

Key Points
The protein-tyrosine phosphatases Shp1 and Shp2 are critical regulators of megakaryocyte development, platelet production, and function.
Shp1 and Shp2 perform mainly distinct functions in megakaryocytes and platelets, with little functional overlap.
Abstract
The SH2 domain-containing protein-tyrosine phosphatases Shp1 and Shp2 have been implicated in regulating signaling from a variety of platelet and megakaryocyte receptors. In this study, we investigate the functions of Shp1 and Shp2 in megakaryocytes and platelets. Megakaryocyte/platelet (MP)-specific deletion of Shp1 in mice resulted in platelets being less responsive to collagen-related peptide due to reduced GPVI expression and signaling via the Src family kinase (SFK)-Syk-PLCγ2 pathway, and fibrinogen due to reduced SFK activity. By contrast, deletion of Shp2 in the MP lineage resulted in macrothrombocytopenia and platelets being hyper-responsive to anti-CLEC-2 antibody and fibrinogen. Shp1- and Shp2-deficient megakaryocytes had partial blocks at 2N/4N ploidy; however, only the latter exhibited reduced proplatelet formation, thrombopoietin, and integrin signaling. Mice deficient in both Shp1 and Shp2 were severely macrothrombocytopenic and had reduced platelet surface glycoprotein expression, including GPVI, αIIbβ3, and GPIbα. Megakaryocytes from these mice were blocked at 2N/4N ploidy and did not survive ex vivo. Deletion of the immunoreceptor tyrosine-based inhibition motif-containing receptor G6b-B in the MP lineage phenocopied multiple features of Shp1/2-deficient mice, suggesting G6b-B is a critical regulator of Shp1 and Shp2. This study establishes Shp1 and Shp2 as major regulators of megakaryocyte development, platelet production, and function.
Introduction
Although much is known about the agonists and receptors that control megakaryocyte development and platelet production, less is understood about how downstream signals are regulated. The SH2 domain-containing non-transmembrane protein-tyrosine phosphatases (PTPs) Shp1 and Shp2 have been demonstrated to regulate signaling from a variety of tyrosine kinase-linked receptors, including cytokine and growth factor receptors, immunoreceptor tyrosine-based activation motif (ITAM)-containing immune receptors, and integrins.1 Shp1, encoded by Ptpn6, is expressed in hematopoietic and epithelial cells, whereas Shp2, encoded by Ptpn11, is expressed widely. Although structurally similar, Shp1 is generally recognized as a negative regulator of signaling and Shp2 as a positive regulator. A well-characterized function of Shp1 is the inhibition of ITAM receptor signaling.1 Central to this function is the interaction of Shp1 with tandem tyrosine phosphorylated immunoreceptor tyrosine-based inhibition motifs (ITIMs) in the cytoplasmic tail of inhibitory immunoreceptors, including CD22, CD72, and platelet endothelial cell adhesion molecule-1 (PECAM-1), which activate and bring Shp1 into proximity with components of ITAM receptors (supplemental Figure 1A).2 A well-characterized function of Shp2 is as a positive regulator of the rat sarcoma (Ras)-mitogen-activated protein kinase (MAPK) pathway downstream of cytokine and growth factor receptors.1 Various models have been proposed of how it does this, including dephosphorylation of binding sites on adapters and receptors that localize C-terminal Src kinase and RasGAP to the plasma membrane, where they can inhibit Src family kinases (SFKs) and Ras activity, respectively (supplemental Figure 1B).1 Shp2 has also been shown to have downstream effects on phosphotidylinositide 3-kinase and the GTPase RhoA that are receptor and cell dependent.3,4 However, the exact mechanism remains controversial. Shp2 also has been implicated in negatively regulating ITAM receptor signaling via interactions with ITIM receptors (supplemental Figure 1C).5,6
Mouse models have revealed distinct physiological functions of Shp1 and Shp2. Mice lacking Shp1, referred to as motheaten due to their patchy hair loss, die 2-3 wk after birth with severe inflammation, immunodeficiency, and autoimmunity.7,8 Motheaten viable mice, which express low levels of catalytically impaired Shp1, die at 9-12 wk.9 Platelets from motheaten viable mice are less reactive to the GPVI-specific agonist, collagen-related peptide (CRP); however, the cause of this defect has not been defined.10 By contrast, Shp2 null embryos die peri-implantation, due at least in part, to a trophoblast stem cell defect.11 Hypomorphic Shp2 mouse models also cause embryonic lethality, but at a later stage than null embryos, presumably due to aberrant compartmentalization and activity of Shp2.11-14
Five ITIM-containing receptors have been identified in megakaryocytes and/or platelets to date, namely PECAM-1, carcinoembryonic antigen-related cell adhesion molecule 1, TREM-like transcript-1, leukocyte-associated immunoglobulin-like receptor-1, and G6b-B, all of which interact with Shp1 and Shp2 upon phosphorylation.15-19 Unique among these is G6b-B, which is constitutively phosphorylated by SFKs and associated with Shp1 and Shp2 (supplemental Figure 1C).20,21 Thus, G6b-B is thought to maintain active Shp1 and Shp2 at the plasma membrane, where they inhibit signaling from various receptors. It is well established that occupancy of both SH2 domains of Shp2 with tandem phosphotyrosine peptides increases Shp2 activity.22-25 Structural and enzymological similarities between Shp1 and Shp2 suggest that Shp1 is regulated in a similar manner.26,27 Targeted deletion of G6b causes severe macrothrombocytopenia and a bleeding diathesis due to enhanced platelet clearance, and aberrant platelet production and function.19
In this study, we investigated the functions of Shp1 and Shp2 in megakaryocytes and platelets through the use of megakaryocyte/platelet (MP)-specific Ptpn6 and Ptpn11 single and double conditional knockout (KO) mouse models. The distinct phenotypes exhibited by the Shp1 and Shp2 conditional KO mice highlight the disparate physiological functions of these structurally related PTPs in megakaryocytes and platelets. Mechanistically, Shp1 regulates GPVI surface expression and signaling via the SFK-Syk-PLCγ2 signaling pathway in platelets, whereas Shp2 is a critical positive regulator of Mpl and αIIbβ3 signaling in megakaryocytes and a negative regulator of CLEC-2- and αIIbβ3-mediated responses in platelets. Ptpn6/Ptpn11 double-KO (DKO) mice were severely macrothrombocytopenic with impaired megakaryocyte development and survival ex vivo. All major surface receptors were severely reduced in DKO-deficient platelets, making them irresponsive to all agonists tested. A similar phenotype was seen in G6b conditional KO mice, suggesting G6b-B signals via and is a major regulator of Shp1 and Shp2 in megakaryocytes and platelets.
Materials and methods
Mice
Ptpn6fl/fl, Ptpn11fl/fl, G6bfl/fl, and PF4-Cre+ mice were generated, as previously described.19,28-30 MP-specific Shp1, Shp2, and G6b KO mice were generated by crossing Ptpn6fl/fl, Ptpn11fl/fl, and G6bfl/fl with PF4-Cre+ mice. Wild-type (WT) mice were PF4-Cre-. All mice were on a C57BL/6 background. All procedures were undertaken with United Kingdom Home Office approval in accordance with the Animals (Scientific Procedures) Act of 1986.
Antibodies and chemicals
DyLight488-conjugated anti-mouse GPIbβ antibody (X488) was purchased from Emfret (Eibelstadt, Germany). Anti-mouse thrombopoietin (Tpo) receptor was purchased from R&D System. All other antibodies and chemicals were either purchased or generated as previously described.19
Serum thrombopoietin measurement
Serum thrombopoietin (Tpo) was measured using the Quantikine murine Tpo Immunoassay kit (R&D System).
Platelet half-life
Platelet half-life was measured as previously described.31
Immune thrombocytopenia
Thrombocytopenia was induced in 8- to 12-wk-old KO and litter-matched WT mice by intraperitoneal injection of anti-mouse GPIbα antibody (2 μg/g of mouse) as previously described.32 Blood samples were collected at indicated times. Platelet counts were measured using an ABX Pentra 60 Hematology Analyzer (Block Scientific, Inc).
Immunohistochemistry
Spleens from KO and litter-matched WT mice were fixed in buffered formalin and embedded in paraffin. Sections (5 μm) were H&E and reticulin stained and examined by light microscopy using a 40× objective.
Platelet aggregation and secretion
Blood was collected from the descending thoracic aorta of CO2-asphyxiated mice into 1/10 (v/v) acid-citrate-dextrose anticoagulant, and washed platelets (2 × 108/mL) were prepared as previously described.33 Platelet aggregation and adenosine triphosphate (ATP) secretion were simultaneously measured using a lumi-aggregometer (Chrono-Log, Havertown, PA).
Flow cytometry
Surface glycoprotein expression was measured in whole blood by flow cytometry using fluorescein isothiocyanate (FITC)-conjugated antibodies. Resting and activated platelets were fixed and stained with anti-P-selectin antibody or fibrinogen.
Platelet biochemistry
Washed mouse platelet whole cell lysates were prepared and western blotted as previously described.34
Platelet spreading
Washed platelets (2 × 107/mL) from KO and litter-matched WT mice were preincubated with or without 0.1 U/mL thrombin for 5 min, placed on 100 μg/mL fibrinogen-coated cover-slips for 45 min at 37°C, and imaged as previously described.35
Platelet adhesion under flow
Blood was collected from CO2-asphyxiated mice into 5 U/mL heparin and 40 μM D-phenylalanyl-L-prolyl-L-arginine chloromethylketone (PPACK) and perfused through collagen-coated (100 μg/mL) glass microslide capillaries (1 × 0.1 mm) at 1000 s−1 for 4 min at 37°C as previously described.33
Tail bleeding assay
Experiments were performed on 8- to 10-wk-old KO and litter-matched WT mice as previously described.36
In vivo thrombosis assay
Laser-induced injury of arterioles in the cremaster muscle of KO and litter-matched WT mice was performed as previously described,36 with the exception that mice were injected via the carotid artery with 0.1 μg/g body weight of X488.
Megakaryocyte culture and functional assays
Mature megakaryocytes from mouse bone marrow were cultured and analyzed as previously described.37
Statistical analysis
Data were analyzed using the χ2 test, Mann-Whitney, or Student t test.
Results
Generation of Shp1 and Shp2 conditional KO mice
Shp1 and Shp2 are expressed throughout megakaryocyte development and platelet production (supplemental Figure 2A). To study their functions in megakaryocytes and platelets, we generated MP-specific Ptpn6 and Ptpn11 conditional KO mice (Ptpn6fl/fl;PF4-Cre+ [MP-Shp1 KO] and Ptpn11fl/fl;PF4-Cre+ [MP-Shp2 KO]). MP-Shp1 KO mice were born slightly below the expected Mendelian frequency, whereas MP-Shp2 KO mice were born at the predicted ratio (supplemental Tables 1 and 2). Shp1 and Shp2 were not detected in megakaryocytes and platelets from MP-Shp1 KO and MP-Shp2 KO mice, respectively (supplemental Figure 2B). By contrast, megakaryocytes and platelets from litter-matched control mice lacking the PF4-Cre transgene (Ptpn6fl/fl;PF4-Cre- and Ptpn11fl/fl;PF4-Cre-, referred to as MP-Shp1 WT and MP-Shp2 WT, respectively) contained normal levels of Shp1 and Shp2 (supplemental Figure 2B). There was no effect of megakaryocyte deletion of Shp1 on Shp2 expression and vice versa (supplemental Figure 2B). MP-Shp1/2 conditional DKO mice were also generated to investigate functional redundancy between the PTPs. As expected, neither Shp1 nor Shp2 were detected in platelets from MP-specific Ptpn6/Ptpn11 conditional DKO mice (Ptpn6fl/fl;Ptpn11fl/fl;PF4-Cre+, referred to as MP-Shp1/2 DKO) (supplemental Figure 2B). We were unable to check Shp1 and Shp2 levels in MP-Shp1/2 DKO megakaryocytes due to the poor growth and survival of these megakaryocytes ex vivo.
Macrothrombocytopenia and aberrant platelet surface glycoprotein expression
Platelet counts were normal and platelet volume was marginally increased (5.8%) in MP-Shp1 KO mice (Figure 1; supplemental Table 3). By contrast, MP-Shp2 KO mice were mildly macrothrombocytopenic (30% reduction in platelet counts and 16% increase in platelet volume) and MP-Shp1/2 DKO mice were severely macrothrombocytopenic (93% reduction in platelet counts and 57% increase in platelet volume) (Figure 1A). MP-Shp1/2 DKO mice had additional hematologic anomalies, including a 26% reduction in red blood cells and a 4.2-fold increase in white blood cells, suggesting defects in hematopoiesis and immunity (supplemental Tables 3-5).
Figure 1.



Aberrant function of Shp1- and Shp2-deficient platelets. (A) Platelet counts and volumes from Shp1 KO (n = 37), Shp2 KO (n = 43), Shp1/2 DKO (n = 11), and litter-matched wild-type mice (Shp1 WT, n = 36; Shp2 WT, n = 39; Shp1/2 WT, n = 7). Mean ± SEM; **P < .01, ***P < .001. (B-C) Aggregation and ATP secretion of washed platelets were measured by lumi-aggregometery in response to agonists indicated. (i) Representative traces, n = 4-8 mice/genotype per condition. (ii) Data presented are means (±SEM) of 3 independent experiments (***P < .001). (D) P-selectin expression and fibrinogen binding of (i) Shp1/2 DKO, (ii) Shp1 KO, and (iii) Shp2 KO platelets in whole blood in response to 3 μg/mL CRP, 3 μg/mL convulxin, and 500 μM PAR-4 peptide (PAR4). Geometric mean fluorescence intensity (MFI) ± SD, n = 4-8 mice/genotype per condition, except convulxin-stimulated Shp1/2 DKO conditions, which are n = 2; **P < .01.
Surface expression of the collagen activation receptor GPVI and the α2 subunit of the collagen integrin α2β1 were reduced by 28% and 16%, respectively, in Shp1-deficient platelets (supplemental Table 6). By contrast, surface expression of the GPIbα subunit of the von Willebrand factor (VWF) receptor complex GPIb-IX-V was increased by 45%, and expression of the integrin αIIbβ3, which binds fibrinogen and VWF, was normal (supplemental Table 6). Surface glycoprotein expression was normal in Shp2-deficient platelets (supplemental Table 7); however, all major surface glycoproteins analyzed were severely reduced in Shp1/2-deficient platelets, except for the metalloprotease ADAM10, which was elevated by 51% (supplemental Table 8). ADAM10 is an established sheddase of GPVI and GPIbα in platelets.38
Collectively, these findings demonstrate differences in platelet counts, volumes, and surface glycoprotein expression in MP-Shp1 and MP-Shp2 KO mice that were severely exacerbated in DKO mice, suggesting Shp1 and Shp2 regulate platelet production.
Aberrant functional responses of Shp1- and Shp2-deficient platelets
We next investigated functional responses of Shp1- and Shp2-deficient platelets. Consistent with reduced GPVI surface expression, Shp1-deficient platelets exhibited a marginal reduction in aggregation and ATP secretion to a low concentration of the GPVI-specific agonist CRP (0.3 μg/mL) (Figure 1Bi-ii). However, they responded normally to the snake toxin convulxin (3 μg/mL) that signals via GPVI but also binds to GPIbα,39 which was up-regulated in these platelets (supplemental Table 6), and collagen (3 μg/mL), which signals via GPVI and binds with high affinity to the integrin α2β1 (Figure 1Bi-ii). They also responded normally to antibody-mediated cross-linking of the hem-ITAM-containing podoplanin receptor CLEC-2 (3 μg/mL) (Figure 1Bi-ii). By contrast, Shp2-deficient platelets responded normally to 0.3 μg/mL CRP, 3 μg/mL convulxin, and 3 μg/mL collagen (Figure 1Ci-ii). However, they responded more rapidly to 3 μg/mL anti-CLEC-2 antibody compared with control platelets (Figure 1Ci-ii), suggesting Shp2 negatively regulates CLEC-2 signaling. Shp1- and Shp2-deficient platelets responded normally to G protein-coupled receptor agonists, including 3 μM U46619 (thromboxane A2 [TxA2] analog), 0.06 U/mL thrombin, and 10 μM ADP (supplemental Figure 3).
Due to the extremely low platelet counts in MP-Shp1/2 DKO mice, we were unable to perform standard aggregation/ATP secretion assays on platelets from these mice. As an alternative, we measured P-selectin surface expression and fibrinogen binding in whole blood, which are indicators of α-granule secretion and integrin αIIbβ3 function, respectively. As expected, Shp1/2-deficient platelets did not respond to 3 μg/mL CRP, 3 μg/mL convulxin, or 500 μM PAR-4 peptide, most likely due to reduced surface glycoprotein expression (Figure 1Di). Shp1- and Shp2-deficient platelet responses were consistent with aggregation/ATP secretion results, ie, Shp1-deficient platelets responded less well than control platelets to CRP but normally to convulxin and PAR-4 peptide (Figure 1Dii), and Shp2-deficient platelets responded normally to all 3 agonists (Figure 1Diii).
Aberrant integrin-mediated responses of Shp1- and Shp2-deficient platelets
We next investigated the ability of Shp1- and Shp2-deficient platelets to spread on a fibrinogen-coated surface. This is an important aspect of platelet function that is dependent on the integrin αIIbβ3 for adhesion and cytoskeletal remodeling.40 Shp1-deficient platelets exhibited a marked reduction in spreading on fibrinogen, whereas Shp2-deficient platelets spread to a greater extent than control platelets (Figure 2Ai-ii), suggesting that Shp1 positively regulates this process, whereas Shp2 negatively regulates it. However, both Shp1- and Shp2-deficient platelets spread normally when preactivated with 0.1 U/mL thrombin (Figure 2Ai-ii). Shp1/2-deficient platelets did not spread on a fibrinogen-coated surface and failed to spread to the same extent as control platelets when preactivated with thrombin (Figure 2Ai-ii).
Figure 2.



Functional roles of Shp1 and Shp2 in hemostasis and thrombosis. (A) Platelet spreading on fibrinogen. Basal and thrombin (0.1 U/mL)-activated platelets were plated on a fibrinogen-coated surface. (i) Representative differential interference contrast (DIC) and phalloidin-stained images of platelets; n = 3-5 mice/genotype. (ii) Surface area of individual platelets in DIC images was measured; n = 250-500 platelets/condition; n = 3 mice/genotype; mean ± SD; *P < .05; **P < .01; bar represents 5 μm. (B) Platelet adhesion to collagen under flow. Anticoagulated blood was flowed through collagen-coated capillary tubes at 1000 s−1. (i) Representative DIC images of adherent platelets; n = 3-5 mice/genotype; bar represents 10 μm. (ii) Percentage of surface coverage was measured. Data presented are means (± SEM) of 3 independent experiments. (C) Tail bleeding assay. Shp1/2 DKO (n = 13) mice bled excessively compared with litter-matched Shp1/2 WT mice (n = 20). Symbols represent individual mice; horizontal lines represent means. Mann-Whitney test was used to compare sample medians and determine statistical significance. (D) Laser injury-induced thrombus formation in vivo. Mice were injected with Dylight488-conjugated anti-GPIbβ antibody (X488). Arterioles in cremaster muscles of recipients were subsequently injured by laser and the accumulation of platelets (green) into the thrombi was assessed. (i) Representative composite brightfield and fluorescence images from X488-labeled platelets after laser injury of arteriole are shown. Bar represents 10 μm. (ii) Each curve represents the mean integrated thrombus fluorescence intensity (± SEM, gray; WT, top; KO, bottom) in arbitrary units (a.u.); n = 19-27 thrombi induced in 5-7 mice/genotype. See also supplemental Figure 2 video 1, Figure 2 video 2, Figure 2 video 3, and Figure 2 video 4.
We also asked whether functional defects caused by Shp1 and Shp2 deficiency affected platelet aggregation to collagen under arterial shear. This is a multi-step process that is dependent on the GPIb-XI-V for tethering to collagen-bound VWF, GPVI for platelet activation, and the integrins α2β1 and αIIbβ3 for firm adhesion and aggregation.41 Anti-coagulated blood from Shp1- and Shp2-deficient mice was flowed through collagen-coated capillary tubes at a shear rate of 1000 s−1, which mimics flow conditions in small arteries. Images of adherent platelet aggregates were taken by differential interference contrast microscopy and the surface area was quantified. Surprisingly, Shp1- and Shp2-deficient platelets responded normally under these conditions (Figure 2Bi-ii), demonstrating that the net effect of the defects described above was not sufficient to abrogate platelet adhesion and aggregate formation under these conditions. Blood from MP-Shp1/2 DKO was not tested in this assay due to the severely low platelet counts.
Hemostasis and thrombosis in mutant mice
Consistent with the in vitro findings described above, MP-Shp1 and MP-Shp2 KO mice did not bleed excessively following tail injury (data not shown), whereas MP-Shp1/2 DKO mice did (Figure 2C). This correlated with severely reduced platelet counts and functional responses of Shp1/2-deficient platelets. Because the tail bleeding assay is relatively insensitive of hemostatic function, we also measured thrombus formation in vivo following laser-induced injury of the endothelial lining of arterioles in the cremaster muscle of mutant mice. The rate and size of thrombus formation were reduced in MP-Shp1 KO mice but not in MP-Shp2 KO mice (Figure 2Di-ii; supplemental videos 1-4). Collectively, these findings demonstrate that deficiency of either Shp1 or Shp2 in platelets does not result in a bleeding diathesis and a significant reduction in thrombus formation in MP-Shp1 KO mice.
Aberrant signaling in Shp1- and Shp2-deficient platelets
Because Shp1 and Shp2 have been implicated in regulating ITAM and integrin signaling, we investigated GPVI and αIIbβ3 signaling in Shp1- and Shp2-deficient platelets, with the caveat that GPVI surface expression was reduced in Shp1-deficient platelets. Western blotting reveled that total GPVI was reduced in resting Shp1-deficient platelets, whereas the sheddase-generated C-terminal tail and FcR γ-chain were normal (Figure 3Ai-iv). There was a general reduction in tyrosine phosphorylation in Shp1-deficient platelets in response to 3 μg/mL CRP (Figure 3B) and 3 μg/mL convulxin at early time points (supplemental Figure 4), whereas no major alterations were observed in Shp2-deficient platelets in response to 3 μg/mL CRP (Figure 3C). Further investigation revealed reduced phosphorylation of Src and Syk family tyrosine kinases on their activation loops (Src Tyr-418 and Syk Tyr-519/520) in response to 3 μg/mL CRP and decreased phosphorylation of the downstream target PLCγ2 Tyr-1217, all of which are indicators of enzymatic activity (Figure 3B). Of particular note, the FcR γ-chain and Syk Tyr-519/520 were disproportionately hypo-phosphorylated compared with Src Tyr-418 30 s post-CRP stimulation, supporting the concept that aberrant GPVI proximal signaling contributes to reduced reactivity of Shp1-deficient platelets to CRP (Figure 3B). A similar observation was made in convulxin-stimulated Shp1-deficient platelets at early time points (supplemental Figure 4).
Figure 3.
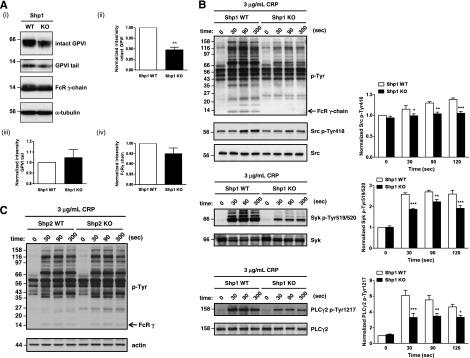
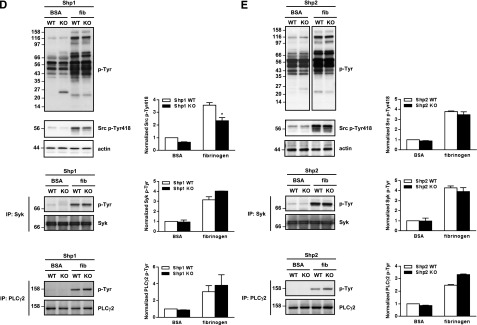
Aberrant tyrosine phosphorylation in Shp1- and Shp2-deficient platelets. (A) (i) Whole cell lysates (WCLs) from Shp1 KO and Shp1 WT were western blotted with anti-GPVI antibody that recognize both intact GPVI and the sheddase-generated C-terminal tail, with anti-FcRγ chain and α-tubulin antibodies. (i) Representative western blots from n = 3 independent experiments and (ii-iv) quantification of intact GPVI, GPVI tail, and FcRγ chain are shown. Data are mean ± SEM, **P < .01. (B) WCLs of resting and CRP-stimulated platelets from Shp1 KO and Shp1 WT mice were western blotted with anti-phosphotyrosine (p-Tyr), -Src p-Tyr418, -Syk p-Tyr519/520, and -PLCγ2p-Tyr1217 antibodies. Membranes were stripped and reblotted with anti-pan Syk, pan PLCγ2, and actin antibodies. Representative blots and densitometry quantification from n = 3-7 independent experiments/genotype (mean ± SEM; *P < .05, **P < .01, ***P < .001). (C) WCLs of resting and CRP-stimulated platelets from Shp2 KO and Shp2 WT mice were western blotted with anti-p-Tyr antibody, stripped, and reblotted with anti-actin antibody. Representative blots from n = 5 independent experiments. (D-E) WCLs of washed platelets from bovine serum albumin (BSA) nonadherent and fibrinogen (fib)-adhered (D) Shp1 KO, (E) Shp2 KO, and litter-matched WT mice in the presence of 10 μM indomethacin and 2 U/mL apyrase were western blotted with anti-p-Tyr and -Src p-Tyr418 antibodies. (E top panels) Equal amounts of total protein were resolved on the same membrane, but different exposure times are shown to highlight differentially phosphorylated proteins in BSA nonadherent and fib-adhered platelets. Syk and PLCγ2 were immunoprecipitated (IP) from WCLs and western blotted with anti-p-Tyr antibody. Membranes were reblotted with anti-pan Syk and -pan PLCγ2 antibodies. Representative blots and densitometry quantification of n = 3 independent experiments/genotype (mean ± SEM; *P < .05).
We next investigated the molecular mechanism underlying aberrant spreading of Shp1- and Shp2-deficient platelets on fibrinogen. Findings suggested Shp1 positively regulates outside-in integrin αIIbβ3 signaling, whereas Shp2 negatively regulates it (Figure 2A). These defects could not be explained by altered αIIbβ3 expression (supplemental Tables 6-7). Several co-migrating bands at 116, 98, 43, 35, 25, and 22 kDa were either hypo- or hyper-tyrosine phosphorylated in BSA nonadherent and fibrinogen-adhered Shp1- and Shp2-deficient platelets compared with control platelets (Figure 3D-E), possibly representing direct or indirect downstream targets of Shp1 and Shp2 and underlying functional defects in these platelets. Reduced tyrosine phosphorylation of the SFK activation loop in fibrinogen-adhered Shp1-deficient platelets (Figure 3D) supported the concept of Shp1 as a positive regulator of SFK activity. Phosphorylation of the SFK activation loop was, however, normal in Shp2-deficient platelets (Figure 3E), demonstrating disparate functions of Shp1 and Shp2 in regulating SFKs downstream of αIIbβ3 in platelets. Downstream tyrosine phosphorylation of Syk and PLCγ2 was normal in both Shp1- and Shp2-deficient platelets (Figure 3D-E); thus, the spreading defects exhibited by these platelets cannot be explained by aberrant signaling via the SFK-Syk-PLCγ2 pathway.
Aberrant platelet kinetics in Shp2 conditional KO mice
Circulating platelet counts are the net result of the rate of platelet clearance and production. To determine the cause of the macrothrombocytopenia in MP-Shp2 KO and MP-Shp1/2 DKO mice, we measured platelet half-life and recovery following antibody-mediated depletion as an indicator of platelet production in vivo. Platelet half-life was normal in MP-Shp1 KO mice (Figure 4A) but reduced by 33% and 57% in MP-Shp2 KO and MP-Shp1/2 DKO mice, respectively (Figure 4B-C). In addition, the initial rate of platelet recovery following anti-GPIbα antibody-mediated platelet depletion was normal in MP-Shp1 KO mice but reduced in MP-Shp2 KO mice (Figure 4D-E), suggesting reduced rate of platelet production in these mice. The rate of platelet recovery was not measured in MP-Shp1/2 DKO mice due to severe macrothrombocytopenia in these mice. Serum Tpo levels, which are generally inversely related to platelet counts, were elevated in MP-Shp2 KO and MP-Shp1/2 DKO mice but not in MP-Shp1 KO mice, as expected (Figure 4F). These findings demonstrate that macrothrombocytopenia in MP-Shp2 KO and MP-Shp1/2 DKO mice is partially due to enhanced platelet clearance and, in the case of MP-Shp2 KO mice, reduced rate of platelet production.
Figure 4.

Impaired platelet clearance and platelet production. (A-C) Platelet half-life. The number of biotin+αIIbβ3+ platelets in the circulation of (A) Shp1 KO, (B) Shp2 KO, (C) Shp1/2 DKO, and Shp1 WT, Shp2 WT, and Shp1/2 WT mice were quantified daily pre-/postinjection of biotin-N-hydroxysuccinamide by flow cytometry (n = 4-8 mice/genotype per time point; mean ± SEM; ***P < .001). (D-E) Platelet recovery following immune-mediated thrombocytopenia. (D) Shp1 KO, (E) Shp2 KO, and litter-matched WT mice were given a single bolus injection of 2 μg/g anti-GPIbα antibody and platelet counts measured daily pre-/postinjection (n = 3-5 mice/genotype per time point; mean ± SEM; **P < .01, ***P < .001). (F) Serum Tpo concentrations (pg/mL) were measured for Shp1 KO (n = 9), Shp2 KO (n = 8), Shp1/2 DKO (n = 15), and litter-matched control mice (Shp1 WT, n = 9; Shp2 WT, n = 8; and Shp1/2 WT n = 6; *P < .05, **P < .01).
Because the spleen acts as a site of platelet clearance and production following bone marrow damage, such as myelofibrosis, we checked for evidence of spleen and bone marrow dysfunction in mutant mice. All KO mice had mild to severe splenomegaly (supplemental Figure 5) and clusters of megakaryocytes in the red pulp (supplemental Figure 6A,C), suggesting extramedullary hematopoiesis was taking place. Megakaryocyte counts were also elevated in the bone marrow of MP-Shp2 KO and MP-Shp1/2 DKO mice but not in MP-Shp1 KO mice (supplemental Figure 6B-C). Reticulin staining revealed evidence of tissue damage in the spleen and bone marrow of MP-Shp2 KO and MP-Shp1/2 DKO mice but not MP-Shp1 KO mice (supplemental Figure 7A-B).
Impaired megakaryocyte polyploidization and function
To avoid nonspecific effects caused by bone marrow dysfunction, we investigated megakaryocyte development and function ex vivo. Shp1 and Shp2 deficiency resulted in an increase in the number of 2N/4N ploidy megakaryocytes and a reduction in the number of >4N ploidy megakaryocytes, demonstrating a partial block in polyploidization (Figure 5A-B). This was dramatically exacerbated in Shp1/2-deficient megakaryocytes, which grew poorly ex vivo and rarely developed past 4N (Figure 5A-B), suggesting defects in their development and survival.
Figure 5.


Impaired megakaryocyte development and proplatelet formation. (A) Mature bone marrow-derived megakaryocytes from Shp1 KO, Shp2 KO, Shp1/2 DKO, and litter-matched WT (Shp1 WT, Shp2 WT, and Shp1/2 WT) mice were stained with propidium iodide and ploidy of cells was quantified by flow cytometry. Representative profiles of n = 4-6 mice/genotype. (B) The percentage of 2-4N and 8-128N ploidy cells was quantified (n = 4-6 mice/genotype; mean ± SEM; **P < .01, ***P < .001). (C-F) Mature bone marrow-derived megakaryocytes from Shp1 KO, Shp2 KO, and corresponding litter-matched WT mice were platelet on fibrinogen-, fibronectin-, and collagen-coated surfaces. (C-D) Representative images of spread megakaryocytes (Alexa Fluor 488-phalloidin stained) and (E-F) megakaryocytes undergoing proplatelet formation (FITC-αIIb stained; n = 4-6 mice/genotype). (C-D) Megakaryocyte surface area, (E-F) proplatelet surface area, and percentage of megakaryocytes undergoing proplatelet formation (n = 4-6 mice/genotype; mean ± SEM; ***P < .001; bar represents 20 μm).
Functional responses of Shp1- and Shp2-deficient megakaryocytes were also analyzed ex vivo. We focused on integrin-mediated spreading as an indicator of integrin function and proplatelet formation, which is a critical step in platelet production.42 Spreading and proplatelet formation were analyzed on fibrinogen-, fibronectin-, and collagen-coated surfaces, all of which are components of the bone marrow microenvironment. Despite defects in polyploidization and reduced αIIbβ3 expression (supplemental Table 9), Shp1-deficient megakaryocytes spread normally on all 3 surfaces (Figure 5C) and formed normal proplatelets on fibrinogen- and fibronectin-coated surfaces (Figure 5E). Neither control nor Shp1-deficient megakaryocytes formed proplatelets on collagen (Figure 5E), which has previously been shown to inhibit proplatelet formation ex vivo.43 By contrast, Shp2-deficient megakaryocytes exhibited reduced spreading (Figure 5D) and proplatelet formation (Figure 5F). Surface area of spread megakaryocytes, proplatelet surface area, and the number of megakaryocytes undergoing proplatelet formation were significantly reduced in Shp2-deficient megakaryocytes (Figure 5D,F). Reduced surface glycoprotein expression may have partially contributed to these defects (supplemental Table 10). These findings are consistent with reduced rate of platelet production in MP-Shp2 KO mice (Figure 4E), as integrin-mediated proplatelet formation is a critical step in this process.32 Similar studies were not performed with Shp1/2-deficient megakaryocytes due to their poor growth and survival ex vivo.
Reduced Tpo and integrin signaling in Shp2-deficient megakaryocytes
Having found reduced ploidy in Shp1- and Shp2-deficient megakaryocytes and reduced functional responses in the latter, we investigated the molecular basis of these defects. Reduced spreading of Shp2-deficient megakaryocytes could not be explained by reduced αIIbβ3 expression, as αIIbβ3 levels were similarly reduced in Shp1-deficient megakaryocytes (supplemental Tables 9 and 10). We focused on Tpo- and integrin-mediated Erk1/2 phosphorylation and SFK activation, as we have previously demonstrated that Erk1/2 plays a critical role in primary megakaryocyte differentiation, motility, and proplatelet formation37 and that SFKs regulate megakaryocyte differentiation and platelet production.44 Moreover, Shp2 is an established positive regulator of the Ras-MAPK pathway and has also been implicated in regulating SFK activity.1 Phospho-specific antibodies were used as indirect indicators of Erk1/2 and SFK activation. Tpo-mediated Erk1/2 activation was normal in Shp1-deficient megakaryocytes (Figure 6A) and significantly reduced in Shp2-deficient megakaryocytes (Figure 6B). SFK activation, which lies upstream of Erk1/2, was, however, normal in Tpo-stimulated Shp2-deficient megakaryocytes (Figure 6B). Moreover, this was not due to reduced Mpl expression (Figure 6C). Integrin αIIbβ3-mediated Erk1/2 activation was also normal in fibrinogen-adhered Shp1-deficient megakaryocytes (Figure 6D) but was significantly reduced in Shp2-deficient megakaryocytes (Figure 6E). SFK activation was also reduced in fibrinogen-adhered Shp2-deficient megakaryocytes (Figure 6E), consistent with a previous report that Shp2 is a positive regulator of integrin signaling.45 Findings from these studies provide mechanistic insight into the cause of developmental and functional defects in Shp2-deficient megakaryocytes. Our findings are consistent with previous results demonstrating the importance of Erk1/2 and SFKs in regulating megakaryocyte development and function.32,37,44 Moreover, they suggest that Shp1 and Shp2 have distinct functions in regulating these processes.
Figure 6.

Impaired thrombopoietin and integrin signaling in Shp2-deficient megakaryocytes. Mature bone marrow-derived megakaryocytes from (A) Shp1 KO, (B) Shp2 KO, and litter-matched WT (Shp1 WT and Shp2 WT) mice were stimulated with 50 ng/mL thrombopoietin (Tpo) for 10 min at 37°C. Whole cell lysates (WCLs) were western blotted with anti-phospho-Erk1/2 (p-Erk1/2), -pan Erk1/2 (Erk1/2), and -Src p-Tyr418 antibodies. Representative blots from n = 3 independent experiments/genotype. (C) WCLs of Shp2-deficient megakaryocytes were western blotted with anti-Mpl receptor and -Erk1/2 antibodies (n = 3 independent experiments/genotype). WCLs prepared of bovine serum albumin (BSA) nonadherent and fibrinogen (fib)-adhered (D) Shp1 KO, (E) Shp2 KO, and litter-matched WT megakaryocytes were western blotted with anti-p-Erk1/2, -Erk1/2, and -Src p-Tyr418 antibodies. Representative blots from n = 3 independent experiments/genotype.
G6b conditional KO mice partially phenocopy Shp1/2 conditional DKO mice
Having established the importance of Shp1 and Shp2 in megakaryocyte and platelets, we next investigated how Shp1 and Shp2 are regulated. We focused on the ITIM-containing receptor G6b-B that is highly expressed in mature megakaryocytes and platelets (supplemental Figures 2A and 8A)20,46 and is constitutively tyrosine phosphorylated and associated with Shp1 and Shp2.19 This suggests that G6b-B anchors active Shp1 and Shp2 at the plasma membrane (supplemental Figure 1C). To determine whether G6b-B lies upstream of Shp1 and Shp2, we compared the phenotypes of MP-specific G6b conditional KO mice (MP-G6b KO) with MP-Shp1 and MP-Shp2 KO mice. This allows direct comparison of all 4 mouse models on the same PF4-Cre+ background. MP-G6b KO mice were born at Mendelian frequencies (supplemental Table 11), and megakaryocytes and platelets from these mice lacked G6b-B (supplemental Figure 8A).
Consistent with G6b-B signaling via and regulating Shp1 and Shp2, MP-G6b KO mice phenocopied multiple aspects of MP-Shp1/2 DKO mice, including: severe macrothrombocytopenia (Figure 7A; supplemental Table 12); reduced platelet surface glycoprotein expression, except for ADAM10, which was significantly elevated (supplemental Table 13); reduced megakaryocyte surface glycoprotein expression (supplemental Table 14); and reduced platelet half-life and platelet recovery following antibody-mediated depletion (Figure 7B-C). In contrast to Shp1 and Shp2 deficiency, G6b-B-deficiency did not result in aberrant megakaryocyte polyploidization ex vivo (supplemental Figure 8B). However, proplatelet formation was severely abrogated in G6b-B–deficient megakaryocytes (Figure 7D), similar to Shp2-deficient megakaryocytes (Figure 5F). Erk1/2 phosphorylation was normal in Tpo-stimulated G6b-B–deficient megakaryocytes (Figure 7E) but reduced in fibrinogen-adhered megakaryocytes (Figure 7F), consistent with normal polyploidization and reduced proplatelet formation. Collectively, these findings suggest that G6b-B signals via and regulates Shp1 and Shp2, but it does not underlie the regulatory role of Shp2 in Mpl-mediated signaling and megakaryocyte development.
Figure 7.

G6b conditional KO mice partially phenocopy Shp1/2 conditional DKO mice. (A) Platelet counts and volumes of G6b KO (n = 35) and G6b WT (n = 20) mice (mean ± SEM; ***P < .001). (B) Platelet half-life and (C) platelet recovery following immune-induced thrombocytopenia in G6b KO and WT mice (n = 3-5 mice/time point/genotype; mean ± SEM; **P < .01, ***P < .001). (D) Proplatelet formation of G6b KO megakaryocytes on fibrinogen-, fibronectin-, and collagen-coated surfaces. Representative images and quantification of megakaryocyte surface area, proplatelet mean area, and percentage of megakaryocytes forming proplatelets are shown (n = 4-6 mice/genotype/condition; mean ± SEM; **P < .01, ***P < .001; scale bar: 20 μm). (E-F) Thrombopoietin (Tpo)- and integrin-mediated Erk1/2 phosphorylation in G6b KO and WT megakaryocytes. Whole cell lysates of (E) Tpo-stimulated (50 ng/mL Tpo, 10 min) and (F) bovine serum albumin (BSA) nonadherent and fibrinogen (fib)-adhered megakaryocytes were western blotted with anti-phospho-Erk1/2 (p-Erk1/2) and -pan Erk1/2 (Erk1/2) antibodies. Representative blots from n = 3 independent experiments/genotype per condition.
Discussion
In this study, we establish that Shp1 and Shp2 are major regulators of megakaryocyte development, platelet production, and function and that they do so by distinct mechanisms. It also represents the first study in which these 2 PTPs have been simultaneously deleted in any cell lineage. This was done to investigate functional redundancy between Shp1 and Shp2 and to test the hypothesis that Shp1 and Shp2 have opposing functions in some instances. Our findings suggest both situations, as some defects were clearly exacerbated in DKO mice, such as the block in megakaryocyte polyploidization, whereas others were the opposite in the single KO models, such as integrin-mediated platelet spreading. The minimal overlap of MP-Shp1 and MP-Shp2 KO phenotypes demonstrates the disparate functional roles of Shp1 and Shp2 in the megakaryocyte lineage. Although the developmental defects caused by Shp1 and Shp2 deficiency complicate interpretation of functions of Shp1 and Shp2 in platelets, some of our findings point to these structurally related PTPs having opposing functions in platelets. Furthermore, Shp1 and Shp2 appear to have different roles in regulating integrin-mediated functions in megakaryocytes and platelets. Mice lacking the ITIM-containing receptor G6b-B in the megakaryocyte lineage phenocopied many of the features of MP-Shp1/2 DKO mice, providing genetic evidence that G6b-B signals through, and is a major regulator of, Shp1 and Shp2.
Shp1 and Shp2 have been hypothesized to be negative regulators of platelet activation through their interaction with the ITIM-containing receptor PECAM-1.47,48 However, direct evidence of this was lacking due to the absence of Shp1- and Shp2-specific inhibitors and the severity of the phenotypes exhibited by Shp1 and Shp2 KO mouse models. Pasquet et al10 previously reported that motheaten-viable mice, which express low levels of catalytically impaired Shp1, have reduced platelet reactivity to CRP. Our study now shows that this is caused by reduced GPVI expression and signaling via the SFK-Syk-PLCγ2 pathway in these platelets. Our attempt to address the question of the functions of Shp1 and Shp2 in platelets through the use of conditional KO mice provides mechanistic insights into their function in platelets but must be interpreted with caution, as targeted deletion of Shp1 and Shp2 in megakaryocytes resulted in developmental defects in megakaryocytes that are likely to affect platelets that are produced. Nevertheless, functional and biochemical defects exhibited by Shp1- and Shp2-deficient platelets suggest that Shp1 and Shp2 have opposite functions. For example, Shp1-deficient platelets were less responsive than their control counterparts to collagen and fibrinogen, whereas Shp2-deficient platelets were hyper-responsive to fibrinogen and anti-CLEC-2 antibody. Based on these results, Shp1 appears to positively regulate ITAM- and integrin-mediated responses, whereas Shp2 negatively regulates integrin- and hem-ITAM–mediated responses (supplemental Figure 9). Shp1 defects can be partially explained by reduced GPVI surface expression and SFK and Syk activity, but this is not the case for Shp2. The cause of enhanced reactivity of Shp2-deficient platelets to fibrinogen remains undefined; however, enhanced reactivity to anti-CLEC-2 antibody likely involves G6b-B.19
Functional differences exhibited by Shp1- and Shp2-deficient megakaryocytes highlight the different roles mediated by Shp1 and Shp2. The only overlapping phenotype was the block in 2N/4N ploidy and reduced αIIbβ3 surface expression, which likely reflects less mature megakaryocytes. However, these defects did not culminate in functional defects in Shp1-deficient megakaryocytes. Defects in Shp2-deficient megakaryocytes and macrothrombocytopenia in MP-Shp2 KO mice can be explained by reduced Mpl and integrin signaling (supplemental Figure 10), which were normal in Shp1-deficient megakaryocytes. The cause of the block in ploidy in Shp1-deficient megakaryocytes remains undefined but suggests a defect in the formation of the cleavage furrow and/or cytokinesis at the 2N/4N transition during the endomitotic process.49 Myosin IIA and the small GTPase RhoA are critical regulators of contractile ring assembly and cytokinesis that might be regulated by Shp1.49
Intriguingly, differences in integrin-mediated functions exhibited by Shp1- and Shp2-deficient megakaryocytes and platelets suggest that Shp1 and Shp2 have different functions in the 2 cell types. For example, Shp1-deficient megakaryocytes spread normally on fibrinogen-coated surfaces, whereas Shp1-deficient platelets did not spread to the same extent as control platelets on the same surface. In contrast, Shp2-deficient megakaryocytes exhibited markedly reduced spreading on fibrinogen, whereas Shp2-deficient platelets spread to a greater extent than control platelets on the same surface. These findings suggest that Shp1 plays little or no role in regulating αIIbβ3 integrin-mediated megakaryocyte spreading, and it positively regulates this response in platelets. In contrast, Shp2 positively regulates αIIbβ3 integrin-mediated megakaryocyte spreading and negatively regulates this response in platelets. Mechanistically, Shp1 and Shp2 positively regulate SFKs in platelets and megakaryocytes, respectively (supplemental Figures 9 and 10). However, the mechanism underlying how Shp1 and Shp2 switch functions in megakaryocytes and platelets remains undefined but likely involves interacting partners, such as G6b-B, that are differentially expressed at different stages of development and modulate Shp1 and Shp2 compartmentalization and activity.
Based on our findings, we suspect the more severe phenotype exhibited by DKO mice compared with single KO mice is mainly due to disruption of multiple signaling pathways rather than a single pathway. Deletion of Shp1 mainly affected the SFK-Syk-PLCγ2signaling pathway in platelets, whereas deletion of Shp2 affected the Ras-MAPK pathway and SFK activation in megakaryocytes. Surprisingly, functional defects did not always correlate between megakaryocytes and platelets, raising the possibility that Shp1 and Shp2 have different roles in the 2 cell types. As a consequence, deletion of G6b-B, which interacts with both Shp1 and Shp2, partially phenocopied aspects of MP-Shp1 and MP-Shp2 KO mice. For example, G6b-B- and Shp1-deficient platelets had reduced GPVI expression, and G6b-B- and Shp2-deficient platelets respond better to anti-CLEC-2 antibody. However, deletion of G6b-B did not affect Mpl-mediated Erk1/2 activation, demonstrating specificity in terms of which receptors G6b-B regulates. This partially explains why the phenotype of DKO mice is more severe than that of G6b KO mice. Another explanation is that Shp1 and Shp2 are expressed much earlier than G6b-B in megakaryocytes, and functional redundancy between G6b-B and other megakaryocyte ITIM receptors may partially compensate in its absence.
In conclusion, Shp1 and Shp2 perform mainly distinct functions in megakaryocytes and platelets, with little functional overlap. Together, they are essential for megakaryocyte development, platelet production, and function. Shp1 regulates megakaryocyte ploidy, GPVI expression, and SFK-Syk-PLCγ2 signaling in platelets, whereas Shp2 regulates Mpl and integrin signaling in megakaryocytes and integrin- and hem-ITAM–mediated responses in platelets. Differences in their functions in megakaryocytes and platelets likely reflect differences in compartmentalization, mediated by receptors and adaptors, and substrate specificity. Findings from this study suggest that the ITIM-containing receptor G6b-B likely signals through, and is a major regulator of, Shp1 and Shp2 in mature megakaryocytes and platelets.
Supplementary Material
Acknowledgments
The authors thank all members of the Biomedical Services Unit for exceptional mouse husbandry, and Alice Rees for assistance with analysis of spread platelets.
This work was supported by British Heart Foundation (BHF) Project Grants PG/10/73/28420 and PG/07/034/22775. B.G.N. is a Canada Research Chair, Tier 1, and is partially supported by funds from the Ontario Ministry of Health and Long Term Care, the Princess Margaret Foundation, and National Cancer Institute at the National Institutes of Health R37CA49132. A.M. and J.M. are BHF Postdoctoral Researchers, S.P.W. is a BHF Chair, and Y.A.S. is a BHF Senior Research Fellow (FS/13/1/29894).
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.M. performed experiments, analyzed data, and wrote and revised the paper; J.M. performed experiments, analyzed data, and wrote parts of and revised the paper; Y.-J.W. performed experiments and analyzed data; S.H. maintained mouse colonies, performed experiments, and analyzed data; B.G.N. provided mouse models, contributed intellectually and revised the paper; S.P.W. contributed intellectually and revised the paper; Y.A.S. performed experiments, analyzed data, and wrote and revised the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for Y.-J.W. is Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Headington, Oxford, United Kingdom.
Correspondence: Yotis A. Senis, Centre for Cardiovascular Sciences, Institute of Biomedical Research, School of Clinical and Experimental Medicine, College of Medical and Dental Sciences, University of Birmingham, Birmingham, B15 2TT, United Kingdom; e-mail: y.senis@bham.ac.uk.
References
- 1.Pao LI, Badour K, Siminovitch KA, Neel BG. Nonreceptor protein-tyrosine phosphatases in immune cell signaling. Annu Rev Immunol. 2007;25:473–523. doi: 10.1146/annurev.immunol.23.021704.115647. [DOI] [PubMed] [Google Scholar]
- 2.Daëron M, Jaeger S, Du Pasquier L, Vivier E. Immunoreceptor tyrosine-based inhibition motifs: a quest in the past and future. Immunol Rev. 2008;224:11–43. doi: 10.1111/j.1600-065X.2008.00666.x. [DOI] [PubMed] [Google Scholar]
- 3.Ivins Zito C, Kontaridis MI, Fornaro M, Feng GS, Bennett AM. SHP-2 regulates the phosphatidylinositide 3′-kinase/Akt pathway and suppresses caspase 3-mediated apoptosis. J Cell Physiol. 2004;199(2):227–236. doi: 10.1002/jcp.10446. [DOI] [PubMed] [Google Scholar]
- 4.Kontaridis MI, Eminaga S, Fornaro M, Zito CI, Sordella R, Settleman J, Bennett AM. SHP-2 positively regulates myogenesis by coupling to the Rho GTPase signaling pathway. Mol Cell Biol. 2004;24(12):5340–5352. doi: 10.1128/MCB.24.12.5340-5352.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamashita T, Suzuki R, Backlund PS, Yamashita Y, Yergey AL, Rivera J. Differential dephosphorylation of the FcRgamma immunoreceptor tyrosine-based activation motif tyrosines with dissimilar potential for activating Syk. J Biol Chem. 2008;283(42):28584–28594. doi: 10.1074/jbc.M802679200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yusa S, Campbell KS. Src homology region 2-containing protein tyrosine phosphatase-2 (SHP-2) can play a direct role in the inhibitory function of killer cell Ig-like receptors in human NK cells. J Immunol. 2003;170(9):4539–4547. doi: 10.4049/jimmunol.170.9.4539. [DOI] [PubMed] [Google Scholar]
- 7.Shultz LD, Schweitzer PA, Rajan TV, et al. Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell. 1993;73(7):1445–1454. doi: 10.1016/0092-8674(93)90369-2. [DOI] [PubMed] [Google Scholar]
- 8.Wo, Tsui H, Siminovitch KA, de Souza L, Tsui FW. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet. 1993;4(2):124–129. doi: 10.1038/ng0693-124. [DOI] [PubMed] [Google Scholar]
- 9.Kozlowski M, Mlinaric-Rascan I, Feng GS, Shen R, Pawson T, Siminovitch KA. Expression and catalytic activity of the tyrosine phosphatase PTP1C is severely impaired in motheaten and viable motheaten mice. J Exp Med. 1993;178(6):2157–2163. doi: 10.1084/jem.178.6.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pasquet JM, Quek L, Pasquet S, Poole A, Matthews JR, Lowell C, Watson SP. Evidence of a role for SHP-1 in platelet activation by the collagen receptor glycoprotein VI. J Biol Chem. 2000;275(37):28526–28531. doi: 10.1074/jbc.M001531200. [DOI] [PubMed] [Google Scholar]
- 11.Yang W, Klaman LD, Chen B, et al. An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev Cell. 2006;10(3):317–327. doi: 10.1016/j.devcel.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 12.Arrandale JM, Gore-Willse A, Rocks S, et al. Insulin signaling in mice expressing reduced levels of Syp. J Biol Chem. 1996;271(35):21353–21358. doi: 10.1074/jbc.271.35.21353. [DOI] [PubMed] [Google Scholar]
- 13.Qu CK, Shi ZQ, Shen R, Tsai FY, Orkin SH, Feng GS. A deletion mutation in the SH2-N domain of Shp-2 severely suppresses hematopoietic cell development. Mol Cell Biol. 1997;17(9):5499–5507. doi: 10.1128/mcb.17.9.5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saxton TM, Ciruna BG, Holmyard D, et al. The SH2 tyrosine phosphatase shp2 is required for mammalian limb development. Nat Genet. 2000;24(4):420–423. doi: 10.1038/74279. [DOI] [PubMed] [Google Scholar]
- 15.Dhanjal TS, Pendaries C, Ross EA, et al. A novel role for PECAM-1 in megakaryocytokinesis and recovery of platelet counts in thrombocytopenic mice. Blood. 2007;109(10):4237–4244. doi: 10.1182/blood-2006-10-050740. [DOI] [PubMed] [Google Scholar]
- 16.Wong C, Liu Y, Yip J, et al. CEACAM1 negatively regulates platelet-collagen interactions and thrombus growth in vitro and in vivo. Blood. 2009;113(8):1818–1828. doi: 10.1182/blood-2008-06-165043. [DOI] [PubMed] [Google Scholar]
- 17.Washington AV, Schubert RL, Quigley L, et al. A TREM family member, TLT-1, is found exclusively in the alpha-granules of megakaryocytes and platelets. Blood. 2004;104(4):1042–1047. doi: 10.1182/blood-2004-01-0315. [DOI] [PubMed] [Google Scholar]
- 18.Xue J, Zhang X, Zhao H, et al. Leukocyte-associated immunoglobulin-like receptor-1 is expressed on human megakaryocytes and negatively regulates the maturation of primary megakaryocytic progenitors and cell line. Biochem Biophys Res Commun. 2011;405(1):128–133. doi: 10.1016/j.bbrc.2010.12.140. [DOI] [PubMed] [Google Scholar]
- 19.Mazharian A, Wang YJ, Mori J, et al. Mice lacking the ITIM-containing receptor G6b-B exhibit macrothrombocytopenia and aberrant platelet function. Sci Signal. 2012;5(248):ra78. doi: 10.1126/scisignal.2002936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Senis YA, Tomlinson MG, García A, et al. A comprehensive proteomics and genomics analysis reveals novel transmembrane proteins in human platelets and mouse megakaryocytes including G6b-B, a novel immunoreceptor tyrosine-based inhibitory motif protein. Mol Cell Proteomics. 2007;6(3):548–564. doi: 10.1074/mcp.D600007-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Senis YA, Antrobus R, Severin S, et al. Proteomic analysis of integrin alphaIIbbeta3 outside-in signaling reveals Src-kinase-independent phosphorylation of Dok-1 and Dok-3 leading to SHIP-1 interactions. J Thromb Haemost. 2009;7(10):1718–1726. doi: 10.1111/j.1538-7836.2009.03565.x. [DOI] [PubMed] [Google Scholar]
- 22.Barford D, Neel BG. Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure. 1998;6(3):249–254. doi: 10.1016/s0969-2126(98)00027-6. [DOI] [PubMed] [Google Scholar]
- 23.Eck MJ, Pluskey S, Trüb T, Harrison SC, Shoelson SE. Spatial constraints on the recognition of phosphoproteins by the tandem SH2 domains of the phosphatase SH-PTP2. Nature. 1996;379(6562):277–280. doi: 10.1038/379277a0. [DOI] [PubMed] [Google Scholar]
- 24.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92(4):441–450. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 25.Pluskey S, Wandless TJ, Walsh CT, Shoelson SE. Potent stimulation of SH-PTP2 phosphatase activity by simultaneous occupancy of both SH2 domains. J Biol Chem. 1995;270(7):2897–2900. doi: 10.1074/jbc.270.7.2897. [DOI] [PubMed] [Google Scholar]
- 26.Yang J, Cheng Z, Niu T, Liang X, Zhao ZJ, Zhou GW. Structural basis for substrate specificity of protein-tyrosine phosphatase SHP-1. J Biol Chem. 2000;275(6):4066–4071. doi: 10.1074/jbc.275.6.4066. [DOI] [PubMed] [Google Scholar]
- 27.Yang J, Liu L, He D, et al. Crystal structure of human protein-tyrosine phosphatase SHP-1. J Biol Chem. 2003;278(8):6516–6520. doi: 10.1074/jbc.M210430200. [DOI] [PubMed] [Google Scholar]
- 28.Pao LI, Lam KP, Henderson JM, et al. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity. 2007;27(1):35–48. doi: 10.1016/j.immuni.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 29.Fornaro M, Burch PM, Yang W, et al. SHP-2 activates signaling of the nuclear factor of activated T cells to promote skeletal muscle growth. J Cell Biol. 2006;175(1):87–97. doi: 10.1083/jcb.200602029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tiedt R, Schomber T, Hao-Shen H, Skoda RC. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood. 2007;109(4):1503–1506. doi: 10.1182/blood-2006-04-020362. [DOI] [PubMed] [Google Scholar]
- 31.Ault KA, Knowles C. In vivo biotinylation demonstrates that reticulated platelets are the youngest platelets in circulation. Exp Hematol. 1995;23(9):996–1001. [PubMed] [Google Scholar]
- 32.Mazharian A, Thomas SG, Dhanjal TS, Buckley CD, Watson SP. Critical role of Src-Syk-PLCγ2 signaling in megakaryocyte migration and thrombopoiesis. Blood. 2010;116(5):793–800. doi: 10.1182/blood-2010-03-275990. [DOI] [PubMed] [Google Scholar]
- 33.Senis YA, Atkinson BT, Pearce AC, et al. Role of the p110delta PI 3-kinase in integrin and ITAM receptor signalling in platelets. Platelets. 2005;16(3-4):191–202. doi: 10.1080/09537100400016711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pearce AC, Senis YA, Billadeau DD, Turner M, Watson SP, Vigorito E. Vav1 and vav3 have critical but redundant roles in mediating platelet activation by collagen. J Biol Chem. 2004;279(52):53955–53962. doi: 10.1074/jbc.M410355200. [DOI] [PubMed] [Google Scholar]
- 35.McCarty OJ, Larson MK, Auger JM, et al. Rac1 is essential for platelet lamellipodia formation and aggregate stability under flow. J Biol Chem. 2005;280(47):39474–39484. doi: 10.1074/jbc.M504672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Senis YA, Tomlinson MG, Ellison S, et al. The tyrosine phosphatase CD148 is an essential positive regulator of platelet activation and thrombosis. Blood. 2009;113(20):4942–4954. doi: 10.1182/blood-2008-08-174318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mazharian A, Watson SP, Severin S. Critical role for ERK1/2 in bone marrow and fetal liver-derived primary megakaryocyte differentiation, motility, and proplatelet formation. Exp Hematol. 2009;37(10):1238–1249 e5. doi: 10.1016/j.exphem.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bender M, Hofmann S, Stegner D, et al. Differentially regulated GPVI ectodomain shedding by multiple platelet-expressed proteinases. Blood. 2010;116(17):3347–3355. doi: 10.1182/blood-2010-06-289108. [DOI] [PubMed] [Google Scholar]
- 39.Kanaji S, Kanaji T, Furihata K, Kato K, Ware JL, Kunicki TJ. Convulxin binds to native, human glycoprotein Ib α. J Biol Chem. 2003;278(41):39452–39460. doi: 10.1074/jbc.M300199200. [DOI] [PubMed] [Google Scholar]
- 40.Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104(6):1606–1615. doi: 10.1182/blood-2004-04-1257. [DOI] [PubMed] [Google Scholar]
- 41.Auger JM, Kuijpers MJ, Senis YA, Watson SP, Heemskerk JW. Adhesion of human and mouse platelets to collagen under shear: a unifying model. FASEB J. 2005;19(7):825–827. doi: 10.1096/fj.04-1940fje. [DOI] [PubMed] [Google Scholar]
- 42.Thon JN, Montalvo A, Patel-Hett S, et al. Cytoskeletal mechanics of proplatelet maturation and platelet release. J Cell Biol. 2010;191(4):861–874. doi: 10.1083/jcb.201006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sabri S, Jandrot-Perrus M, Bertoglio J, et al. Differential regulation of actin stress fiber assembly and proplatelet formation by α2β1 integrin and GPVI in human megakaryocytes. Blood. 2004;104(10):3117–3125. doi: 10.1182/blood-2003-12-4398. [DOI] [PubMed] [Google Scholar]
- 44.Mazharian A, Ghevaert C, Zhang L, Massberg S, Watson SP. Dasatinib enhances megakaryocyte differentiation but inhibits platelet formation. Blood. 2011;117(19):5198–5206. doi: 10.1182/blood-2010-12-326850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oh ES, Gu H, Saxton TM, et al. Regulation of early events in integrin signaling by protein tyrosine phosphatase SHP-2. Mol Cell Biol. 1999;19(4):3205–3215. doi: 10.1128/mcb.19.4.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Macaulay IC, Tijssen MR, Thijssen-Timmer DC, et al. Comparative gene expression profiling of in vitro differentiated megakaryocytes and erythroblasts identifies novel activatory and inhibitory platelet membrane proteins. Blood. 2007;109(8):3260–3269. doi: 10.1182/blood-2006-07-036269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cicmil M, Thomas JM, Leduc M, Bon C, Gibbins JM. Platelet endothelial cell adhesion molecule-1 signaling inhibits the activation of human platelets. Blood. 2002;99(1):137–144. doi: 10.1182/blood.v99.1.137. [DOI] [PubMed] [Google Scholar]
- 48.Jones KL, Hughan SC, Dopheide SM, Farndale RW, Jackson SP, Jackson DE. Platelet endothelial cell adhesion molecule-1 is a negative regulator of platelet-collagen interactions. Blood. 2001;98(5):1456–1463. doi: 10.1182/blood.v98.5.1456. [DOI] [PubMed] [Google Scholar]
- 49.Bluteau D, Lordier L, Di Stefano A, et al. Regulation of megakaryocyte maturation and platelet formation. J Thromb Haemost. 2009;7(Suppl 1):227–234. doi: 10.1111/j.1538-7836.2009.03398.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.