

Background: MYC is rapidly degraded in cells, and its accumulation is associated with many human malignancies.
Results: Sequential phosphorylation of MYC by protein kinase A (PKA) and polo-like kinase 1 (PLK1) protects MYC from proteasome-mediated degradation.
Conclusion: A MYC-PKA-PLK1 signaling loop exists in cells.
Significance: We highlight the importance of considering possible regulatory feedback loops while targeting molecules occupying hub positions in signaling pathways.
Keywords: Cancer Biology, MYC, Phosphorylation, Post-translational Modification, Protein Kinase A (PKA), Polo-like Kinase 1 (PLK1)
Abstract
MYC levels are tightly regulated in cells, and deregulation is associated with many cancers. In this report, we describe the existence of a MYC-protein kinase A (PKA)-polo-like kinase 1 (PLK1) signaling loop in cells. We report that sequential MYC phosphorylation by PKA and PLK1 protects MYC from proteasome-mediated degradation. Interestingly, short term pan-PKA inhibition diminishes MYC level, whereas prolonged PKA catalytic subunit α (PKACα) knockdown, but not PKA catalytic subunit β (PKACβ) knockdown, increases MYC. We show that the short term effect of pan-PKA inhibition on MYC is post-translational and the PKACα-specific long term effect on MYC is transcriptional. These data also reveal distinct functional roles among PKA catalytic isoforms in MYC regulation. We attribute this effect to differential phosphorylation selectivity among PKA catalytic subunits, which we demonstrate for multiple substrates. Further, we also show that MYC up-regulates PKACβ, transcriptionally forming a proximate positive feedback loop. These results establish PKA as a regulator of MYC and highlight the distinct biological roles of the different PKA catalytic subunits.
Introduction
MYC is a basic helix-loop-helix leucine zipper transcription factor that regulates a large number of target genes important in cell growth, metabolism, differentiation, proliferation, and apoptosis (1–6). Complete loss of MYC function results in embryonic lethality, whereas its overexpression predisposes cells to malignant transformation (7–9). MYC overexpression is an early and consistent feature of many human malignancies, where it is suspected to regulate key events in tumorigenesis.
Because MYC drives the transcription of genes important in multiple cellular processes, precise temporal regulation of MYC is required. Tight regulation of MYC is achieved through multiple mechanisms at the transcriptional, post-transcriptional, translational, and post-translational levels (10–14). Once translated, MYC is rapidly turned over in normal cells. The MYC steady-state level is regulated at the post-translational level through a series of exquisitely orchestrated phosphorylation events. Ras-mediated activation of MAPK stabilizes MYC by phosphorylation at Ser-62 within the evolutionarily conserved MYC box I region (11). Ser-62-phosphorylated MYC is recognized by GSK3β, which phosphorylates Ser-62-primed MYC at Thr-58. Peptidylprolyl isomerase 1 then catalyzes a cis to trans isomerization of the bond preceding Ser-62, thereby allowing the trans-specific protein phosphatase 2A to remove the stabilizing phosphorylation at Ser-62 (15, 16). MYC phosphorylated at Thr-58, but not Ser-62, is recognized by the E3 ligase Fbw7, which ubiquitinates MYC at the N terminus and targets it for proteasome-dependent degradation (11, 17). Recently, another E3 ligase, βtrcp, was shown to interact through a previously unknown phospho-degron and oppose the Fbw7-mediated ubiquitination at the N terminus. PLK13 phosphorylation within the phospho-degron was reported to be critical for βtrcp binding (18).
In this study, we demonstrate the existence of a MYC-PKA-PLK1 signaling loop and show that MYC is regulated both through transcriptional and post-translational mechanisms by PKA, in a PKA catalytic subunit isoform-specific manner. This work also highlights both the promise and potential pitfalls of global kinase inhibition and emphasizes the need to develop next generation therapeutic strategies capable of disrupting specific kinase-substrate interactions.
EXPERIMENTAL PROCEDURES
Antibodies, Reagents, and Vectors
Antibodies were as follows: c-MYC (N-term) (Epitomics, 1472-1); pan-PKA antibody (BD Biosciences, 610980); Thr-197 phospho-PKA (Cell Signaling, 4781S); HA antibody (Roche Applied Science, 11867423001); donkey anti-rabbit, donkey anti-mouse, and goat anti-rat HRP-linked (Jackson Laboratories). Reagents were as follows: H89 Insolution (Calbiochem), BI2536 (Selleck Chemicals, S1109), myristoylated PKI (14–22) amide (Biomol, P-210), and MG132 (Sigma). For vector constructs, all proteins purified from Escherichia coli were expressed using pQE80 vectors (Qiagen). The pCDNA3.1 vector (Invitrogen) was used for mammalian cell transfections.
Expression and Purification of Recombinant Proteins
All proteins were cloned with an N-terminal His6 tag and expressed in either E. coli or mammalian cells (COS cells or PC3 cells). Recombinant proteins were purified under native conditions using Ni-affinity chromatography. The purified proteins were further enriched by ion-exchange chromatography whenever necessary. The final buffer condition for all proteins used in in vitro kinase assay is 50 mm NaH2PO4, 300 mm NaCl, 250 mm imidazole, and 0.1% Tween 20.
Cell Culture and Transfection
PC3, LNCaP, and 22RV1 cells (Invitrogen) were cultured in RPMI 1640 medium containing l-glutamine and supplemented with 10% heat-inactivated fetal bovine serum in 5% CO2 at 37 °C. Cells were transfected using LipoD293 (SignaGen) according to the manufacturer's instructions. Lipofectamine 2000 (Invitrogen) was used to transfect siRNA as described previously (19, 20). The siRNA sequences used for PKACα knockdown are 5′-AAGCUCCCUUCAUACCAAAGU-3′ and 5′-ACUUUGGUAUGAAGGGAGCUU-3′. The siRNA sequences used for PKACβ knockdown are 5′-AAGGUCCGAUUCCCAUCCCAC-3′ and 5′-GUGGGAUGGGAAUCGGACCUU-3′.
Western Blotting and Immunoprecipitation
For Western blot analysis, mammalian cells were harvested 24–48 h post-transfection and lysed using 150 mm NaCl, 50 mm Tris (pH 7.5), 0.5% Nonidet P-40, and Complete protease inhibitor (Roche Applied Science). Western blotting was performed as described previously (21). Total protein from mouse prostate was extracted during RNA extraction using the protocol described in the RNeasy Kit (Qiagen). Western blotting was performed as described (22). PKACβ2 protein containing the N-terminal HA and His6 tag was purified from transiently transfected COS and PC3 cells either by immunoprecipitation using an antibody against the HA tag or by Ni-affinity chromatography (further enriched by anion-exchange chromatography).
Quantitative PCR Analysis
Total RNA from mammalian cells was extracted using the RNeasy Kit (Qiagen) and reverse-transcribed using the Superscript cDNA synthesis kit (Bio-Rad). qRT-PCR was performed as per the instruction in the iScript qRT-PCR kit (Bio-Rad). -Fold difference in gene expression was determined after normalizing against GAPDH or TATA-binding protein (TBP) (as mentioned in the legends). Primers used for qRT-PCR are: MYC (134 bp), 5′-GCTCTCCTCGACGGAGTCC-3′ and 5′-CCACAGAAACAACATCGATTTCTT-3′; PKACβ(240 bp), 5′-TGGTGGGCATTAGGAG-3′ and 5′-CTTGTGAGTTTTTATATCACTGAC-3′; PKACβ2 (191 bp), 5′-AACCACCTTGTAACCAGTAT-3′ and 5′-TTTGGCTAGAAACTCTTTCA-3′; TBP (100 bp), 5′-GCCAGCTTCGGAGAGTTCTG-3′ and 5′-GCACGAAGTGCAATGGTCTTT-3′; GAPDH (226 bp), 5′-GAAGGTGAAGGTCGGAGT-3′ and 5′-GAAGATGGTGATGGGATTTC-3′.
In Vitro Kinase Assay
In vitro kinase assay was performed using purified recombinant proteins as described previously (21, 23). Kinase assay was carried out for 2 h at room temperature and stopped using 2× Laemmli buffer. After in vitro kinase assay, the proteins were analyzed by SDS-PAGE, transferred to a PVDF membrane, and analyzed by autoradiography. The PVDF membrane was also stained with Coomassie Brilliant Blue to reveal protein loading.
Reverse In-gel Kinase Assay (RIKA)
50 mg/ml PKACα in 8 m urea, 50 mm NaH2PO4, 300 mm NaCl, 250 mm imidazole, and 0.1% Tween 20 was co-polymerized in the gel, and RIKA was performed as described previously (23). RIKA was performed on a PVDF membrane for PLK1-priming experiments (on-membrane kinase assay). Purified recombinant MYC and MYC mutants were resolved by SDS-PAGE and transferred onto a PVDF membrane. The proteins on the membrane were refolded using the buffer conditions used in RIKA. The membrane was incubated with purified active recombinant PLK1 (PLK1T210D) for 2 h in a buffer containing 20 mm Tris (pH 7.6), 20 mm MgCl2, 50 mm DTT) and [γ-32P]ATP. The membrane was washed with water overnight to remove nonspecifically bound ATP. Phosphorylation of MYC by PLK1 was detected by autoradiography.
Protein Identification and Phosphosite Mapping Mass Spectrometry
In-gel tryptic digestion was performed as described previously (23). Desalted tryptic peptides were analyzed by nano-LC-tandem MS on a linear ion-trap mass spectrometer (LTQ; Thermo Fisher). Acquired data were searched against a Homo sapiens protein database or phosphoproteome database using the TurboSEQUEST algorithm (Thermo Fisher).
Immunofluorescence
Cells were plated on poly-l-lysine-treated glass coverslips in 6-well plates. PC3 cells were grown on coverslips and transfected using lipoD293T (Signagen) according to the manufacturer's instructions. Transfected cells were fixed using 2% paraformaldehyde in PBS. Immunofluorescence analysis was performed as described previously (22).
RESULTS
PKA Activity Influences the Steady-state Levels of MYC
During our efforts to identify PKA substrates in prostate cancer cells, we identified α-enolase as a PKACα substrate (Table 1). An alternative in-frame internal translation initiating at +291 of the α-enolase mRNA yields an alternate product termed MYC promoter-binding protein 1 (MBP1) (24). We confirmed both ENO1 and MBP1 to be substrates of PKACα by in vitro kinase assay (Fig. 4E). Because MBP1 is a known transcriptional repressor of MYC, we sought to determine whether phosphorylation of MBP1 by PKA has an effect on MBP1 function and steady-state MYC level (25). We monitored changes in endogenous MYC level in prostate cancer cells treated with the PKA-selective small molecule inhibitor H89 by Western blotting. Pan-PKA inhibition using H89 resulted in decreased MYC accumulation in prostate cancer cells (Fig. 1A). This was found to be consistent in HeLa and K562 cells as well (Fig. 1B). Inhibition of PKA using the PKA-specific peptide inhibitor PKI also destabilized MYC (Fig. 1C). Conversely, activation of PKA using forskolin increased MYC levels (Fig. 1D). Although these results were consistent with our initial hypothesis, the rapid kinetics suggested that the effect could be the result of a direct interaction between PKA and MYC. To determine whether the effect of PKA on MYC is transcriptional, we evaluated changes in MYC mRNA levels upon PKA inhibition by qRT-PCR. The MYC mRNA level did not change upon H89 treatment, suggesting the effect of PKA inhibition on MYC to be post-transcriptional (Fig. 1E). Because MYC is degraded in cells via the 26S proteasome, we determined whether inhibition of proteasome activity rescues the effect of PKA inhibition on MYC accumulation. MG132 rescued the effect of PKA inhibition, thereby demonstrating that PKA protects MYC from 26S proteasome-mediated degradation (Fig. 1F).
TABLE 1.
PKACα substrates identified by mass spectrometry
PKA substrates identified by RIKA were excised from the gel, and protein identity was determined by mass spectrometry.
| Protein name | Accession number | Rank | Mascot score | Peptides matched | Sequence coverage | Predicted mass kDa/pI | Known PKA substrate? |
|---|---|---|---|---|---|---|---|
| % | |||||||
| ATP synthase subunit β (ATPB) | P06576 | 1 | 124 | 14 | 38 | 56.5/5.2 | No |
| Actin, cytoplasmic 1 (β-actin) | P60709 | 1 | 58 | 7 | 24 | 41.7/5.3 | Yes |
| Triose phosphate isomerase I (TPI I) | P60174 | 1 | 153 | 12 | 63 | 26.6/6.4 | No |
| Elongation factor 1δ | P29692 | 1 | 131 | 10 | 38 | 31.1/4.9 | No |
| Hetrogeneous nuclear ribonucleoprotein H (hnRNP H) | P31943 | 2 | 90 | 9 | 21 | 49.1/5.8 | No |
| 40S ribosomal protein SA (p40) | P08865 | 1 | 70 | 9 | 38 | 32.8/4.7 | No |
| Thyroid receptor-interacting protein 13 (TRIP-13) | Q15645 | 1 | 51 | 12 | 31 | 48.5/5.7 | No |
| Ubiquitin carboxyl-terminal hydrolase isozyme L5 (UCHL5) | Q9Y5K5 | 1 | 59 | 19 | 52 | 37.5/5.2 | No |
| Complement component 1Q subcomponent-binding protein | Q07021 | 1 | 65 | 6 | 38 | 31.3/4.7 | No |
| Ubiquitin carboxyl-terminal Hydrolase isozyme L3 (UCHL3) | P15374 | 1 | 125 | 9 | 51 | 26.1/4.8 | No |
| 78-kDa glucose-regulated protein precursor (GRP 78) | P11021 | 1 | 155 | 15 | 26 | 72.2/5.0 | No |
| T-complex protein 1 subunit θ | P50990 | 1 | 144 | 25 | 34 | 59.5/5.4 | No |
| α-Enolase (ENO1) | P06733 | 1 | 81 | 24 | 54 | 47.1/7.0 | No |
| Histone-binding protein (RBBP4) | Q09028 | 1 | 74 | 7 | 19 | 47.6/4.7 | No |
| Peptidyl-prolyl cis-trans isomerase B precursor | P23284 | 1 | 148 | 12 | 52 | 22.7/9.3 | No |
| Glucosidase 2 subunit β Precursor | P14314 | 1 | 72 | 10 | 21 | 59.3/4.3 | No |
| Methyl-CpG-binding domain protein 4 | Q95243 | 1 | 52 | 20 | 27 | 66/9.1 | No |
| Histone H2B | CAB02545 | 1 | NAa | 15 | NAa | 13.9/10.3 | Yes |
| Family with sequence similarity 82 | EAW91638 | 1 | 85 | 10 | 35 | 31/6.21 | No |
a NA, not applicable.
FIGURE 4.

PKA phosphorylation primes MYC for PLK1 phosphorylation. A, Ser-279 lies within a phospho-degron region important in the stability of MYC. The canonical degron sequence (D/ES*GXXS*) requires Ser-279 to be phosphorylated for efficient binding of the E3 ligase βtrcp (18). Phospho-acceptor sites within the phospho-degron are designated by asterisks. TAD, transcription activation domain DBD, DNA binding domain. B, inhibition of both PKA (20 μm H89, 3 h) and PLK1 (50 nm BI2536, 3 h) destabilized MYC. The effect of combined inhibition of PKA and PLK1 was more profound compared with inhibiting either kinase alone. Data from multiple experiments were quantified and are presented ± S.E. (error bars). DMSO, dimethyl sulfoxide. C, scheme illustrates the three possible mechanisms through which PKA and PLK1 could regulate MYC stability by phosphorylation within the phospho-degron. D, MYC phosphorylation by PKA primes phosphorylation by PLK1. Prephosphorylation of recombinant MYC by PKA (using nonradiolabeled ATP) prior to phosphorylation by PLK1 resulted in enhanced PLK1 phosphorylation (using [γ-32P]ATP) (top). Addition of H89 completely blocks subsequent phosphorylation of MYC by PKA. Coomassie staining shows protein loading (bottom). E, in vitro kinase assay shows enhanced phosphorylation of MYCS279D by PLK1 compared with MYCWT. F, on-membrane kinase assay shows enhanced phosphorylation of MYCS279D by PLK1 compared with MYCWT and other MYC mutants. G, PLK1 phosphorylates MYC at Ser-281. PLK1 phosphorylation of MYCS279D was diminished upon mutation of Ser-281 to alanine (MYCS279D/S281A).
FIGURE 1.

PKA stabilizes MYC in cells. A and B, PKA inhibition using H89 (10 mm, 1 h) decreased the MYC steady-state level in cells. C, PKA-specific peptide inhibitor PKI (20 mm, 1 h) destabilized MYC. D, PKA activation by forskolin (1 h) results in MYC accumulation. E, MYC mRNA level does not change upon H89 treatment (n = 3). F, PKA protects MYC from proteasome-mediated degradation. MG132 rescued the effect of PKA inhibition on MYC stability. DMSO, dimethyl sulfoxide.
MYC Phosphorylation at Ser-279 by PKA Stabilizes MYC
To determine whether PKA can phosphorylate MYC, we performed in vitro kinase reactions using recombinant PKACα and MYC in the presence of [γ-32P]ATP. PKA efficiently phosphorylated MYC, and the phosphorylation was inhibited by H89 (Fig. 2, A and C). LC-MS2 analysis identified Ser-279 as a PKA phospho-acceptor site on MYC (Fig. 2B and supplemental Tables S1 and S2). The ability of PKA to phosphorylate a MYCS279A mutant in vitro was nearly extinguished compared with WT-MYC (MYCWT), demonstrating Ser-279 as the major PKA phospho-acceptor site on MYC (Fig. 2C). Ser-279 is a predicted PKA phosphorylation site and is situated within an evolutionarily conserved region between the N-terminal transactivation and C-terminal DNA binding domains in MYC (Fig. 2, D and E). Furthermore, the region spanning Ser-279 is predicted to be solvent-exposed and intrinsically disordered (data not shown). Such intrinsically disordered domains have been implicated in the regulation of protein stability (26).
FIGURE 2.

PKA phosphorylates MYC at Ser-279. A, PKACα phosphorylates MYC in vitro. B, PKACα phosphorylates MYC at Ser-279. Top, LC-MS2 spectra of the peptide SESGSPSAGGHSKPPHSPLVLK from nonphosphorylated recombinant MYC are shown. A region spanning Ser-279 is shown to reveal the nonphosphorylated state at Ser-279. The b ions corresponding to Glu-278 and Ser-279 are labeled using red arrows. The corresponding amino acid in the peptide is also labeled in red. The mass difference between these ions is also indicated. A complete list of ions for the given peptide is listed in supplemental Table S1. Bottom panels, LC-MS2 spectra of the peptide RSESGSPSAGGHSKPPHSPLVLK from PKA-phosphorylated recombinant MYC is shown. A region spanning Ser-279 is shown to reveal the phosphorylated state at Ser-279.The b ions corresponding to Ser-277 and phosphorylated Ser-279 (also designated by an asterisk) are labeled using a red arrow. The corresponding amino acid in the peptide is also labeled in red. The positions of the hypothetical Glu-278 and nonphosphorylated Ser-279 ion are indicated by the black arrow. The mass difference between these ions is also indicated. A mass increase of 80 Da was observed in the phosphorylated peptide, demonstrating that Ser-279 is phosphorylated. A complete list of ions for the given peptide is listed in supplemental Table S2. C, mutating Ser-279 to alanine diminished MYC phosphorylation by PKA in vitro. Addition of H89 completely abolished phosphorylation of MYC by PKA. Arrowheads indicate phosphorylated MYC. D, PKA phospho-acceptor sites on MYC are predicted by in silico phosphorylation prediction tool GPS 2.0 (43). Ser-279 is a predicted phospho-acceptor site of PKA on MYC (highlighted in yellow). E, the region containing Ser-279 in MYC is evolutionarily conserved across species.
To determine whether the site phosphorylated by PKA on MYC is accessible and able to be phosphorylated in vivo, we transfected His6-3×HA-tagged MYC in PC3 cells. The cells were harvested 36 h post-transfection, lysed (using 150 mm NaCl, 50 mm Tris (pH 7.5), 0.5% Nonidet P-40 and Complete protease inhibitor), and MYC was purified using Ni-affinity chromatography. The purified MYC was divided in half. One half was completely dephosphorylated by hydrogen fluoride (HF) treatment as described (27). The remaining half was left untreated and contained the pool of MYC molecules that retained their in vivo phosphorylation status. We performed a RIKA with PKA in the gel on the HF-treated and untreated purified MYC samples. If MYC is phosphorylated in vivo at the site phosphorylated by PKA in vitro (in a RIKA), then we would expect an increase in phosphorylation signal in the HF-treated samples (because these sites would be dephosphorylated upon HF treatment and become available to be phosphorylated during the RIKA). Total levels of MYC loaded in these samples were quantified by anti-HA Western blotting, and the change in phosphorylation signal was calculated after normalization as described previously (27). This process is illustrated in Fig. 3A. We observed ∼50% increase in MYC phosphorylation upon HF treatment, suggesting that 50% of the in vivo MYC population in the cells was phosphorylated at the PKA site under the given culture conditions (Fig. 3B). Importantly, endogenous PKA and MYC co-localized to the nucleus (Fig. 3C).
FIGURE 3.

PKA phosphorylation at Ser-279 stabilizes MYC. A, scheme illustrates the RIKA-based approach used to determine whether the PKA phosphorylation site is phosphorylated in vivo. B, the PKA RIKA gel shows phosphorylation of untreated and HF-treated MYC purified from PC3 cells (top); Unphosphorylated MYC refers to the fraction of MYC not phosphorylated at a PKA site in vivo at the time of purification (bottom). A corresponding Western blot showing MYC levels (bottom) was used to normalize the phosphorylation signal on RIKA gel. C, immunofluorescence analysis co-localization of endogenous PKACα and MYC in PC3 cells is shown. D, ectopically expressed MYCWT is destabilized upon PKA inhibition whereas MYCS279A levels are not affected by H89. HA-tagged MYCWT and MYCS279A were transfected into PC3 cells. Anti-HA immunoblotting was performed to detect levels of transfected MYC following H89 treatment (20 μm, 1 h). Tubulin was used as loading control.
To ascertain whether MYC phosphorylation at Ser-279 by PKA affected the MYC steady-state level, HA-tagged MYCWT and MYCS279A were transfected into PC3 cells, and the effect of PKA inhibition on their accumulation was analyzed. Whereas MYCWT was destabilized upon H89 treatment, the steady-state level of MYCS279A was unresponsive to PKA inhibition (Fig. 3D). Further, the MYCS279A steady-state level was considerably lower compared with MYCWT (Fig. 3D). These data demonstrate that phosphorylation at Ser-279 by PKA plays a role in stabilizing MYC in cells. No difference in subcellular localization between the endogenous MYC, HA-tagged MYCWT and HA-tagged MYCS279A mutant was observed (data not shown).
PKA Phosphorylation at Ser-279 Primes MYC for PLK1 Phosphorylation
A recent report showed the region spanning Ser-279 to constitute a phospho-degron (Fig. 4A). PLK1-mediated βtrcp binding within this region was shown to stabilize MYC (18). However, neither the events preceding PLK1 phosphorylation nor the PLK1 phospho-acceptor site(s) on MYC is known. Because both PLK1 and PKA phosphorylate residues within this phospho-degron, we sought to clarify their roles. We found that the effect of combined inhibition of PKA and PLK1 on MYC stability is more profound compared with inhibiting either kinase alone (Fig. 4B). We hypothesized three possible scenarios: (i) PKA and PLK1 phosphorylate the same residue on MYC (i.e. Ser-279) and hence elicit a similar response; (ii) PKA and PLK1 phosphorylate mutually independent residues on MYC within the phospho-degron; and (iii) PKA phosphorylates MYC at Ser-279, which primes PLK1 to phosphorylate an adjacent residue within the phospho-degron by relieving the inhibitory effect of the polo-box domain or by making an adjacent site a more favorable PLK1 phospho-acceptor (Fig. 4C). To distinguish among these models, we performed an in vitro kinase reaction using recombinant PLK1T210D and recombinant MYC, which was either prephosphorylated by PKA in vitro using nonradiolabeled ATP (primed) or not prephosphorylated by PKA (unprimed). His6-tagged MYC purified from E. coli was prephosphorylated by in vitro kinase assay using recombinant PKACα and nonradiolabeled ATP. Nonphosphorylated MYC served as the unprimed control. Subsequently, H89 (30 μm), was added to all reactions to inhibit PKA activity. The absence of radiolabeling upon further incubation (1 h at room temperature) of the reactions containing MYC, PKA and H89 with [γ-32P]ATP confirmed complete inactivation of PKA by H89. Recombinant PLK1T210D and [γ-32P]ATP was then added to these tubes containing either primed or unprimed MYC. The in vitro kinase reaction was allowed to proceed for 1 h at room temperature. Radioactive labeling of MYC by PLK1T210D was assessed by running the reaction on a SDS-polyacrylamide gel, transferring to a PVDF membrane followed by autoradiography. We used the PLK1T210D mutant version of PLK1 because it was previously shown to be constitutively active. The mutation at Thr-210 mimics the phosphorylation of PLK1 at this site within the activation loop, and phosphorylation at Thr-210 is necessary for PLK1 activation (28). We observed that the phosphorylation signal of MYC by PLK1T210D (using [γ-32P]ATP) was higher when MYC was primed by PKA (using nonradiolabeled ATP) compared with unprimed MYC (Fig. 4D). This result suggested that PKA phosphorylation at Ser-279 on MYC primes subsequent MYC phosphorylation by PLK1. To confirm this, we generated the phospho-mimetic mutant at Ser-279 (MYCS279D) and performed an in vitro kinase assay using PLK1T210D. We observed that PLK1 phosphorylation of the MYCS279D mutant was dramatically higher compared with MYCWT and other MYC mutants (Fig. 4E). We also compared the ability of PLK1T210D to phosphorylate other MYC substitution mutants in the region and found MYCS279D to be a more efficient PLK1 substrate compared with MYCWT and other mutants (Fig. 4F). These results showed that MYC phosphorylation at Ser-279 increases the efficiency of subsequent PLK1T210D phosphorylation of MYC, possibly at an adjacent site. LC-MS3 analysis of MYCS279D phosphorylated in vitro using PLK1T210D revealed Ser-281 as the PLK1 phospho-acceptor site on MYC (Fig. 5). MYCS279D phosphorylation by PLK1T210D was significantly compromised in the MYCS279D:S281A double mutant (Fig. 4G), thus confirming Ser-281 as the major PLK1 phospho-acceptor site on MYC that is primed by Ser-279 phosphorylation.
FIGURE 5.
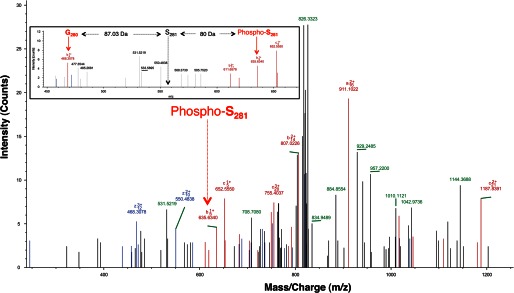
PLK1 phosphorylates MYC at S281. LC-MS3 spectra of PLK1 phosphorylated MYCS279D. The peak indicating phosphorylation at Ser-281 is indicated using a red arrow. Inset, LC-MS3 spectra of the peptide RSEDGSPSAGGHSKPPHSPLVLK from PLK1 phosphorylated recombinant MYC. Region spanning Ser-281 is shown to reveal the phosphorylated state at Ser-281.The b ions corresponding to phosphorylated Ser-281 and Gly-280 are labeled using red arrows. The position of the hypothetical nonphosphorylated Ser-281 ion is indicated using a black arrow. The mass difference between these ions is also indicated. A mass increase of 80 Da was observed in the phosphorylated peptide, demonstrating that Ser-281 is phosphorylated.
PKA Isoform-specific Transcriptional Repression of MYC
Three different genes encode PKA catalytic subunits in humans: Cα, Cβ, and Cγ. The Cα and Cβ isoforms have multiple splice variants. To validate the effect of PKA inhibition on MYC and to dissect the role of individual PKA catalytic subunit isoforms in stabilizing MYC, we knocked down either PKACα or PKACβ using siRNA in PC3 cells. Analysis of siRNA-treated samples yielded an unexpected but intriguing result. Contrary to our expectation based on chemical PKA inhibition, knockdown of PKACα increased MYC steady-state level in PC3 cells (Fig. 6A). In contrast, PKACβ knockdown caused a slight decrease in MYC levels. Because siRNA knockdown occurs over an extended time period, we hypothesized that the result of PKACα knockdown on MYC could be through an indirect mechanism. Comparison of MYC mRNA after siRNA-mediated knockdown of PKA catalytic subunits revealed that prolonged knockdown of PKACα, but not PKACβ, increased the steady-state MYC mRNA level (Fig. 6B). These data reveal a link between PKACα activity and MYC transcription and, for the first time, show that the different PKA catalytic subunits perform distinct biological functions.
FIGURE 6.

PKA catalytic subunits regulate MYC differentially. A, long term knockdown of PKACα by siRNA (48–72 h) resulted in increased MYC protein level whereas knockdown of PKACβ subunit caused a slight decrease in MYC protein level. Western blotting using pan-PKAC antibody confirmed knockdown of PKACα. PKACβ knockdown was confirmed by semiquantitative PCR (Fig. 6B). The change in MYC level was quantified using ImageJ software and normalized to the corresponding β-actin level. B, the effect of PKACα knockdown on MYC is transcriptional. Semiquantitative PCR and Northern blot analysis reveal an increase in MYC mRNA upon siRNA knockdown of PKACα but not PKACβ.
PKA Catalytic Subunit Isoforms Have Both Overlapping and Distinct Substrates
The ability of PKA catalytic subunits to have a distinct effect on the transcription of MYC suggested exclusive, nonredundant roles for PKA isoforms. We hypothesized that the different PKA catalytic subunit isoforms have distinct substrate profiles and hence elicit different responses upon activation. To test this hypothesis, we profiled and compared the substrates of PKA catalytic subunit isoforms by RIKA (23). To identify PKACα substrates, the PKACα RIKA was first standardized (Fig. 7A). LNCaP cell lysate was fractionated by anion-exchange chromatography, and a one-dimensional PKACα RIKA on the enriched fractions was performed to identify fractions containing PKA substrates (data not shown). Fractions containing PKACα substrates were used for two-dimensional PKACα RIKAs. The signal on RIKA gels was aligned with parallel silver-stained gels (Fig. 7B). PKA substrates were excised from the silver-stained gels and identified by mass spectrometry (Table 1). We validated a subset of the identified Cα substrates by in vitro kinase reaction using purified recombinant proteins (Fig. 7C).
FIGURE 7.

PKA catalytic subunits have distinct substrate selectivity. A, two-dimensional PKACα RIKA using an LNCaP whole-cell protein extract. Numbers on the top left and right corners indicate pH of the IPG strip. The autoradiograms from gel containing catalytically active PKACα (top) and the kinase-dead PKACα control (bottom) are shown. PKA substrates appear as signals on the autoradiograms. The signal on the control gel is likely due to autophosphorylation of endogenous kinases or phosphorylation of the co-polymerized kinase-dead PKACα or co-migrating proteins by endogenous kinases. B, two-dimensional RIKA on anion exchange fraction. A parallel gel from the same fraction (containing no kinase) was silver-stained to enable substrate identification (bottom). Arrows and closed circles represent several substrates used for alignment. Red-dashed circle represents an abundant non-PKACα substrate protein, which was used as an internal marker to align the gel. C, in vitro kinase assay using recombinant substrates and PKACα. Substrates identified by RIKA are phosphorylated by PKACα in vitro. Arrowheads indicate phosphorylated substrates. D, PKACβ1 has a substrate profile similar to that of PKACα. In vitro kinase assay was performed using recombinant PKACβ1 and PKACα substrates. PKACβ1 phosphorylated all tested PKACα substrates in vitro. Arrowheads indicate phosphorylated substrates. E, PKACβ2 has a distinct substrate profile compared with PKACα and PKACβ1. F, H89 inhibits phosphorylation of the telomerase-binding protein (TEBP) by PKACβ2. G, amino acid sequence alignment of N-terminal region PKACα and PKACβ2 shows the presence of an additional N-terminal arm (62 amino acids) in PKACβ2 isoform. The Yaspin secondary structure prediction program was used to predict the α-helical region in the N-terminal 62 amino acids in PKACβ2, and the helical wheel program available from the University of Virginia was used to reveal the amphipathic nature of the predicted helix region (indicated by purple helix over the corresponding sequence).
To address whether substrate diversity contributes to the observed functional nonredundancy among PKA catalytic subunits, we tested the ability of PKACβ1 and PKACβ2 isoforms to phosphorylate these identified PKACα substrates in vitro. Catalytically active PKACβ2 was expressed and purified from mammalian cells (COS cells or PC3 cells) because PKACβ2 purified from E. coli was catalytically inactive (data not shown). We observed that whereas PKAβ1 phosphorylated all of the tested PKACα substrates (Fig. 7D), PKACβ2 phosphorylated only the telomerase-binding protein among the PKACα substrates tested (Fig. 7E). Addition of the pan-PKA inhibitor H89 diminished the phosphorylation of the telomerase-binding protein by PKACβ2 (Fig. 7F). These differences in substrate selectivity were not totally unexpected because PKACα and PKACβ1 share a very high sequence similarity (92%), whereas PKACβ2 has an additional 62 amino acids at its N terminus not present in PKACβ1 (Fig. 7G). In silico secondary structure prediction of PKACβ2 N terminus revealed an amphipathic helix region within this unique N terminus (Fig. 7G). We believe that this region might play a role in the differential substrate selectivity of PKACβ2.
Increased PKACβ Expression in MYC-overexpressing Prostate Epithelial Cells
PKACβ is known to be a direct transcriptional target of MYC in rat fibroblasts and lymphocytes (29). In these cells, it was further suggested that PKACβ plays an important role in MYC-mediated transformation (29). MYC overexpression is a consistent and key early event in prostate cancer, where it drives proliferation (30). Rapidly proliferating prostate epithelial cells have also been reported to have higher levels of the PKACβ2 isoform (31). These results prompted us to determine whether MYC overexpression in prostate cancer cells influences the PKACβ level (and the PKACβ2 splice variant), thereby forming a proximate positive feedback loop. Ectopic expression of MYC in PC3 cells resulted in increased PKACβ and PKACβ2 expression (Fig. 8A). We then compared the levels of PKACβ2 protein in the prostates of mice overexpressing MYC specifically in the prostate to their age-matched controls. We observed a positive correlation between MYC expression and PKCβ2 levels in the prostates of multiple mouse models in which MYC expression was controlled by distinct prostate-specific promoters (Fig. 8B). We could not compare levels of PKACβ1 protein in these experiments due to the unavailability of an antibody that would allow us to differentiate between the PKACβ1 and PKACα subunits, which have the same apparent molecular mass (∼42 kDa). Oncomine analyses of previously published human prostate cancer microarray datasets revealed that both MYC and PKACβ are overexpressed in human prostate cancer cases (Fig. 8C) (32, 33). Oncomine analyses of multicancer datasets also revealed elevated PKACβ in prostate cancer (data not shown) (34, 35).
FIGURE 8.

MYC up-regulates PKACβ in prostate epithelial cells. A, ectopic MYC expression in LNCaP cells (left), increasing Cβ and Cβ2 mRNA (right). Values are normalized to TBP mRNA level. B, comparison of Cβ2 protein level in the prostate of FVB and transgenic mice overexpressing MYC. Western blot analysis (top) of lysate from the prostate of age-matched FVB, Hi-MYC (MYC-driven using the Probasin promoter), and NK70-MYC (MYC driven by a 70-kb region of the NKX3.1 promoter) reveal higher Cβ2 levels (47 kDa) in the prostate of MYC-overexpressing mice (bottom). C, Oncomine box plots comparing MYC, and PKACβ expression levels between normal and prostate cancer samples in multiple datasets (32, 33) show a positive correlation between levels of MYC and PKACβ in prostate cancer. D, MYC-PKA-PLK1 signaling loop. MYC positively regulates the level of the PKACβ isoform. Phosphorylation of MYC by PKA primes MYC for PLK1 phosphorylation. Together these phospho-transfer reactions stabilizes MYC. Prolonged knockdown of PKACα (but not PKACβ) transcriptionally up-regulates MYC. The different PKA catalytic subunits have distinct substrate specificity, which enables them to perform nonredundant functions.
DISCUSSION
Diverse factors and signaling pathways influence MYC accumulation. In this study, we identified a role for PKA in MYC regulation and demonstrated the existence of a MYC-PKA-PLK1 signaling loop in cells. Our data show that MYC transcriptionally up-regulates PKACβ, and PKA in turn protects MYC from proteasome-mediated degradation through phosphorylation at Ser-279, establishing a proximate positive feedback interaction. Ser-279 lies within a phospho-degron recently shown to be important in MYC stability (18). These studies demonstrated this region to mediate binding of the E3 ligase βtrcp, which competes with Fbw7 (recruited through Thr-58 phosphorylation) for ubiquitination at the MYC N terminus (18). Phosphorylation within the degron was shown to be important for βtrcp recruitment, and PLK1 was suggested to phosphorylate MYC within this region (18). Our data demonstrate that MYC phosphorylation at Ser-279 by PKA primes subsequent PLK1 phosphorylation at Ser-281. Thus, through sequential phosphorylation, PKA and PLK1 cooperatively stabilize MYC.
Interestingly, whereas brief pharmacologic pan-PKA inhibition diminished MYC level, prolonged PKACα knockdown resulted in transcriptional up-regulation of MYC. These results provide an example of contrasting functional responses between short term and long term kinase inhibition. Furthermore, unlike in the case of PKACα knockdown, we observed no change in MYC mRNA level upon PKACβ knockdown, demonstrating functionally distinct roles for PKA catalytic isoforms in the transcriptional regulation of MYC. We hypothesized this transcriptional response to be mediated through a PKACα-specific substrate, possibly a transcription factor that regulates MYC transcription. Although the PKACα target(s) critical for this response are unknown, CBP/p300 is a plausible candidate. Recruitment of CBP/p300 at the MYC promoter is known to down-regulate MYC transcription (36–38). CBP/p300 is a known PKACα substrate, but the effect of PKA phosphorylation on the DNA binding ability of CBP or recruitment to the MYC promoter region has not been investigated (39, 40). In this regard, it will also be interesting to investigate the role of MBP-1 phosphorylation by PKA. During the course of this study we had identified MBP-1 as a PKACα (but not PKACβ2) substrate.
Comparison of substrate selectivity of the different PKA isoforms revealed PKACβ2 to have distinct phosphorylation selectivity compared with PKACα and PKACβ1. We attribute this difference to a predicted amphipathic helix in the N terminus of PKACβ2, which could mediate substrate recognition and binding. Except for this unique N terminus region, PKACβ2 is identical to PKACβ1, and PKACβ1 was shown to phosphorylate all PKACα substrates tested. Substrate docking domains distant from catalytic sites have been reported for multiple protein kinases (41). In addition, amphipathic helices have been reported to mediate several kinase-substrate interactions (42). We suggest that the solvent-exposed N-terminal amphipathic helix of PKACβ2 acts as a substrate-docking site and hence accounts for the differences in its substrate selectivity. These studies reveal the possibility of antithetical roles for closely related protein kinase isoforms and highlight the risk of therapeutic strategies aimed at global kinase inhibition. PKACα overexpression in LNCaP cells was reported to induce trans-differentiation of these cells into neuroendocrine-like cells, which divide slowly. It has also been reported that PKACβ2 is overexpressed in rapidly proliferating prostate cancer cells. Such discordance in the phenotype of cells overexpressing the different PKA isoforms argued for a significant functional difference between these proteins. The data reported here provide a mechanistic premise for the distinct functional roles of PKA catalytic subunits in prostate cancer. Although PKACα has been extensively studied and many of its substrates are known, the role of other PKA catalytic isoforms has received less attention. This report is the first detailed analysis of differences in substrate selectivity among PKA catalytic isoforms and the functional implications of these differences. Collectively, our data demonstrate that differential substrate selectivity and functional diversity among protein kinase isoforms are critical for modulation and precise signaling regulation in response to stimuli of varying intensity and perdurance.
Our model suggests that short PKA activity bursts in the cell stabilize MYC through a post-translational mechanism, whereas prolonged activation of PKACα transcriptionally represses MYC (Fig. 8D). This establishes a regulatory loop and protects the cells from downstream effects of post-translational MYC stabilization as a result of prolonged elevated PKACα activity. This mechanism could contribute to the slow proliferative rate of LNCaP cells trans-differentiated to a neuroendocrine-like phenotype by PKACα overexpression. MYC accumulation in turn activates a positive feedback loop that transcriptionally activates PKACβ. PKACβ protects MYC from proteasomal degradation, but does not repress MYC transcriptionally. This is consistent with the higher expression of PKACβ subunits in rapidly proliferating, MYC-overexpressing prostate cancer cells (31–33). Thus, through differential substrate selectivity, PKA catalytic isoforms regulate MYC differentially.
It is clear from the results presented here that the short and long term effects of PKA inhibition differ substantially. These observations underscore the need to establish kinase inhibition strategies that interfere with the ability of the kinase to interact with specific target substrates without impairing the function of the kinase in general. It may be possible to achieve this pharmacologically using peptides or peptidomimetics that interfere with interaction of protein kinases and specific substrates. Clearly, this strategy, which must be predicated upon an in-depth understanding of the structural aspects of kinase-substrate interactions, could enable subtle, yet functionally significant pathway disruption with fewer side effects and reduced toxicity compared with global inhibition strategies.
Acknowledgments
We thank Anu Divakaruni and Brandon McClary for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant RCA155568A (to C. J. B.).

This article contains supplemental Tables S1 and S2.
- PLK1
- polo-like kinase 1
- MBP-1
- MYC promoter-binding protein 1
- qRT
- quantitative RT
- RIKA
- reverse in-gel kinase assay
- TBP
- TATA-binding protein.
REFERENCES
- 1. Bernard S., Eilers M. (2006) Control of cell proliferation and growth by Myc proteins. Results Probl. Cell Differ. 42, 329–342 [DOI] [PubMed] [Google Scholar]
- 2. Dang C. V. (1999) c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 19, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoffman B., Liebermann D. A. (2008) Apoptotic signaling by c-MYC. Oncogene 27, 6462–6472 [DOI] [PubMed] [Google Scholar]
- 4. Jiang G., Albihn A., Tang T., Tian Z., Henriksson M. (2008) Role of Myc in differentiation and apoptosis in HL60 cells after exposure to arsenic trioxide or all-trans-retinoic acid. Leuk. Res. 32, 297–307 [DOI] [PubMed] [Google Scholar]
- 5. Gao P., Tchernyshyov I., Chang T. C., Lee Y. S., Kita K., Ochi T., Zeller K. I., De Marzo A. M., Van Eyk J. E., Mendell J. T., Dang C. V. (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schmidt E. V. (1999) The role of c-myc in cellular growth control. Oncogene 18, 2988–2996 [DOI] [PubMed] [Google Scholar]
- 7. Davis A. C., Wims M., Spotts G. D., Hann S. R., Bradley A. (1993) A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 7, 671–682 [DOI] [PubMed] [Google Scholar]
- 8. Lee C. M., Reddy E. P. (1999) The v-myc oncogene. Oncogene 18, 2997–3003 [DOI] [PubMed] [Google Scholar]
- 9. Ramsay G. M., Moscovici G., Moscovici C., Bishop J. M. (1990) Neoplastic transformation and tumorigenesis by the human protooncogene MYC. Proc. Natl. Acad. Sci. U.S.A. 87, 2102–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dani C., Blanchard J. M., Piechaczyk M., El Sabouty S., Marty L., Jeanteur P. (1984) Extreme instability of myc mRNA in normal and transformed human cells. Proc. Natl. Acad. Sci. U.S.A. 81, 7046–7050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sears R. C. (2004) The life cycle of c-Myc: from synthesis to degradation. Cell Cycle 3, 1133–1137 [PubMed] [Google Scholar]
- 12. Lemm I., Ross J. (2002) Regulation of c-myc mRNA decay by translational pausing in a coding region instability determinant. Mol. Cell. Biol. 22, 3959–3969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wall M., Poortinga G., Hannan K. M., Pearson R. B., Hannan R. D., McArthur G. A. (2008) Translational control of c-MYC by rapamycin promotes terminal myeloid differentiation. Blood 112, 2305–2317 [DOI] [PubMed] [Google Scholar]
- 14. Malempati S., Tibbitts D., Cunningham M., Akkari Y., Olson S., Fan G., Sears R. C. (2006) Aberrant stabilization of c-Myc protein in some lymphoblastic leukemias. Leukemia 20, 1572–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yeh E., Cunningham M., Arnold H., Chasse D., Monteith T., Ivaldi G., Hahn W. C., Stukenberg P. T., Shenolikar S., Uchida T., Counter C. M., Nevins J. R., Means A. R., Sears R. (2004) A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 6, 308–318 [DOI] [PubMed] [Google Scholar]
- 16. Arnold H. K., Sears R. C. (2006) Protein phosphatase 2A regulatory subunit B56α associates with c-myc and negatively regulates c-myc accumulation. Mol. Cell. Biol. 26, 2832–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Welcker M., Orian A., Jin J., Grim J. E., Grim J. A., Harper J. W., Eisenman R. N., Clurman B. E. (2004) The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. U.S.A. 101, 9085–9090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Popov N., Schülein C., Jaenicke L. A., Eilers M. (2010) Ubiquitylation of the amino terminus of Myc by SCF(β-TrCP) antagonizes SCF(Fbw7)-mediated turnover. Nat. Cell Biol. 12, 973–981 [DOI] [PubMed] [Google Scholar]
- 19. Liu J., Schuff-Werner P., Steiner M. (2004) Double transfection improves small interfering RNA-induced thrombin receptor (PAR-1) gene silencing in DU 145 prostate cancer cells. FEBS Lett. 577, 175–180 [DOI] [PubMed] [Google Scholar]
- 20. Amarzguioui M. (2004) Improved siRNA-mediated silencing in refractory adherent cell lines by detachment and transfection in suspension. BioTechniques 36, 766–768, 770 [DOI] [PubMed] [Google Scholar]
- 21. Padmanabhan A., Gosc E. B., Bieberich C. J. (2013) Stabilization of the prostate-specific tumor suppressor NKX3.1 by the oncogenic protein kinase Pim-1 in prostate cancer cells. J. Cell. Biochem. 114, 1050–1057 [DOI] [PubMed] [Google Scholar]
- 22. Guan B., Pungaliya P., Li X., Uquillas C., Mutton L. N., Rubin E. H., Bieberich C. J. (2008) Ubiquitination by TOPORS regulates the prostate tumor suppressor NKX3.1. J. Biol. Chem. 283, 4834–4840 [DOI] [PubMed] [Google Scholar]
- 23. Li X., Guan B., Srivastava M. K., Padmanabhan A., Hampton B. S., Bieberich C. J. (2007) The reverse in-gel kinase assay to profile physiological kinase substrates. Nat. Methods 4, 957–962 [DOI] [PubMed] [Google Scholar]
- 24. Feo S., Arcuri D., Piddini E., Passantino R., Giallongo A. (2000) ENO1 gene product binds to the c-myc promoter and acts as a transcriptional repressor: relationship with Myc promoter-binding protein 1 (MBP-1). FEBS Lett. 473, 47–52 [DOI] [PubMed] [Google Scholar]
- 25. Chaudhary D., Miller D. M. (1995) The c-myc promoter-binding protein (MBP-1) and TBP bind simultaneously in the minor groove of the c-myc P2 promoter. Biochemistry 34, 3438–3445 [DOI] [PubMed] [Google Scholar]
- 26. Dyson H. J., Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208 [DOI] [PubMed] [Google Scholar]
- 27. Li X., Rao V., Jin J., Guan B., Anderes K. L., Bieberich C. J. (2012) Identification and validation of inhibitor-responsive kinase substrates using a new paradigm to measure kinase-specific protein phosphorylation index. J. Proteome Res. 11, 3637–3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Smits V. A., Klompmaker R., Arnaud L., Rijksen G., Nigg E. A., Medema R. H. (2000) Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat. Cell Biol. 2, 672–676 [DOI] [PubMed] [Google Scholar]
- 29. Wu K. J., Mattioli M., Morse H. C., 3rd, Dalla-Favera R. (2002) c-MYC activates protein kinase A (PKA) by direct transcriptional activation of the PKA catalytic subunit β (PKA-Cβ) gene. Oncogene 21, 7872–7882 [DOI] [PubMed] [Google Scholar]
- 30. Gurel B., Iwata T., Koh C. M., Jenkins R. B., Lan F., Van Dang C., Hicks J. L., Morgan J., Cornish T. C., Sutcliffe S., Isaacs W. B., Luo J., De Marzo A. M. (2008) Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod. Pathol. 21, 1156–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kvissel A. K., Ramberg H., Eide T., Svindland A., Skålhegg B. S., Taskén K. A. (2007) Androgen dependent regulation of protein kinase A subunits in prostate cancer cells. Cell. Signal. 19, 401–409 [DOI] [PubMed] [Google Scholar]
- 32. Tomlins S. A., Mehra R., Rhodes D. R., Cao X., Wang L., Dhanasekaran S. M., Kalyana-Sundaram S., Wei J. T., Rubin M. A., Pienta K. J., Shah R. B., Chinnaiyan A. M. (2007) Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 39, 41–51 [DOI] [PubMed] [Google Scholar]
- 33. Varambally S., Yu J., Laxman B., Rhodes D. R., Mehra R., Tomlins S. A., Shah R. B., Chandran U., Monzon F. A., Becich M. J., Wei J. T., Pienta K. J., Ghosh D., Rubin M. A., Chinnaiyan A. M. (2005) Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 8, 393–406 [DOI] [PubMed] [Google Scholar]
- 34. Su A. I., Welsh J. B., Sapinoso L. M., Kern S. G., Dimitrov P., Lapp H., Schultz P. G., Powell S. M., Moskaluk C. A., Frierson H. F., Jr., Hampton G. M. (2001) Molecular classification of human carcinomas by use of gene expression signatures. Cancer Res. 61, 7388–7393 [PubMed] [Google Scholar]
- 35. Ramaswamy S., Ross K. N., Lander E. S., Golub T. R. (2003) A molecular signature of metastasis in primary solid tumors. Nat. Genet. 33, 49–54 [DOI] [PubMed] [Google Scholar]
- 36. Kolli S., Buchmann A. M., Williams J., Weitzman S., Thimmapaya B. (2001) Antisense-mediated depletion of p300 in human cells leads to premature G1 exit and up-regulation of c-MYC. Proc. Natl. Acad. Sci. U.S.A. 98, 4646–4651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rajabi H. N., Baluchamy S., Kolli S., Nag A., Srinivas R., Raychaudhuri P., Thimmapaya B. (2005) Effects of depletion of CREB-binding protein on c-Myc regulation and cell cycle G1-S transition. J. Biol. Chem. 280, 361–374 [DOI] [PubMed] [Google Scholar]
- 38. Baluchamy S., Sankar N., Navaraj A., Moran E., Thimmapaya B. (2007) Relationship between E1A binding to cellular proteins, c-myc activation, and S-phase induction. Oncogene 26, 781–787 [DOI] [PubMed] [Google Scholar]
- 39. Xu L., Lavinsky R. M., Dasen J. S., Flynn S. E., McInerney E. M., Mullen T. M., Heinzel T., Szeto D., Korzus E., Kurokawa R., Aggarwal A. K., Rose D. W., Glass C. K., Rosenfeld M. G. (1998) Signal-specific co-activator domain requirements for Pit-1 activation. Nature 395, 301–306 [DOI] [PubMed] [Google Scholar]
- 40. Swope D. L., Mueller C. L., Chrivia J. C. (1996) CREB-binding protein activates transcription through multiple domains. J. Biol. Chem. 271, 28138–28145 [DOI] [PubMed] [Google Scholar]
- 41. Dimitri C. A., Dowdle W., MacKeigan J. P., Blenis J., Murphy L. O. (2005) Spatially separate docking sites on ERK2 regulate distinct signaling events in vivo. Curr. Biol. 15, 1319–1324 [DOI] [PubMed] [Google Scholar]
- 42. Scott J. W., Norman D. G., Hawley S. A., Kontogiannis L., Hardie D. G. (2002) Protein kinase substrate recognition studied using the recombinant catalytic domain of AMP-activated protein kinase and a model substrate. J. Mol. Biol. 317, 309–323 [DOI] [PubMed] [Google Scholar]
- 43. Xue Y., Ren J., Gao X., Jin C., Wen L., Yao X. (2008) GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics 7, 1598–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]