

Background: Nur77 expression is regulated by MEF2D, which is found to be associated with the calpain-CDK5-MEF2D neuronal death pathway.
Results: Nur77 deficiency results in hypersensitivity to neuronal toxicity with Nur77 expression rescuing this loss.
Conclusion: Nur77 reduces toxic neuronal insult regulated by the calpain-CDK5-MEF2 pathway.
Significance: Previously reported calpain-CDK5-MEF2 signaling is now further elucidated with regulation of Nur77 in dopaminergic neuronal loss.
Keywords: Cell Death, Dopamine, Neurodegeneration, Neurotoxin, Parkinson Disease, Cdk5, MEF2, MPTP, Nur77
Abstract
We have earlier reported the critical nature of calpain-CDK5-MEF2 signaling in governing dopaminergic neuronal loss in vivo. CDK5 mediates phosphorylation of the neuronal survival factor myocyte enhancer factor 2 (MEF2) leading to its inactivation and loss. However, the downstream factors that mediate MEF2-regulated survival are unknown. Presently, we define Nur77 as one such critical downstream survival effector. Following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) treatment in vivo, Nur77 expression in the nigrostriatal region is dramatically reduced. This loss is attenuated by expression of MEF2. Importantly, MEF2 constitutively binds to the Nur77 promoter in neurons under basal conditions. This binding is lost following 1-methyl-4-phenylpyridinium treatment. Nur77 deficiency results in significant sensitization to dopaminergic loss following 1-methyl-4-phenylpyridinium/MPTP treatment, in vitro and in vivo. Furthermore, Nur77-deficient MPTP-treated mice displayed significantly reduced levels of dopamine and 3,4-Dihydroxyphenylacetic acid in the striatum as well as elevated post synaptic FosB activity, indicative of increased nigrostriatal damage when compared with WT MPTP-treated controls. Importantly, this sensitization in Nur77-deficient mice was rescued with ectopic Nur77 expression in the nigrostriatal system. These results indicate that the inactivation of Nur77, induced by loss of MEF2 activity, plays a critical role in nigrostriatal degeneration in vivo.
Introduction
Parkinson disease is characterized by progressive clinical hallmarks, including tremor, bradykinesia, rigidity, and postural instability (1) and loss of dopamine (DA)3 neurons in the substantia nigra pars compacta (SNc). Current therapies are mainly directed toward replacing dopamine levels in the brain and, as such, provide only symptomatic relief (2). Attenuation of the underlying degeneration in Parkinson disease is a potentially more effective therapeutic strategy, and the mechanism(s) controlling this loss is not clear.
CDK5, a member of the cyclin-dependent kinase family (CDKs) (3) is primarily involved in regulation of neuronal function (4, 5). CDK5 is involved in regulation of normal physiological function with evidence, suggesting that this protein also mediates death signaling (6–9). Importantly, we have previously provided evidence for the importance of CDK5 in MPTP-induced dopaminergic death (6). For example, CDK inhibitors, dominant negative CDK5 expression, and the deficiency of the CDK5 regulatory activating partner p35 (10) block dopaminergic loss in vivo. MPTP induces calpain-dependent cleavage of p35 to form p25, resulting in increased CDK5 activity in the SNc (7).
We identified a number of potential CDK5 substrates that mediate dopaminergic cell loss. The first substrate identified was myocyte enhancer factor 2 (MEF2), a transcription factor with a critical role in muscle development (11), neuronal differentiation (12), and neuronal survival (13–15). In this regard, we observed calpain-mediated cleavage of p35 and consequent activation of CDK5 increased phosphorylation and inactivation of MEF2D in an MPTP model of dopamine degeneration in vivo (7). In addition, expression of constitutively active MEF2D protects dopamine neurons from MPTP-induced insult. However, the critical question of how MEF2 regulates survival is not completely clear.
Recent evidence suggests Nur77 as a potential MEF2 transcriptional target gene (16–18). The Nur77 promoter contains putative binding sites for MEF2 (19). Nur77 was originally identified as an inducer of apoptosis in thymocyte selection (16, 20, 21). However, it is now known to show remarkable functional versatility. Nur77 has also been found to act as a survival factor in several apoptotic paradigms outside the CNS (22, 23). Interestingly, evidence suggests Nur77 expression regulates dopaminergic cell biochemistry and dopamine metabolism (24).
A potential link between MEF2D and Nur77 in dopaminergic cell loss and the critical need to understand how MEF2 may regulate dopaminergic survival, and we examined whether Nur77 might be the mechanistic link driving a calpain-CDK5-MEF2D-mediated pathway of death. We provide evidence that Nur77 is both regulated by CDK5-MEF2D and plays a critical role in the survival response to dopaminergic cell death.
EXPERIMENTAL PROCEDURES
Animals
Male Nur77 KO mice (generously provided by Dr. Jeff Milbrandt, University of Washington, St. Louis, MO) (21, 25) on a C57BK/6J background were further backcrossed for six generations with C57BK/6J strain mice (The Jackson Laboratory). Animals were maintained on a 12-h light/dark cycle with lights on between 06:00 and 18:00 and were permitted an ad libitum diet of Ralston Purina mouse chow. The room temperature was kept at 21 °C. All experimental procedures meet the guidelines set out by the Canadian Council on animal care and were approved by the University of Ottawa Committee for Animal Care.
MPTP Administration
Intraperitoneal injections of MPTP (Sigma; 25 mg/kg, measured free base; MPTP-HCl) were administered to mice (8–10 weeks old) once a day for five consecutive days (26). Control animals received an equivalent volume of saline (0.9%). Mice were sacrificed 14 days following initial MPTP/saline treatment or at indicated time points.
Immunohistochemistry
Following overnight post-fixation in 4% paraformaldehyde, brains were cryoprotected in 10% sucrose with free-floating sections obtained as described previously (26, 27). Immunolabeling was performed as described previously using anti-tyrosine hydroxylase (TH) (ImmunoStar, 1:10,000), rat anti-DA transporter (DAT) (Millipore; 1:2000), or rabbit anti-ΔfosB (Santa Cruz; 1:1000). Primary antibodies were visualized using diaminobenzidine.
Assessment of Neuronal Survival
Dopaminergic neurons in the SNc of treated mice were assessed for survival using the dopaminergic cell marker TH. A total of six to eight animals per group were analyzed. Estimates of total TH+ stained neurons in the SNc were analyzed by unbiased stereological estimates, applying optical fractionation (28) using Stereo Investigator (version 6; MicroBrightField, Williston, VT), as described previously (26, 29). In brief, 40-nm brain sections were examined within the rostral and caudal limits of the SNc (bregma, −2.54 to −3.88 mm) (65). For each brain, six coronal sections were examined. Following immunohistochemistry, mounting, defatting, and coverslipping, the mean section thickness, as measured with a z axis microdissector, was 18 μm. Sections were analyzed using a 100× lens.
Quantification of Striatal Immunohistochemistry
Striatal dopaminergic (TH and DAT) fiber density and FosB-positive nuclei were quantified using NIH ImageJ densitometry analysis (26). Each tissue quantified was initialed to its own non-stained background.
Adenovirus Delivery
Adenoviral constructs expressing MEF2D-S444A (7, 30) and Nur77 (pcDNA generously provided by Dr. Jeff Milbrandt) (31) were constructed using the pAdEasy system as described previously (8, 32, 33). Adenoviruses were delivered unilaterally to the right striatum using coordinates as described previously (7), 7 days prior to initiation of MPTP/saline treatment. A GFP-containing construct was used as a control for all adenoviral experiments. A single unilateral injection of virus was given to each animal (2 μl, 1 × 107 particles per μl), delivered to the right striatum (0.5 mm rostral, 2.2 mm right of the bregma, and 3.4 mm below the skull surface). Each adenovirus injection was given at a constant rate of 0.5 μl/min using a syringe pump system (Harvard Apparatus). Brains were extracted at indicated times following the first MPTP injection.
Brain Microdissection
Following decapitation, brains were sectioned into serial 2.0-mm coronal slices using a plastic dissecting block. Employing a 2-mm diameter biopsy needle, the SNc and striatum were obtained by punch biopsy. Brain tissue samples were taken according to the Franklin and Paxinos mouse brain atlas (26).
Amine Analyses
HPLC analysis was used to evaluate the levels of DA and its metabolite DOPAC in brain microdissections, 14 days following initial MPTP treatment. Levels were determined by HPLC as described previously (26).
Real-time PCR
RNA was extracted from mouse SNc microdissections using TRIzol reagent (Invitrogen). The primer sequences were as follows: Nur77, 5′-TGATGTTCCCGCCTTTGC-3′ (forward) and 5′-CAATGCGATTCTGCAGCTCTT-3′ (reverse); GAPDH, 5′-CTGCACCACCAACTGCTTAG-3′ (forward) and 5′-GGGCCATCCACAGTCTTCT-3′ (reverse). Nur77 and GAPDH primers used for gene expression and PCR conditions have been described previously (34), with the following cycling parameters: 55 °C for 10 min, 95 °C for 5 min, and 40 cycles of 95 °C for 30 s and 60 °C for 60 s.
Mesencephalic Embryonic Survival Cell Culture
Mesencephalic neurons were collected from day 13.5 embryos, adapted as described previously (35). 7 days in vitro cultures were treated with 20 μm 1-methyl-4-phenylpyridinium (MPP+; Sigma) for 24 and 36 h. Cultures were fixed and stained for TH (1:2500) and Hoescht. Survival was assessed by quantifying live TH+ cells.
RNA Interference
To disrupt Nur77 expression, 3 days following plating, siRNA, against Nur77 (Santa Cruz Biotechnology, sc-36109), as well as control siRNA (Santa Cruz Biotechnology, sc-37007) was transfected in cortical neuron cultures 1 day prior to MPP+ (20 μm) treatment (36). At select time points, cells were lysed and assessed for survival (36).
In Situ Hybridization
In situ hybridization was performed as described previously (24).
Western Blot Analysis
Western blot was performed as described previously (37), using antibodies against Nur77 (1:1000; BD Pharmingen) and β-actin (1:5000; Sigma) as a loading control.
Chromatin Immunoprecipitation
ChIP was performed as described previously with embryonic day 15 mouse cortical neurons, following a 0-, 24-, 36-, and 48-h MPP+ time course (37). The primers used for PCR amplification of the mouse Nur77 promoter region were 5′-CTGCGGGCACGGATTTACAACACC-3′ and 5′-GGCGAGCCCGACCCACATCTT-5′.
Behavioral Analysis
Behavioral analysis was carried out by utilizing the computer-assisted beam break, MicroMax system (Accuscan, Columbia, OH). Animals were monitored for a 24 h at 8 weeks of age as described (38).
Statistical Analysis
Histochemical and monoamine data analysis was carried out using one-way ANOVA, followed by a Newman-Keuls post hoc test.
RESULTS
Nur77 Expression Following MPTP and Relationship with MEF2
Our previous evidence indicated that loss of MEF2 activity, mediated by CDK5, was critical for dopaminergic loss induced by MPTP in vivo (7). This evidence suggested that loss of MEF2 mediated expression of downstream transcriptional targets may lead to neuronal loss. Several reports suggested Nur77 as a candidate for MEF2 regulation (17, 39, 40). Therefore, we initially ascertained whether loss of Nur77 expression could be detected in normal WT animals with MPTP treatment. We first assessed the levels of Nur77 transcripts from nigral extracts from an MPTP time course by quantitative real-time PCR. Previous reports have shown Nur77/TH+ colocalization in the SNc following haloperidol treatment, whereas quantitative analysis by in situ hybridization did not display detectable levels under basal conditions (24, 41). However, employing quantitative real-time PCR, we observed a dramatic loss of Nur77 transcript expression in comparison with basal levels at 6 h to 7 days post initial MPTP treatment, ranging from a 44 to 80% reduction in Nur77 mRNA (Fig. 1A). These results were corroborated via in situ hybridization analysis in the dorsolateral and ventrolateral striatum (Fig. 1, B–D). We next assessed whether MEF2D binding on the endogenous Nur77 promoter may be affected by conditions that lead to neuronal loss using ChIP analysis. Because of the need for a more homogenous population of neurons, we utilized the MPP+ treatment paradigm in cultured cortical neurons. We and others (8, 9, 42, 43) have shown that MPP+ can induce death in non-dopaminergic neurons, such as ceberebellar granule and cortical neurons, by non-dopamine transporter related mechanisms. Importantly, we observed that there was detectable MEF2D binding at the Nur77 promoter basally in neurons (Fig. 1F) and that this binding diminished following MPP+ exposure (Fig. 1G).
FIGURE 1.

Nur77 expression and regulation following MPTP and MPP+ treatment. A, expression of Nur77 mRNA in an MPTP time course, at 0, 6, 12, and 24 h and 3 and 7 days (d) post-initial MPTP treatment. B, representative photomicrographs illustrating Nur77 in situ hybridization of striatal tissue, following 0 and 12 h and 3 days MPTP. Black boxes designate dorsolateral (upper) and ventrolateral (lower) striatal quantified regions. C and D, quantification of striatal Nur77 in situ hybridization following no treatment, 12 h, and 1 and 3 days post-initial MPTP treatment. C, striatal dorsolateral. D, striatal ventrolateral. E, attenuation of Nur77 loss as determined by quantitative real-time PCR (qPCR) following adenoviral (AV) MEF2-S444A expression and 1 day of MPTP treatment in vivo. F, ChIP assay for MEF2 binding to Nur77 promoter in untreated cortical cultures. G, ChIP assay for MEF2 binding to Nur77 promoter of cultures untreated (0 h) or treated with MPP+ for 12 and 24 h. Error bars represent mean ± S.E. ANOVA, *, p < 0.05; **, p < 0.01; n = three to four animals per group.
We next examined whether the reduction in Nur77 might relate to loss of MEF2 as previously reported to occur following MPTP treatment. In this regard, Smith et al. (7) showed that expression of non-phosphorylatable MEF2D attenuate DA cell death. Accordingly, we determined whether expression of this construct might also attenuate the loss of Nur77 expression in the SNc in vivo. Nur77 levels were examined in WT mice virally expressing MEF2D-S444A or a GFP control as reported previously (7). Adenoviral MEF2D-S444A significantly attenuated the decrease in Nur77 at 1 day post-MPTP (Fig. 1E).
Nur77 siRNA Elevates Neuronal Cell Death in Vitro
The above results indicate that Nur77 expression decreases in a MEF2D-dependent manner following MPTP treatment in vivo. However, whether the loss of Nur77 plays any functional role in DA loss is unknown. Accordingly, we examined this question and reasoned that if our logic was true, loss of Nur77 should reduce neuronal survival either basally or in the presence of stress.
We first evaluated whether Nur77-deficient mesencephalic TH+ neurons might display sensitivity to MPP+ treatment in culture. Midbrain neurons from WT and Nur77-deficient mice were cultured and treated with MPP+. Survival of TH+ neurons was then evaluated. Nur77-deficient TH+ neurons showed a significant hypersensitivity at 24 h exposure to MPP+ (13% survival with Nur77 deficiency versus 39% survival in WT; Fig. 2A). As an interesting side note, we also examined how other neuron types might respond to loss of Nur77. In this case, we examined cortical neurons treated with siRNA for Nur77. Importantly, treatment of cultured neurons to control siRNA to Nur77 siRNA induced significant toxicity even in the absence of any stress (91% survival with control siRNA versus 49% survival with Nur77 siRNA) (Fig. 2B). MPP+ treatment also diminished survival. However, the survival ratio remained relatively stable between the control and Nur77 siRNA-treated cultures at the various time points. This suggested that acute loss in Nur77 expression in neurons produces a basal detrimental effect that can be further exaggerated by toxin exposure. Nur77 siRNA down-regulation was confirmed by Western blot analysis (Fig. 2B). These results indicated that Nur77 can play a role in neuronal loss and provided the rational to proceed further to examine the role of Nur77 in vivo.
FIGURE 2.
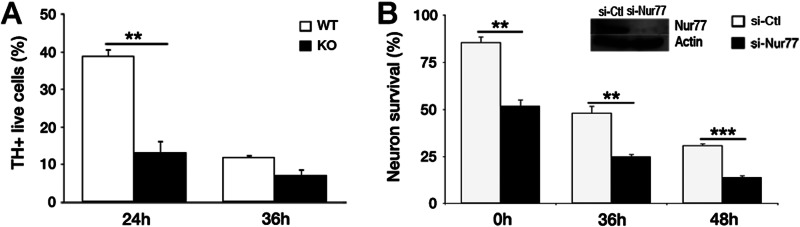
In vitro neuronal cultures display sensitization to toxic insult with loss in Nur77 expression. A, mesencephalic WT and Nur77-deficient neuronal cultures treated with MPP+ for 24 and 36 h. B, cortical neuronal cultures treated with Nur77 and control siRNA (si-Ctl) and MPTP for 36 and 48 h. Error bars represent mean ± S.E. ANOVA, *, p < 0.05; **, p < 0.01; ***, p < 0.001; n = three embryos per group.
Nur77-deficient Mice Exhibit Attenuated MPTP-induced Degeneration of Dopaminergic Cell Bodies in the SNc
We next assessed dopaminergic neuron survival following saline or MPTP treatment of WT and Nur77 KO mice on a C57BK/6J background following a subchronic MPTP dosing regime (Fig. 3). These animals were analyzed by stereological assessment for surviving TH+ neurons over the entire SNc region. Interestingly, unlike in vitro, with acute down-regulation of Nur77 in cortical neurons, no statistical difference in the number of TH+ neurons in the SNc could be detected between saline-treated WT and Nur77 KO mice (Fig. 3, A–C). Similarly, no statistical difference in basal striatal TH or DAT fiber density was detected by immunohistochemistry in saline-treated animals (Fig. 3, D–G). DAT is critical for the reuptake of DA and MPP+, the active MPTP metabolite in dopaminergic neurons (44). Finally, no differences in levels of striatal DA or its metabolite DOPAC were detected in these mutant mice under basal conditions (Fig. 4, C and D). Furthermore, in our study, Nur77-deficient mice displayed no significant differences in 24 h of home cage activity (Fig. 3H). Similar groups of animals were also treated and assessed for potential differences in DA cell survival following MPTP. Following MPTP administration, there was a significant reduction (36.7%; p < 0.001) in TH+ SNc neurons in WT mice (Fig. 3B). Interestingly, and consistent with results observed in culture, Nur77-deficient mice displayed a significantly greater loss of TH+ cells than WT mice treated with MPTP (68.6%; p < 0.001). This observation of increased dopaminergic neuron loss was corroborated morphologically using cresyl violet staining, assessing neurons at the level of the medial terminal nucleus (bregma, −3.16 mm) within the SNc (Fig. 3C). Analysis was consistent with that of TH survival, showing no difference between saline-treated groups, a significant loss of TH+ neurons in WT MPTP-treated mice (28.7%; p < 0.001), and a significant increased loss in DA neurons in Nur77-deficient MPTP-treated animals (56.7%; p < 0.001).
FIGURE 3.

Nur77-deficient mice display increased dopamine cell bodies and striatal terminals of MPTP-induced degeneration of SNc dopaminergic neurons. A, representative photomicrographs illustrating TH immunoreactivity in the ventral midbrain SNc following indicated treatments. B, quantification of TH+ neurons using stereological analysis, as described under “Experimental Procedures.” C, quantification of cresyl violet-stained (CV) cells of the SNc (medial terminal nucleus (MTN) level). D, representative photomicrographs of striatal TH-immunoreactive sections from treated animal groups in A. E, quantification of striatal TH fiber optical density. F, representative photomicrographs of striatal DAT-immunoreactive sections from treated animal groups in A. G, quantification of striatal DAT fiber optical density. H, analysis of 24-h home cage locomotor activity for WT and Nur77-deficient mice. Error bars represent mean ± S.E. ANOVA, *, p < 0.05; **, p < 0.01; ***, p < 0.001; n = 6–8 animals per group. CV, cresyl-violet; OD, optical density.
FIGURE 4.

Nur77 KO mice treated with MPTP display increased striatal FosB, a marker for postsynaptic changes in the denervated striatum, as well as further reduced DA and DOPAC in comparison with WT MPTP-treated mice. A, representative photomicrographs showing FosB staining in the striatum of mice treated as indicated (inset, black arrow, positive FosB; white arrow, negative FosB. B, quantification of FosB+ cells/nuclei striatal photomicrographs. C and D, levels of DA (C) and its metabolite DOPAC (D) in the striatum were analyzed using HPLC on 14-day-old tissue after saline/MPTP injection. Error bars represent mean ± S.E. ANOVA, *, p < 0.05; **, p < 0.01; ***, p < 0.001; n = 5–8 animals per group.
MPTP derives its toxic effects once it crosses the blood-brain barrier and is converted to MPP+, the active dopaminergic neural toxin, by monoamine oxidase B (45). The hypersensitivity observed in Nur77-deficient animals may be attributed to altered MPTP metabolism and therefore significant increased MPP+. To examine this possibility, striatal tissue was evaluated from mice 90 min after a single injection of MPTP, and levels of MPP+ in the striatum were determined using HPLC. It was observed that MPP+ levels did not significantly differ between WT and Nur77-deficient animals (66.0 ± 5.4 and 74.5 ± 3.2, respectively, p = 0.225), suggesting that the increased sensitivity afforded by Nur77 deficiency was not due to impaired MPTP metabolism.
Nur77-deficient Mice Exhibit Increased Degeneration of DA Terminals, Loss of Amines, and Markers of Deregulated Basal Ganglia Following MPTP Treatment
Our results above indicate that MPTP-induced loss of dopaminergic cell bodies in the SNc is increased in Nur77-deficient mice. To determine whether the striatal projections of the SNc neurons also exhibit hypersensitivity in Nur77-deficient mice, striatal dopaminergic terminal fiber density was assessed in response to MPTP. This analysis is important because this is the region where dopaminergic neurons express their activity through DA release. Densitometry analysis of TH striatal fiber staining showed a significant reduction in WT MPTP-treated animals compared with saline controls (69.9% versus 89.6%; p < 0.001) (Fig. 3, D and E) Examination of DAT levels also confirm these findings. Densitometric analysis of striatal DAT stained fibers showed a significant reduction (62%; p < 0.001) in WT MPTP-treated animals compared with saline controls (Fig. 3). Nur77 KO animals exhibited a hypersensitivity to MPTP-induced reduction in striatal DAT density compared with WT-treated mice (80.2%; p < 0.001). These results indicate that striatal terminal fibers were further sensitized by Nur77 deficiency, similar to that exhibited by dopaminergic cell bodies.
Treatment with MPTP previously has been demonstrated to induce expression of the transcription factor ΔFosB postsynaptically within the striatum (6). ΔFosB is suggested to mediate the supersensitivity of striatal DA receptors after denervation (46). Consistent with these reports, WT MPTP-treated mice displayed a significant increase in the number of ΔFosB+ cells (422.1%; p < 0.001) (Fig. 4, A and B) compared with saline controls. Importantly, MPTP-treated Nur77-deficient mice showed consistent hypersensitivity (929.3%; p < 0.001), a significant increase in ΔFosB staining over that observed in WT mice (p < 0.05).
We next examined how DA levels are affected in Nur77-deficient mice after MPTP treatment. Previous analyses report diminished DA and its metabolite DOPAC in the striatum 14 days following MPTP treatment (7, 26). The effects of Nur77 deficiency parallel the observed dopaminergic cell survival results. In the striatum, MPTP significantly reduced the levels of DA in WT mice (43.1%; p < 0.05) with a greater diminishment in the KO animals (70.0%; p < 0.001 ) (Fig. 4C). Striatal DOPAC was significantly reduced in Nur77 KO mice treated with MPTP (Fig. 4D). However, this significant loss was greater in the Nur77 KO MPTP-treated mice than WT (60.5% versus 13.6%; p < 0.05).
Ectopic Expression of Nur77 Rescues Nur77-deficient Hypersensitivity to MPTP
The results described above demonstrate that germ line loss of Nur77 does not lead to degeneration of DA basally but increases these neurons sensitivity to MPTP-induced degeneration in vivo. Because of the germ line nature of Nur77 loss in the model system examined, we included some controls for potential confounding compensatory factors, which might account for the sensitization observed with chronic Nur77 loss. To this end, we explored whether we could rescue the sensitivity observed with Nur77 with ectopic WT Nur77 expression. We expressed Nur77 and control GFP in the SNc using an adenoviral system, in both Nur77-deficient and WT mice. Seven days following intracranial viral injection, the animals followed the same MPTP paradigm as previously employed. GFP expression in WT and Nur77-deficient mice produced similar results to those earlier exhibited in the in vivo survival experiments described above (Fig. 5, A–C). WT, MPTP-treated, GFP-expressing mice showed a 28.4% reduction in TH+ neurons in comparison with the experimental control WT, saline-treated, GFP-expressing mice (p < 0.05). Nur77-deficient, GFP-expressing, MPTP-treated mice showed a similar heightened hypersensitivity, as observed in the initial in vivo experiments, with a 65.0% reduction in TH+ neurons in comparison with control mice. Ectopic Nur77 expression in Nur77-deficient animals dramatically and almost completely reversed the sensitivity observed in comparison with WT animals. These results were corroborated using the morphological cresyl violet analysis (Fig. 5C). Interestingly, Nur77 expression in WT animals produced a slight, but non-significant increase in DA neuron numbers.
FIGURE 5.

Nur77-deficient hypersensitivity to MPTP can be attenuated with ectopic expression of Nur77 in the nigrostriatal system. A, representative photomicrographs illustrating TH immunoreactivity in the ventral midbrain SNc following indicated treatments. B, quantification of TH+ neurons at the medial terminal nucleus (MTN) level of the SNc. C, quantification of cresyl violet-stained cells at the medial terminal nucleus level of the SNc. D, representative photomicrographs of striatal DAT immunoreactive. E, quantification of striatal DAT fiber optical density. Error bars represent mean ± S.E. ANOVA, *, p < 0.05; **, p < 0.01; ***, p < 0.001; n = 5–8 animals per group. OD, optical density; AV, adenoviral; Sal, saline.
Examination of striatal DAT density produced similar results (Fig. 5, D and E). Ectopic expression of Nur77 in Nur77-deficient animals almost completely reversed the sensitization induced by germ line loss of Nur77. Interestingly, Nur77 expression again did not significantly attenuate fiber loss in the WT mouse. This suggests that levels of Nur77 expression was insufficient to promote dramatically increased protection in WT cells but was sufficient to prevent the sensitization observed with full Nur77 deficiency.
DISCUSSION
Previous work has suggested the importance of a calpain-CDK5-MEF2 signaling cascade in the adult in vivo MPTP model of dopaminergic loss (6, 7, 15, 30, 47). How CDK5-mediated repression of MEF2 activity leads to DA loss is unclear. Our findings are of significance as they delineate a novel player, Nur77, in DA loss and link this activity to the calpain-regulated pathway of death. We found that 1) Nur77 is rapidly lost following MPTP treatment and that this loss is attenuated by expression of the transcription factor MEF2D; 2) Nur77 loss results in hypersensitization of the nigrostriatal system to exogenous toxic stress and degeneration; and 3) this hypersensitization can be rescued by re-expression of exogenous Nur77.
Our observations of MEF2-mediated Nur77 regulation is consistent with previous reports suggesting a relationship between the two transcription factors in other systems. For example, MEF2 has been shown to regulate Nur77 expression in thymocyte differentiation (48). In this context, MEF2 regulation of Nur77 is mediated by molecular transcription associating proteins, including HDAC7 (40, 49) and Cabin1 (17, 39). In neurons, depolarization induced up-regulation of MEF2 has also been shown to increase Nur77 levels (50). In addition, CREB-mediated induction of Nur77 is also regulated by MEF2 in PC12 cells (51). Importantly, MEF2 sites exist in the Nur77 promoter (19) with several groups reporting MEF2 binding to the Nur77 promoter (17, 39). Importantly, we presently observed that MEF2 does constitutively occupy these sites endogenously, at least in cultured neurons. Therefore, an important caveat is that this analysis was not performed with SNc neurons. Taken together, we have proposed a model by which neuronal MEF2, known to act as a survival signal, mediates a constitutive level of NUR77 expression. This basal level of NUR77 activity is rapidly lost when calpain-mediated CDK5 activation leads to phosphorylation and inactivation of MEF2.
Several observations support a role for Nur77 loss in facilitating DA loss. We showed that Nur77-deficient animals are hypersensitive to MPTP-induced degeneration. Importantly, we provide support, although not definitive proof, that this sensitivity relates to deficiency of Nur77 in neurons themselves. First, TH+ Nur77-deficient neurons cultured from neuronal midbrain cultures, under conditions where other contaminating cell types are limited, are more sensitive to MPP+ than their WT counterparts. Second, acute suppression of Nur77 by siRNA in cortical neurons leads to their demise even in the absence of stress. It is important to note that this NUR77 basal neuronal loss does not appear to occur with germ line Nur77 loss, suggesting some compensatory events with germ line deficiency. Indeed, there are other Nur family members, including Nor1 and Nurr1 (52, 53). Nevertheless, Nur77-deficient neurons are sensitive to exogenous stress, and this sensitivity can be rescued by ectopic Nur77 expression.
These results are consistent with other reports indicating a role for Nur77 in survival, as seen in TNFα (23) and ceramide-induced cell death (22), and Nur77-NFκB inhibition of apoptosis (54). However, the role of Nur77 appears context dependent. In other cases such as T cell selection (19, 55) and cancer cell death induction (56, 57), Nur77 appears to induce death. Interestingly, NUR77 can localize to the mitochondria where it can interact and inhibit Bcl-2, inducing apoptosis (58, 59). Adding to the complexity is that Nur77 can have functional activity outside of death regulation. In neurons, for example, Nur77 has been associated with synaptic remodeling, response to l-DOPA, and behavioral modifications (41, 60, 61). Taken together, these observations belie the complexity of Nur77 function. At least in DA neurons, however, in response to MPTP, we provide evidence for a unique MEF2-dependent function in DA survival.
What are potential targets of Nur77 that might promote survival? In high metabolic cells such as muscle, Nur77 expression appears to regulate genes involved in glycolysis, glycogenolysis, and the glycerophosphate shuttle (62). Overexpression and shRNA inhibition of Nur77 in muscle cells, respectively, increased and decreased expression of glucose transporter 4 (GLUT4), muscle phosphofructokinase (Phkka1), and glycogen phosphorylase (Pygm). Furthermore, NUR77 was found to bind to the promoter regions of these metabolic controlling genes. Therefore, one attractive hypothesis is that interruption in expression of these metabolic controlling genes under conditions of elevated energy requirements, such as in neurons, may be detrimental. In this regard, maintenance of proper bioenergentics in neurons is critical for their survival, and loss of this capacity may sensitize to exogenous stresses, posing additional metabolic stress onto the cell, as observed with MPTP treatment.
Although our present results suggest Nur77 as one critical effector downstream of MEF2, other MEF2-regulated pathways may also play a role in regulating dopaminergic loss following MPTP treatment. For example, She et al. (15) recently discovered MEF2 localized to the mitochondria, where it is bound to its consensus site on mitochondrial DNA containing and regulating the ND6 gene. ND6, a component of the mitochondrial complex I, controlling oxidative phosphorylation, is key to maintaining energy requiring homeostasis. MPTP-induced MEF2 degradation disrupted the expression of NG6 along with increased hydrogen peroxide concentration, reduced ATP production, and increased DA cell death. MEF2 has also been shown to be degraded in other toxic insult models, including 6-hydroxydopamine (63). Further inhibition of MEF2 contributing to neuronal toxicity is also presented by GSK3β phosphorylation (64). Finally, MEF2D itself acts alongside other regulatory targets of CDK5 to mediate DA loss following MPTP treatment. These additional CDK5 targets include both cytoplasmic and nuclear factors, Prx2 and APE1, respectively (8, 9). Prx2 is critical for handling oxidative stress in this paradigm, although APE1 acts to facilitate DNA repair. With all three targets, CDK5 mediated phosphorylation of these substrates leads to down-regulation of their respective activities, promoting death. In the case of MEF2, however, we have identified an additional critical downstream effector of this transcription factor.
In conclusion, we have shown that mice deficient in Nur77 display a hypersensitization to MPTP-induced dopaminergic cell death and that levels of Nur77 are regulated by MEF2. Our data suggest a new candidate in the calpain-CDK5-MEF2 pathway involving the demise of the nigrostriatal dopaminergic system. We propose that targeting and specifically inhibiting the calpain-CDK5-MEF2-NUR77 pathway may be effective in protecting dopamine neurons in Parkinson disease.
Acknowledgments
We thank Dr. Jeff Milbrandt for generously providing Nur77 knock-out mice and constructs and Dr. Claude Rouillard for technical assistance and discussions, along with Joanie Baillargeon and Brigitte Paquet. We also thank Linda Jui, Carmen Estey, and Hossein Aleyasin for technical assistance and scientific input.
This work was supported by grants from Parkinson Society Canada, the Canadian Institutes of Health Research, Network of Centres of Excellence in Neurodegeneration, the Heart and Stroke Foundation of Ontario, Neuroscience Canada/Krembil Foundation, the Parkinson's Disease Foundation, The Michael J. Fox Foundation for Parkinson's research, the Parkinson Research Consortium, the Canadian Stroke Network, the Heart and Stroke Foundation Centre for Stroke Recovery, and the World Class University program through the National Research Foundation of Korea funded by the Ministry of Education, Science, and Technology (South Korea) (R31-2008-000-20004-0 to D. S. P.).
- DA
- dopamine
- SNc
- substantia nigra pars compacta
- TH
- tyrosine hydroxylase
- DAT
- DA transporter
- MPP+
- 1-methyl-4-phenylpyridinium
- ANOVA
- analysis of variance
- CDK
- cyclin-dependent kinase
- MEF2
- myocyte enhancer factor 2.
REFERENCES
- 1. Przedborski S., Vila M. (2001) The last decade in Parkinson's disease research. Basic sciences. Adv. Neurol. 86, 177–186 [PubMed] [Google Scholar]
- 2. Kostic V., Przedborski S., Flaster E., Sternic N. (1991) Early development of levodopa-induced dyskinesias and response fluctuations in young-onset Parkinson's disease. Neurology 41, 202–205 [DOI] [PubMed] [Google Scholar]
- 3. Pines J. (1993) Cyclins and cyclin-dependent kinases: take your partners. Trends Biochem. Sci. 18, 195–197 [DOI] [PubMed] [Google Scholar]
- 4. Tsai L. H., Takahashi T., Caviness V. S., Jr., Harlow E. (1993) Activity and expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system. Development 119, 1029–1040 [DOI] [PubMed] [Google Scholar]
- 5. Tsai L. H., Delalle I., Caviness V. S., Jr., Chae T., Harlow E. (1994) p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 371, 419–423 [DOI] [PubMed] [Google Scholar]
- 6. Smith P. D., Crocker S. J., Jackson-Lewis V., Jordan-Sciutto K. L., Hayley S., Mount M. P., O'Hare M. J., Callaghan S., Slack R. S., Przedborski S., Anisman H., Park D. S. (2003) Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 100, 13650–13655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith P. D., Mount M. P., Shree R., Callaghan S., Slack R. S., Anisman H., Vincent I., Wang X., Mao Z., Park D. S. (2006) Calpain-regulated p35/cdk5 plays a central role in dopaminergic neuron death through modulation of the transcription factor myocyte enhancer factor 2. J. Neurosci. 26, 440–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Qu D., Rashidian J., Mount M. P., Aleyasin H., Parsanejad M., Lira A., Haque E., Zhang Y., Callaghan S., Daigle M., Rousseaux M. W., Slack R. S., Albert P. R., Vincent I., Woulfe J. M., Park D. S. (2007) Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson's disease. Neuron 55, 37–52 [DOI] [PubMed] [Google Scholar]
- 9. Huang E., Qu D., Zhang Y., Venderova K., Haque M. E., Rousseaux M. W., Slack R. S., Woulfe J. M., Park D. S. (2010) The role of Cdk5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation in neuronal death. Nat. Cell Biol. 12, 563–571 [DOI] [PubMed] [Google Scholar]
- 10. Ko J., Humbert S., Bronson R. T., Takahashi S., Kulkarni A. B., Li E., Tsai L. H. (2001) p35 and p39 are essential for cyclin-dependent kinase 5 function during neurodevelopment. J. Neurosci. 21, 6758–6771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McKinsey T. A., Zhang C. L., Olson E. N. (2001) Control of muscle development by dueling HATs and HDACs. Curr. Opin. Genet. Dev. 11, 497–504 [DOI] [PubMed] [Google Scholar]
- 12. Heidenreich K. A., Linseman D. A. (2004) Myocyte enhancer factor-2 transcription factors in neuronal differentiation and survival. Mol. Neurobiol. 29, 155–166 [DOI] [PubMed] [Google Scholar]
- 13. Mao Z., Bonni A., Xia F., Nadal-Vicens M., Greenberg M. E. (1999) Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 286, 785–790 [DOI] [PubMed] [Google Scholar]
- 14. Wang X., Tang X., Li M., Marshall J., Mao Z. (2005) Regulation of neuroprotective activity of myocyte-enhancer factor 2 by cAMP-protein kinase A signaling pathway in neuronal survival. J. Biol. Chem. 280, 16705–16713 [DOI] [PubMed] [Google Scholar]
- 15. She H., Yang Q., Shepherd K., Smith Y., Miller G., Testa C., Mao Z. (2011) Direct regulation of complex I by mitochondrial MEF2D is disrupted in a mouse model of Parkinson disease and in human patients. J. Clin. Invest. 121, 930–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Youn H. D., Sun L., Prywes R., Liu J. O. (1999) Apoptosis of T cells mediated by Ca2+-induced release of the transcription factor MEF2. Science 286, 790–793 [DOI] [PubMed] [Google Scholar]
- 17. Liu W., Youn H. D., Liu J. O. (2001) Thapsigargin-induced apoptosis involves Cabin1-MEF2-mediated induction of Nur77. Eur. J. Immunol. 31, 1757–1764 [DOI] [PubMed] [Google Scholar]
- 18. Ananieva O., Macdonald A., Wang X., McCoy C. E., McIlrath J., Tournier C., Arthur J. S. (2008) ERK5 regulation in naïve T-cell activation and survival. Eur. J. Immunol. 38, 2534–2547 [DOI] [PubMed] [Google Scholar]
- 19. Woronicz J. D., Lina A., Calnan B. J., Szychowski S., Cheng L., Winoto A. (1995) Regulation of the Nur77 orphan steroid receptor in activation-induced apoptosis. Mol. Cell. Biol. 15, 6364–6376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fujii Y., Matsuda S., Takayama G., Koyasu S. (2008) ERK5 is involved in TCR-induced apoptosis through the modification of Nur77. Genes Cells 13, 411–419 [DOI] [PubMed] [Google Scholar]
- 21. Lee S. L., Wesselschmidt R. L., Linette G. P., Kanagawa O., Russell J. H., Milbrandt J. (1995) Unimpaired thymic and peripheral T cell death in mice lacking the nuclear receptor NGFI-B (Nur77). Science 269, 532–535 [DOI] [PubMed] [Google Scholar]
- 22. Brás A., Albar J. P., Leonardo E., de Buitrago G. G., Martínez-A C. (2000) Ceramide-induced cell death is independent of the Fas/Fas ligand pathway and is prevented by Nur77 overexpression in A20 B cells. Cell Death Differ. 7, 262–271 [DOI] [PubMed] [Google Scholar]
- 23. Suzuki S., Suzuki N., Mirtsos C., Horacek T., Lye E., Noh S. K., Ho A., Bouchard D., Mak T. W., Yeh W. C. (2003) Nur77 as a survival factor in tumor necrosis factor signaling. Proc. Natl. Acad. Sci. U.S.A. 100, 8276–8280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gilbert F., Morissette M., St-Hilaire M., Paquet B., Rouillard C., Di Paolo T., Lévesque D. (2006) Nur77 gene knockout alters dopamine neuron biochemical activity and dopamine turnover. Biol. Psychiatry 60, 538–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee S. L., Tourtellotte L. C., Wesselschmidt R. L., Milbrandt J. (1995) Growth and differentiation proceeds normally in cells deficient in the immediate early gene NGFI-A. J. Biol. Chem. 270, 9971–9977 [DOI] [PubMed] [Google Scholar]
- 26. Mount M. P., Lira A., Grimes D., Smith P. D., Faucher S., Slack R., Anisman H., Hayley S., Park D. S. (2007) Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. J. Neurosci. 27, 3328–3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Crocker S. J., Liston P., Anisman H., Lee C. J., Smith P. D., Earl N., Thompson C. S., Park D. S., Korneluk R. G., Robertson G. S. (2003) Attenuation of MPTP-induced neurotoxicity and behavioural impairment in NSE-XIAP transgenic mice. Neurobiol. Dis. 12, 150–161 [DOI] [PubMed] [Google Scholar]
- 28. Gundersen H. J., Bagger P., Bendtsen T. F., Evans S. M., Korbo L., Marcussen N., Møller A., Nielsen K., Nyengaard J. R., Pakkenberg B. (1988) The new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. APMIS 96, 857–881 [DOI] [PubMed] [Google Scholar]
- 29. Rymar V. V., Sasseville R., Luk K. C., Sadikot A. F. (2004) Neurogenesis and stereological morphometry of calretinin-immunoreactive GABAergic interneurons of the neostriatum. J. Comp. Neurol. 469, 325–339 [DOI] [PubMed] [Google Scholar]
- 30. Gong X., Tang X., Wiedmann M., Wang X., Peng J., Zheng D., Blair L. A., Marshall J., Mao Z. (2003) Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron 38, 33–46 [DOI] [PubMed] [Google Scholar]
- 31. Milbrandt J. (1988) Nerve growth factor induces a gene homologous to the glucocorticoid receptor gene. Neuron 1, 183–188 [DOI] [PubMed] [Google Scholar]
- 32. Sedarous M., Keramaris E., O'Hare M., Melloni E., Slack R. S., Elce J. S., Greer P. A., Park D. S. (2003) Calpains mediate p53 activation and neuronal death evoked by DNA damage. J. Biol. Chem. 278, 26031–26038 [DOI] [PubMed] [Google Scholar]
- 33. O'Hare M. J., Kushwaha N., Zhang Y., Aleyasin H., Callaghan S. M., Slack R. S., Albert P. R., Vincent I., Park D. S. (2005) Differential roles of nuclear and cytoplasmic cyclin-dependent kinase 5 in apoptotic and excitotoxic neuronal death. J. Neurosci. 25, 8954–8966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tsukahara T., Haniu H. (2011) Nanoparticle-mediated intracellular lipid accumulation during C2C12 cell differentiation. Biochem. Biophys. Res. Commun. 406, 558–563 [DOI] [PubMed] [Google Scholar]
- 35. Cheung N. S., Hickling Y. M., Beart P. M. (1997) Development and survival of rat embryonic mesencephalic dopaminergic neurones in serum-free, antioxidant-rich primary cultures. Neurosci. Lett. 233, 13–16 [DOI] [PubMed] [Google Scholar]
- 36. Zhang Y., Qu D., Morris E. J., O'Hare M. J., Callaghan S. M., Slack R. S., Geller H. M., Park D. S. (2006) The Chk1/Cdc25A pathway as activators of the cell cycle in neuronal death induced by camptothecin. J. Neurosci. 26, 8819–8828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang Y., Parsanejad M., Huang E., Qu D., Aleyasin H., Rousseaux M. W., Gonzalez Y. R., Cregan S. P., Slack R. S., Park D. S. (2010) Pim-1 kinase as activator of the cell cycle pathway in neuronal death induced by DNA damage. J. Neurochem. 112, 497–510 [DOI] [PubMed] [Google Scholar]
- 38. Hayley S., Crocker S. J., Smith P. D., Shree T., Jackson-Lewis V., Przedborski S., Mount M., Slack R., Anisman H., Park D. S. (2004) Regulation of dopaminergic loss by Fas in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J. Neurosci. 24, 2045–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Youn H. D., Liu J. O. (2000) Cabin1 represses MEF2-dependent Nur77 expression and T cell apoptosis by controlling association of histone deacetylases and acetylases with MEF2. Immunity 13, 85–94 [DOI] [PubMed] [Google Scholar]
- 40. Dequiedt F., Kasler H., Fischle W., Kiermer V., Weinstein M., Herndier B. G., Verdin E. (2003) HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity 18, 687–698 [DOI] [PubMed] [Google Scholar]
- 41. Mahmoudi S., Samadi P., Gilbert F., Ouattara B., Morissette M., Grégoire L., Rouillard C., Di Paolo T., Lévesque D. (2009) Nur77 mRNA levels and L-Dopa-induced dyskinesias in MPTP monkeys treated with docosahexaenoic acid. Neurobiol. Dis. 36, 213–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Harbison R. A., Ryan K. R., Wilkins H. M., Schroeder E. K., Loucks F. A., Bouchard R. J., Linseman D. A. (2011) Calpain plays a central role in 1-methyl-4-phenylpyridinium (MPP+)-induced neurotoxicity in cerebellar granule neurons. Neurotox. Res. 19, 374–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sheline C. T., Zhu J., Zhang W., Shi C., Cai A. L. (2013) Mitochondrial inhibitor models of Huntington's disease and Parkinson's disease induce zinc accumulation and are attenuated by inhibition of zinc neurotoxicity in vitro or in vivo. Neurodegener. Dis. 11, 49–58 [DOI] [PubMed] [Google Scholar]
- 44. Meissner W., Prunier C., Guilloteau D., Chalon S., Gross C. E., Bezard E. (2003) Time-course of nigrostriatal degeneration in a progressive MPTP-lesioned macaque model of Parkinson's disease. Mol. Neurobiol. 28, 209–218 [DOI] [PubMed] [Google Scholar]
- 45. Jackson-Lewis V., Przedborski S. (2007) Protocol for the MPTP mouse model of Parkinson's disease. Nat Protoc 2, 141–151 [DOI] [PubMed] [Google Scholar]
- 46. Dragunow M., Butterworth N., Waldvogel H., Faull R. L., Nicholson L. F. (1995) Prolonged expression of Fos-related antigens, Jun B and TrkB in dopamine-denervated striatal neurons. Brain Res. Mol. Brain Res. 30, 393–396 [DOI] [PubMed] [Google Scholar]
- 47. Verdaguer E., Alvira D., Jiménez A., Rimbau V., Camins A., Pallàs M. (2005) Inhibition of the cdk5/MEF2 pathway is involved in the antiapoptotic properties of calpain inhibitors in cerebellar neurons. Br. J. Pharmacol. 145, 1103–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. He Y. W. (2002) Orphan nuclear receptors in T lymphocyte development. J. Leukoc. Biol. 72, 440–446 [PubMed] [Google Scholar]
- 49. Verdin E., Dequiedt F., Kasler H. (2004) HDAC7 regulates apoptosis in developing thymocytes. Novartis Found. Symp. 259, 115–129; discussion 129–31, 163–169 [PubMed] [Google Scholar]
- 50. Tian X., Kai L., Hockberger P. E., Wokosin D. L., Surmeier D. J. (2010) MEF-2 regulates activity-dependent spine loss in striatopallidal medium spiny neurons. Mol. Cell. Neurosci. 44, 94–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lam B. Y., Zhang W., Ng D. C., Maruthappu M., Roderick H. L., Chawla S. (2010) CREB-dependent Nur77 induction following depolarization in PC12 cells and neurons is modulated by MEF2 transcription factors. J. Neurochem. 112, 1065–1073 [DOI] [PubMed] [Google Scholar]
- 52. Chao L. C., Bensinger S. J., Villanueva C. J., Wroblewski K., Tontonoz P. (2008) Inhibition of adipocyte differentiation by Nur77, Nurr1, and Nor1. Mol. Endocrinol. 22, 2596–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao Y., Bruemmer D. (2010) NR4A orphan nuclear receptors: transcriptional regulators of gene expression in metabolism and vascular biology. Arterioscler. Thromb. Vasc. Biol. 30, 1535–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. de Léséleuc L., Denis F. (2006) Inhibition of apoptosis by Nur77 through NF-κB activity modulation. Cell Death Differ. 13, 293–300 [DOI] [PubMed] [Google Scholar]
- 55. Green D. R., Droin N., Pinkoski M. (2003) Activation-induced cell death in T cells. Immunol. Rev. 193, 70–81 [DOI] [PubMed] [Google Scholar]
- 56. Cheng Z., Völkers M., Din S., Avitabile D., Khan M., Gude N., Mohsin S., Bo T., Truffa S., Alvarez R., Mason M., Fischer K. M., Konstandin M. H., Zhang X. K., Heller Brown J., Sussman M. A. (2011) Mitochondrial translocation of Nur77 mediates cardiomyocyte apoptosis. Eur. Heart J. 32, 2179–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cho S. D., Yoon K., Chintharlapalli S., Abdelrahim M., Lei P., Hamilton S., Khan S., Ramaiah S. K., Safe S. (2007) Nur77 agonists induce proapoptotic genes and responses in colon cancer cells through nuclear receptor-dependent and nuclear receptor-independent pathways. Cancer Res. 67, 674–683 [DOI] [PubMed] [Google Scholar]
- 58. Lin B., Kolluri S. K., Lin F., Liu W., Han Y. H., Cao X., Dawson M. I., Reed J. C., Zhang X. K. (2004) Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell 116, 527–540 [DOI] [PubMed] [Google Scholar]
- 59. Thompson J., Winoto A. (2008) During negative selection, Nur77 family proteins translocate to mitochondria where they associate with Bcl-2 and expose its proapoptotic BH3 domain. J. Exp. Med. 205, 1029–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lévesque D., Rouillard C. (2007) Nur77 and retinoid X receptors: crucial factors in dopamine-related neuroadaptation. Trends Neurosci. 30, 22–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. St-Hilaire M., Bourhis E., Lévesque D., Rouillard C. (2006) Impaired behavioural and molecular adaptations to dopamine denervation and repeated L-DOPA treatment in Nur77-knockout mice. Eur. J. Neurosci. 24, 795–805 [DOI] [PubMed] [Google Scholar]
- 62. Chao L. C., Zhang Z., Pei L., Saito T., Tontonoz P., Pilch P. F. (2007) Nur77 coordinately regulates expression of genes linked to glucose metabolism in skeletal muscle. Mol. Endocrinol. 21, 2152–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kim M. K., Kim S. C., Kang J. I., Hyun J. H., Boo H. J., Eun S. Y., Park D. B., Yoo E. S., Kang H. K., Kang J. H. (2011) 6-Hydroxydopamine-induced PC12 cell death is mediated by MEF2D down-regulation. Neurochem. Res. 36, 223–231 [DOI] [PubMed] [Google Scholar]
- 64. Wang X., She H., Mao Z. (2009) Phosphorylation of neuronal survival factor MEF2D by glycogen synthase kinase 3β in neuronal apoptosis. J. Biol. Chem. 284, 32619–32626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Paxinos G., Franklin K. B. J. (2001) The Mouse Brain in Stereotaxic Coordinates, Academic Press, San Diego, CA [Google Scholar]