 2002;1(11):845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 5.Whiteaker JR, Zhang HD, Eng JK, Fang RH, Piening BD, Feng LC, Lorentzen TD, Schoenherr RM, Keane JF, Holzman T, Fitzgibbon M, Lin CW, Zhang H, Cooke K, Liu T, Camp DG, Anderson L, Watts J, Smith RD, McIntosh MW, Paulovich AG. J Proteome Res. 2007;6(2):828–836. doi: 10.1021/pr0604920. [DOI] [PubMed] [Google Scholar]
- 6.Gianazza E, Arnaud P. Biochem J. 1982;201(1):129–136. doi: 10.1042/bj2010129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gianazza E, Arnaud P. Biochem J. 1982;203(3):637–641. doi: 10.1042/bj2030637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lollo BA, Harvey S, Liao J, Stevens AC, Wagenknecht R, Sayen R, Whaley J, Sajjadi FG. Electrophoresis. 1999;20(4-5):854–859. doi: 10.1002/(SICI)1522-2683(19990101)20:4/5<854::AID-ELPS854>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 9.Pieper R, Su Q, Gatlin CL, Huang ST, Anderson NL, Steiner S. Proteomics. 2003;3(4):422–432. doi: 10.1002/pmic.200390057. [DOI] [PubMed] [Google Scholar]
- 10.Qian WJ, Kaleta DT, Petritis BO, Jiang HL, Liu T, Zhang X, Mottaz HM, Varnum SM, Camp DG, Huang L, Fang XM, Zhang WW, Smith RD. Mol Cell Proteomics. 2008;7(10):1963–1973. doi: 10.1074/mcp.M800008-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polaskova V, Kapur A, Khan A, Molloy MP, Baker MS. Electrophoresis. 2010;31(3):471–482. doi: 10.1002/elps.200900286. [DOI] [PubMed] [Google Scholar]
- 12.Liu HB, Sadygov RG, Yates JR. Anal Chem. 2004;76(14):4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 13.Kulasingam V, Diamandis EP. Mol Cell Proteomics. 2007;6(11):1997–2011. doi: 10.1074/mcp.M600465-MCP200. [DOI] [PubMed] [Google Scholar]
- 14.Hood BL, Grahovac J, Flint MS, Sun M, Charro N, Becker D, Wells A, Conrads TP. J Proteome Res. 2010;9(7):3656–3663. doi: 10.1021/pr100164x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piersma SR, Fiedler U, Span S, Lingnau A, Pham TV, Hoffmann S, Kubbutat MHG, Jimenez CR. J Proteome Res. 2010;9(4):1913–1922. doi: 10.1021/pr901072h. [DOI] [PubMed] [Google Scholar]
- 16.Kawase H, Fuji K, Miyamoto M, Kubota KC, Hirano S, Kondo S, Inagaki F. J Proteome Res. 2009;8(8):4092–4103. doi: 10.1021/pr900468k. [DOI] [PubMed] [Google Scholar]
- 17.Kawamura T, Nomura M, Tojo H, Fujii K, Hamasaki H, Mikami S, Bando Y, Kato H, Nishimura T. J Proteomics. 2010;73(6):1089–1099. doi: 10.1016/j.jprot.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Gao F, Mo F, Hong X, Wang HY, Zheng SS, Lin BY. Proteom Clin Appl. 2009;3(5):541–551. [Google Scholar]
- 19.Pang JX, Ginanni N, Dongre AR, Hefta SA, Opiteck GJ. J Proteome Res. 2002;1(2):161–169. doi: 10.1021/pr015518w. [DOI] [PubMed] [Google Scholar]
- 20.Rao PV, Reddy AP, Lu X, Dasari S, Krishnaprasad A, Biggs E, Roberts CT, Nagalla SR. J Proteome Res. 2009;8(1):239–245. doi: 10.1021/pr8003776. [DOI] [PubMed] [Google Scholar]
- 21.Pan J, Chen HQ, Sun YH, Zhang JH, Luo XY. Lung. 2008;186(4):255–261. doi: 10.1007/s00408-008-9093-7. [DOI] [PubMed] [Google Scholar]
- 22.Hu XF, Zhang Y, Zhang AL, Li YZ, Zhu ZM, Shao ZM, Zeng R, Xu LX. Omics. 2009;13(4):291–300. doi: 10.1089/omi.2009.0016. [DOI] [PubMed] [Google Scholar]
- 23.Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, Resing KA, Ahn NG. Mol Cell Proteomics. 2005;4(10):1487–1502. doi: 10.1074/mcp.M500084-MCP200. [DOI] [PubMed] [Google Scholar]
- 24.Collier TS, Sarkar P, Franck WL, Rao BM, Dean RA, Muddiman DC. Anal Chem. 2010 doi: 10.1021/ac101978b. In Press. [DOI] [PubMed] [Google Scholar]
- 25.Andrews GL, Shuford CM, Burnett JC, Hawkridge AM, Muddiman DC. J Chromatogr B. 2009;877(10):948–954. doi: 10.1016/j.jchromb.2009.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Putman FW. The Plasma Proteins. Academic Press; New York: 1960. [Google Scholar]
- 27.Mrozinski P, Zolotarjova N, Chen H. Human Serum and Plasma Protein Depletion - Novel High-Capacity Affinity Column for the Removal of the “Top 14” Abundant Proteins. Agilent Technologies, Inc; 2008. [Google Scholar]
- 28.Mukoyama M, Nakao K, Hosoda K, Suga S, Saito Y, Ogawa Y, Shirakami G, Jougasaki M, Obata K, Yasue H, Kambayashi Y, Inouye K, Imura H. J Clin Invest. 1991;87(4):1402–1412. doi: 10.1172/JCI115146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maisel AS, Krishnaswamy P, Nowak RM, McCord J, Hollander JE, Duc P, Omland T, Storrow AB, Abraham WT, Wu AHB, Clopton P, Steg PG, Westheim A, Knudsen CW, Perez A, Kazanegra R, Herrmann HC, McCullough PA. New Engl J Med. 2002;347(3):161–167. doi: 10.1056/NEJMoa020233. [DOI] [PubMed] [Google Scholar]
- 30.Hawkridge AM, Muddiman DC, Helmlein DM, Cataliotti A, Burnett JC. Clin Chem. 2008;54(5):933–934. doi: 10.1373/clinchem.2007.098038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blancovaca F, Via DP, Yang CY, Massey JB, Pownall HJ. J Lipid Res. 1992;33(12):1785–1796. [PubMed] [Google Scholar]
- 32.Tissot JD, Schifferli JA, Hochstrasser DF, Pasquali C, Spertini F, Clement F, Frutiger S, Paquet N, Hughes GJ, Schneider P. J Immunol Methods. 1994;173(1):63–75. doi: 10.1016/0022-1759(94)90284-4. [DOI] [PubMed] [Google Scholar]
- 33.Kato GJ. J Clin Invest. 2009;119(8):2140–2142. doi: 10.1172/JCI40258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jenne DE, Lowin B, Peitsch MC, Bottcher A, Schmitz G, Tschopp J. J Biol Chem. 1991;266(17):11030–11036. [PubMed] [Google Scholar]
- 35.Leung LLK. J Clin Invest. 1986;77(4):1305–1311. doi: 10.1172/JCI112435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis AE, Cai SH, Liu DX. Immunobiology. 2007;212(4-5):313–323. doi: 10.1016/j.imbio.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fearon DT, Austen KF. J Exp Med. 1975;142(4):856–863. doi: 10.1084/jem.142.4.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mrozinski P, Zolotarjova N, Chen H, Martosella J, Liu H. A Novel High-Capacity Affinity Column for the Removal of the Top Seven Abundant Proteins from Human Plasma. Agilent Technologies, Inc; 2006. [Google Scholar]
- 39.Products & Services, Test Menu. Specialty Laboratories; Valencia, CA: 2010. [Google Scholar]
- 40.Stein EA, Dipersio L, Pesce AJ, Kashyap M, Kao JT, Srivastava L, McNerney C. Clin Chem. 1986;32(6):967–971. [PubMed] [Google Scholar]
- 41.Haughton MA, Mason RS. Clin Chem. 1992;38(9):1796–1801. [PubMed] [Google Scholar]
- 42.Hogasen K, Mollnes TE, Tschopp J, Harboe M. J Immunol Methods. 1993;160(1):107–115. doi: 10.1016/0022-1759(93)90014-x. [DOI] [PubMed] [Google Scholar]
- 43.Suhler E, Lin W, Yin HL, Lee WM. Crit Care Med. 1997;25(4):594–598. doi: 10.1097/00003246-199704000-00007. [DOI] [PubMed] [Google Scholar]
- 44.Delanghe JR, Langlois MR. Clin Chim Acta. 2001;312(1-2):13–23. doi: 10.1016/s0009-8981(01)00586-1. [DOI] [PubMed] [Google Scholar]
- 45.Axler O, Ahnstrom J, Dahlback B. J Lipid Res. 2007;48(8):1772–1780. doi: 10.1194/jlr.M700113-JLR200. [DOI] [PubMed] [Google Scholar]
- 46.Tollefsen DM, Majerus DW, Blank MK. J Biol Chem. 1982;257(5):2162–2169. [PubMed] [Google Scholar]
- 47.Davis D, Liyou N, Lockwood D, Johnson A. Clin Genet. 2002;61(5):363–368. doi: 10.1034/j.1399-0004.2002.610508.x. [DOI] [PubMed] [Google Scholar]
- 48.Dieplinger H, Ankerst DP, Burges A, Lenhard M, Lingenhel A, Fineder L, Buchner H, Stieber P. Cancer Epidem Biomar. 2009;18(4):1127–1133. doi: 10.1158/1055-9965.EPI-08-0653. [DOI] [PubMed] [Google Scholar]
- 49.Wuillemin WA, Furlan M, Lammle B. J Immunol Mehtods. 1990;130(1):133–140. doi: 10.1016/0022-1759(90)90307-h. [DOI] [PubMed] [Google Scholar]
- 50.Weivoda S, Andersen JD, Skogen A, Schlievert PM, Fontana D, Schacker T, Tuite P, Dubinsky JM, Jemmerson R. J Immunol Methods. 2008;336(1):22–29. doi: 10.1016/j.jim.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang P, Smit E, Brouwers M, Goossens GH, van der Kallen CJ, van Greevenbroek MMJ, Mariman ECM. Eur J Endocrinol. 2008;159(6):713–718. doi: 10.1530/EJE-08-0521. [DOI] [PubMed] [Google Scholar]
- 52.Mackness B, Mackness MI, Arrol S, Turkie W, Julier K, Abuasha B, Miller JE, Boulton AJM, Durrington PN. Atherosclerosis. 1998;139(2):341–349. doi: 10.1016/s0021-9150(98)00095-1. [DOI] [PubMed] [Google Scholar]
- 53.Thougaard AV, Hogdall CK, Kjaer SK, Blaakaer J, Jaliashvili I, Christiansen M. Clin Chim Acta. 1998;276(1):19–34. doi: 10.1016/s0009-8981(98)00092-8. [DOI] [PubMed] [Google Scholar]
- 54.Aoki N, Sumi Y, Miura O, Hirosawa S. Human Alpha-2-Plasmin Inhibitor. Proteolytic Enzymes in Coagulation, Fibrinolysis, and Complement Activation, Part B. 1993;223:185–197. doi: 10.1016/0076-6879(93)23045-o. [DOI] [PubMed] [Google Scholar]
- 55.Stejskal D, Karpisek M, Reutova H, Stejskal P, Kotolova H, Kollar P. Clin Biochm. 2008;41(4-5):313–316. doi: 10.1016/j.clinbiochem.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 56.Bijl M, Bootsma H, van der Geld Y, Limburg PC, Kallenberg CGM, van Rijswijk MH. Ann Rheum Dis. 2004;63(7):831–835. doi: 10.1136/ard.2002.004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Argraves WS, Tran H, Burgess WH, Dickerson K. J Cell Biol. 1990;111(6):3155–3164. doi: 10.1083/jcb.111.6.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kurtz TA, Fineberg NS, Considine RV, Deeg MA. Metabolism. 2004;53(2):138–139. doi: 10.1016/j.metabol.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 59.Udby L, Cowland JB, Johnsen AH, Sorensen OE, Borregaard N, Kjeldsen L. J Immunol Methods. 2002;263(1-2):43–55. doi: 10.1016/s0022-1759(02)00033-9. [DOI] [PubMed] [Google Scholar]
- 60.Han X, Fiehler R, Broze GJ. Proc Natl Acad Sci USA. 1998;95(16):9250–9255. doi: 10.1073/pnas.95.16.9250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.