
Figure 3.
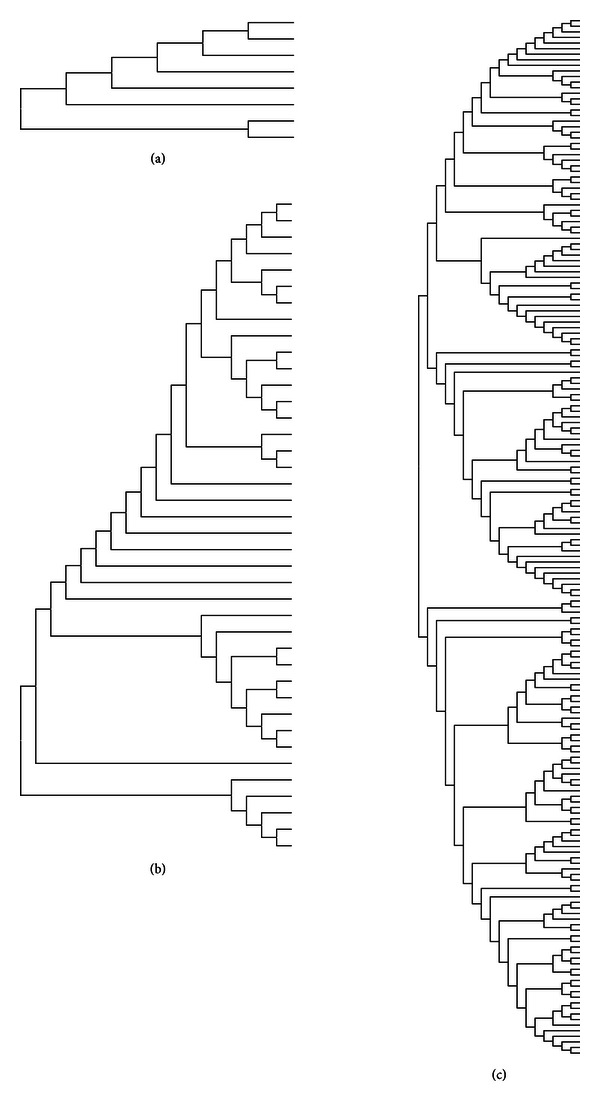
Molecular phylogenetic analysis of HVR1 populations inferred after the application of three different error correction methods. (a) Cut-off method >1% (I), (b) bidirectional coverage method (II), and (c) ShoRAH algorithm (III). The evolutionary history was inferred by using the Maximum Likelihood method based on the Tamura-Nei model [23]. Evolutionary analyses were conducted using MEGA 5.0 [22].