
Fig. 2.
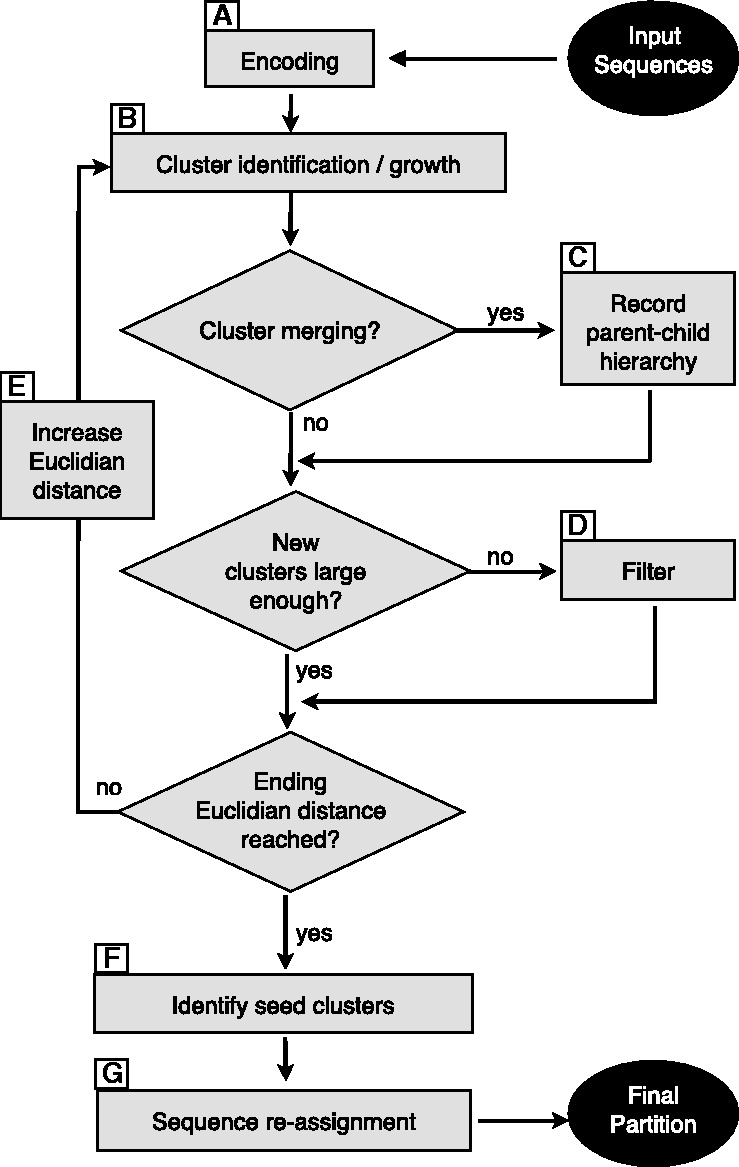
Algorithm flowchart. A: Encoding of the FASTA sequences using dinucleotide counts (see Section 3.1.1 for details). B: Identify new clusters and add sequences to previously identified clusters by single linkage using Euclidian distance cutoff d. C: When any previously identified clusters are merging at the distance cutoff d, record the cluster content before merging, and update the cluster parent–child hierarchy. D: Remove newly identified clusters containing less than N sequences. E: Increase the Euclidian distance cutoff d for the next pass. F: Traverse the cluster parent–child merging tree to re-create each cluster discovered in its last state before its merging with another cluster (identification of seed clusters). G: Re-assign sequences to closest seed clusters (see text for detail)