

Abstract
Two different approaches to synthesize oligonucleotides containing the 2′-deoxyguanosine adducts formed by nitropyrenes are described. A direct reaction of an unmodified oligonucleotide with an activated nitropyrene derivative is a convenient biomimetic approach for generating the major adducts in DNA. A total synthetic approach, by contrast, involves several synthetic steps, including Buchwald-Hartwig Pd-catalyzed coupling, but can be used for incorporating both the major and minor adducts in DNA in high yield.
Keywords: nitropyrene, dinitropyrene, DNA adducts
INTRODUCTION
Nitropyrenes are ubiquitous in the environment and have been detected in urban air particulates, coal fly ash, and automobile exhaust.[1,2] 1-Nitropyrene (1-NP) is one of the major mutagenic components of diesel particulates, whereas the dinitropyrenes (DNPs) (substituted at 1,3-, 1,6-, and 1,8-positions) are present in much lower concentrations.[3,4] 1-NP accounts for 25% of the mutagenicity of diesel exhausts. The DNPs, though present in much smaller quantity in diesel emissions, are much more mutagenic than 1-NP.[4] All the mono- and dinitropyrenes are tumorigenic in experimental animals.[5,6] 1,6-DNP is the most potent carcinogen in this group, whereas 1,8-DNP is the most mutagenic by several assays. The major DNA adducts formed by 1-NP and the 1,6- or 1,8-DNP both in vitro and in vivo are the C8 2′-deoxyguanosine (dG) adducts N-(deoxyguanosine-8-yl)-1-aminopyrene (dG-C8-AP) and N-(deoxyguanosine-8-yl)-1-amino-6(or 8)-nitropyrene (dG-C8-amino-6(or 8)-NP) (SCHEME 1).[4] For 1-NP, two N2 dG adducts have also been isolated in vitro (SCHEME 1).[4]
SCHEME 1.
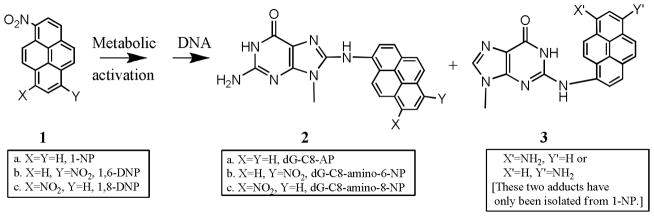
DNA adducts formed by nitropyrenes.
Incorporation of these DNA adducts in desired DNA sequences is an important step for investigation of mutagenicity, repair, and biophysical properties of the lesions. In a prior work, we have successfully synthesized oligonucleotides containing dG-C8-AP by allowing the unmodified oligonucleotides in double stranded form to react with N-hydroxy-1-aminopyrene.[7] However, we could not detect any N2 dG adduct by this biomimetic approach. In recent years Buchwald-Hartwig palladium-catalyzed amination[8,9] has been extensively employed to synthesize a series of carcinogen-DNA adducts. [10–14] We and others have used this key step to generate decent yield of both the major and minor dG adducts of 1-NP.[15,16] In the current work, we have converted these dG adducts into the corresponding phosphoramidites and incorporated them into DNA. We determined that the strategy to introduce the major adducts in preformed oligonucleotides in duplex conditions, as noted in the case of 1-NP,[7] is also highly efficient for dinitropyrenes. We compared the incorporation of the nitropyrene and dinitropyrene adducts in a mutagenic hotspot CpG repetitive sequence d(CGCG*CG) by both approaches.
RESULTS
Postoligomerization Strategy
A biomimetic approach for a direct reaction with an unmodified duplex hexamer is shown in SCHEME 2, in which the low pH and duplex nature of the oligonucleotide are the important criteria for a reaction that occurs almost exclusively with dG. 1-Amino-6-nitropyrene and 1-amino-8-nitropyrene, synthesized as reported by Purohit and Basu,[17] were oxidized to the corresponding nitrosonitropyrenes using m-chloroperoxybenzoic acid. Each nitrosonitropyrene was converted to the N-hydroxy derivative in situ in the presence of ascorbic acid, which was allowed to react with the self-complementary oligonucleotide d(CGCGCG) in aqueous DMF at pH 5.2. DNA adduction was HPLC monitored by removing an aliquot and injecting it onto a reverse-phase column after the organic reactants were removed by chloroform extraction. The adducted hexamers eluted more slowly than the unmodified oligonucleotide, and though three adducted peaks eluted consistent with the presence of three guanines, one of the peaks was clearly dominant. Highest yield of the products (~25%) occurred at 16 h when the products were HPLC purified. FIGURE 1 shows a typical adduction profile after 16 h at 260 and 450 nm (unlike unmodified DNA, the nitro- and/or aminopyrene-containing peaks are detectable at 450 nm).
SCHEME 2.
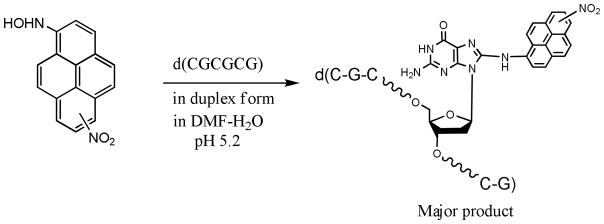
Postoligomerization strategy to introduce DNA adducts.
FIGURE 1.

Reverse-phase HPLC profile of crude reaction mixture of d(CGCGCG) with N-hydroxy-1-amino-6-nitropyrene after stirring for 16 h at pH 5.2 in DMF: H2O (1:9). The excess organic reagents were removed by chloroform extraction. The peaks were monitored at 260 nm (panel A) and 450 nm (panel B). Peak 1 shows the unreacted hexamer, whereas peaks 2, 3, and 4 show the adducted hexanucleotide peaks. Peak 2 was identified as d(CGCG*CG) by piperidine cleavage experiment.
Enzymatic digestion followed by HPLC analysis established that the major adducted hexamers contained the dG-C8-amino-6(or 8)-NP, which co-eluted with an authentic sample. A portion of the major adducted hexamer was 32P-radiolabeled and cleaved at the site of the adduct by hot piperidine, which was compared with Maxam-Gilbert ‘G’ reaction[17] of the unmodified hexamer on a denaturing polyacrylamide gel. With this approach, we determined that the major peak in each case contained the C8 adduct at the second G, where the cleavage occurred. The minor peaks corresponded to the C8 adduct at two other guanines. It is important to emphasize that the presence of N2 guanine adduct, which should be refractory the piperidine cleavage, was not detected. Each adducted hexamer, d(CGCG*CG), also gave a monoisotopic mass of 2051.6 Da by mass spectral analysis with electrospray ionization as expected (TABLE 1).
TABLE 1.
ESI-MS Characterization of the d(CGCG*CG)
| Chemical | Adduct | Synthetic approach | Calculated (M-H) m/z (Da) | Found (M-H) m/z (Da) |
|---|---|---|---|---|
| 1,6-DNP | dG-C8-amino-6-NP | Direct reaction with N-hydroxy-1-amino-6-nitropyrene | 2051.61 | 2051.39 |
| 1,8-DNP | dG-C8-amino-8-NP | Direct reaction with N-hydroxy-1-amino-8-nitropyrene | 2051.61 | 2051.52 |
| 1-NP | dG-C8-AP | Direct reaction with N-hydroxy-1-aminopyrene | 2006.60 | 2006.45 |
| 1-NP | dG-C8-AP | Total synthesis | 2006.60 | 2006.42 |
| 1-NP | dG-N2-8-AP | Total synthesis | 2006.60 | 2006.50 |
We believe that in this reaction non-covalent intercalation played an important role, which was manifested in the most facile reaction at the duplex middle region of the self-complementary hexamer. The initial N7 guanine adduct subsequently rearranged to the C8 guanine adduct.[18]
Total Synthesis by Using a Phosphoramidite Reagent
The Buchwald-Hartwig C-N bond formation strategy[8,9] has been successfully applied by many researchers to synthesize various DNA adducts.[10–14] For the synthesis of the C8 dG adduct, we followed the synthetic strategy of Gillet and Schärer[15] to convert dG (4) to dG-C8-AP (7) in eight steps (SCHEME 3). In our hands, the yield in each step, including the Pd2(dba)3 catalyzed coupling, was similar to the published procedure. The C8 adduct 7 was converted to the protected phosphoramidite monomer in three steps (SCHEME 3).
SCHEME 3.

Synthesis of fully protected dG-C8-AP
Reaction conditions: a. Aqueous NBS, AcCN (78%); b. TBDMS-Cl, imidazole, DMF (90%); c. Bn-OH, PPh3, DIAD, dioxane (70%); d. DMT-Cl, pyridine (90%); e. Pd2(dba)3, BINAP, toluene, NaOtBu, 1-aminopyrene (48%); f. 0.02 M HCl in methanol (90%); g. Acetic acid, TBAF, THF (85%); h. Pd/C, cyclohexane, ethanol (90%); i. iBu anhydride, pyridine, TMCS (75%); j. DMT-Cl, DMAP, pyridine (84%); k. 2-Cyanoethyl-N,N-diisopropylchlorophosphoramidite, TEA, CH2Cl2, r.t., 1h (55%).
Similarly, a protected monomer (9) was subjected to Buchwald-Hartwig amination and the N2 dG adduct derivative (10) was isolated in high yield as we reported in our communication Chakraborti et al.[16] (SCHEME 4). Following removal of the hydroxyl protecting groups, the adduct derivative 11 was converted to fully protected adduct monomer 12 in good yield.
SCHEME 4.

Synthesis of a fully protected monomer that can be converted to the N2-dG adduct after oligonucleotide synthesis.
Reaction conditions: a. Pd(OAc)2, Cs2CO3, BINAP, toluene (88%); b. TBAF, THF (80%); c. DMT-Cl, DMAP, TEA, pyridine (80%); d. 2-Cyanoethyl-N,N-diisopropylchlorophosphoramidite, TEA, CH2Cl2, r.t., 1h (60%).
The protected monomers 8 and 12 were used to synthesize modified hexamers, d(CGCG*CG), by standard protocol in an automated DNA synthesizer, except that the coupling time for these monomers was increased. Deprotection of the oligonucleotides was carried out with concentrated NH4OH in the presence of β-mercaptoethanol. For the dG-C8-AP, the final product d(CGCG*CG) was the major peak in reverse-phase HPLC, which gave a monoisotopic mass of 2006.42 Da (theoretical mass 2006.60 Da) (TABLE 1). For the N2 adduct, on the other hand, the isolated product was the precursor of the desired product, which was treated with palladium black to reduce the 8-nitro group and to deprotect the benzyl protecting group at the O6 position. The final product was isolated by reverse-phase HPLC. As shown in FIGURE 3, the reduced product eluted approximately 4 minutes earlier. ESI-MS analysis of the reduced product at 2006.50 Da was in excellent agreement with the theoretical monoisotopic mass of 2006.60 (TABLE 1). Indeed, as shown in TABLE 1, both the postoligomerization strategy as well as total synthesis generated the hexamers that gave monoisotopic mass by electrospray ionization MS consistent with theoretical calculations.
FIGURE 3.
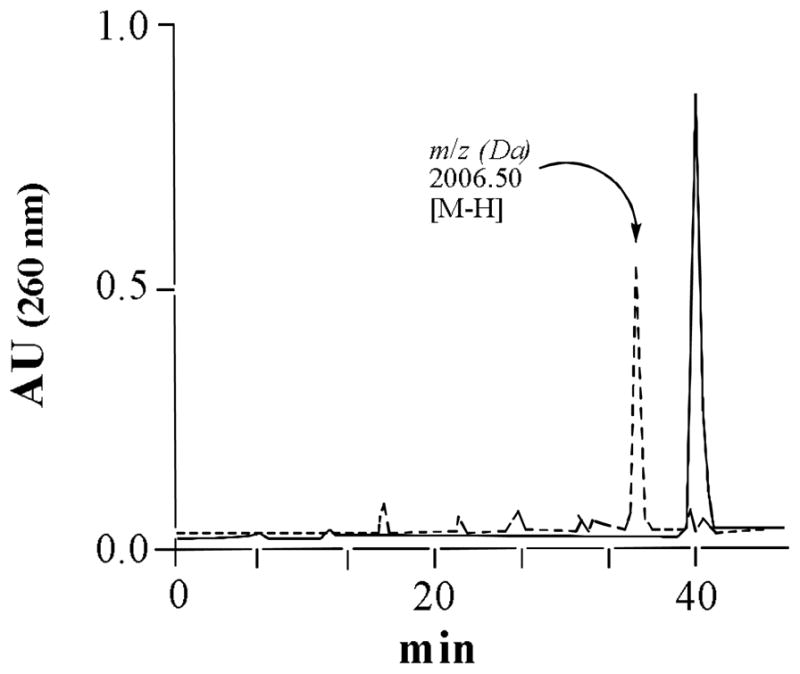
Reverse-phase HPLC chromatograms (monitored at 260 nm): Before (solid line) and after (dotted line) Pd black/cyclohexadiene reduction of d(CGCG*CG) synthesized with monomer 12.
In conclusion, using two different strategies, we synthesized the DNA adducts formed by 1-NP, 1,6-DNP, and 1,8-DNP, three carcinogenic nitropyrenes, in a mutagenic hotspot sequence, d(CGCG*CG). The biomimetic approach of the reaction between the unmodified oligonucleotide with the N-hydroxy pyrene derivatives worked well for preparing the major DNA adducts, whereas a more involved total synthesis approach enabled us to synthesize both the major and minor adducts.
EXPERIMENTAL
General procedures
All starting materials, reagents and solvents were of commercial grade and used as such unless otherwise specified. Anhydrous solvents were purchased from Aldrich and THF was dried by standard methods. NMR (1H and 13C) spectra were recorded on a Bruker AC-400 spectrometer. Samples prepared for NMR analysis were dissolved in CDCl3 or DMSO-d6. Chemical shifts are reported in δ ppm relative to TMS in the proton spectra and to the deuterated solvent in the carbon spectra. LCMS (Electrospray ionization) was recorded on a Micromass Quattro II triple quadrupole mass spectrometer in acetonitrile mobile phase and the cone voltage set to 30 V. High resolution MS spectras were obtained either from Nebraska Center for Mass Spectrometry, University of Nebraska, Lincoln or Yale Cancer Center Mass Spectrometry Source. Thin-layer chromatography (TLC) was performed on silica gel sheets containing fluorescent indicator. Components were visualized by UV light. Chromatographic separations were carried out on silica gel 60 μm (230–400 mesh). Preparative TLC was performed on fluorescent silica gel glass backed plates (250 μm). All experiments dealing with moisture and air-sensitive compounds were conducted under dry nitrogen. All new products showed a single spot on TLC analysis, after purification.
Synthesis of O6-benzyl-8-bromo-3′,5′-O-bis(tert-butyldimethylsilyl)-N2-dimethoxytrityl-2′-deoxyguanosine (5), 8-(N2-aminopyrene)-O6-benzyl-3′,5′-O-bis(tert-butyldimethylsilyl)-2′-deoxyguanosine (6) and N-(deoxyguanosine-8-yl)-1-aminopyrene (7) essentially followed a method reported by Gillet and Schärer.[15]
2,8-Diisobutyryl-8-(1-aminopyrenyl)-5′-O-(4,4′dimethoxytrityl)-3′-O-[N,N′-diisopropylamino(2-cyanoethoxy)phosphinyl]-2′-deoxyguanosine (8)
Compound 7 (60 mg, 0.2 mmol) was twice dried with pyridine and suspended in 2 mL of dry pyridine to which a large excess of chlorotrimethylsilane (0.2 mL, ~2 mmol) was added. The mixture was cooled for 30 min in an ice-water bath. Isobutyric anhydride (0.2 mL, ~1 mmol) was added, and the mixture was left standing at ambient temperature for 2 h. The mixture was cooled again in an ice-cold water bath and quenched with water. A solution of ammonium hydroxide (1 mL) was added and the solvent was removed by evaporation. The residue was dissolved in 2 mL water and extracted with diethyl ether (5 mL). The precipitate appearing in the aqueous phase was collected, dried twice with pyridine, and dissolved in 0.5 mL dry pyridine. Two equivalent 4,4′-dimethoxytrityl chloride (143 mg, 0.4 mmol) and DMAP (0.01 mmol) were added and the solution was stirred for 2 h at room temperature when TLC monitoring showed that the reaction was complete. A saturated NaHCO3 solution was added and the solution was extracted three times with methylene chloride. The combined organic layers were dried over Mg2SO4 and the solvent was removed in vacuo. Purification of the residue by flash chromatography on silica gel with 1% methanol in methylene chloride containing 0.5% triethylamine gave the desired 2,8-diisobutyryl-5′-O-DMT-protected product in 84% yield.
The 2,8-diisobutyryl-8-(1-aminopyrenyl)-5′-dimethoxytrityl-2′-deoxyguanosine (95 mg, 0.1 mmol) was coevaporated with dry toluene (2 ×10 mL), dissolved in dry methylene chloride (5 mL), and treated with triethylamine (0.1 mL) followed by 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (0.035 mL, 0.15 mmol). The reaction mixture was stirred at room temperature for 1 h under nitrogen and then diluted with methylene chloride solution containing 1% TEA (25 mL). The mixture was washed with aqueous saturated NaHCO3 solution, dried over Na2SO4, filtered, concentrated under reduced pressure, and the residue was purified by flash chromatography on silica, eluting with 1% methanol in methylene chloride containing 0.5% pyridine, and provided 8 (62 mg, 55% yield) as a pale yellow solid.
1H NMR (DMSO-d6): δ 8.76 (s, 1H, NH), 8.68-8.01 (m, 9H; pyrene), 7.85-7.20 (m, 9H), 6.38 (d, J=6 Hz, 4H), 6.31 (t, J = 6.5 Hz, 1H; H-1′), 4.32 (m, 1H; H-3′), 4.15 (m, 2H, OCH2), 3.85 (m, 1H; H-4′), 3.75 (s, 6H), 3.62 (m, 1H), 3.51 (m, 2H; H-5′), 3.42 (m, 2H), 2.86 (t, J=6 Hz, 2H), 2.58 (m, 1H; H-2′β), 2.42 (m, 2H), 2.20-2.32 (m, 1H; H-2′α),1.32-1.01 (m, 24H). 31P-NMR: 148.5 (diastereomeric pair).
HRMS: 1124.0627 (M − H) (Calcd. C64H69N8O9P minus H: 1124.2607)
2-(8-nitro-1-pyrenyl)-O6-benzyl-3′, 5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine (10)
Compound 9 (0.1 g, 0.17 mmol), 1-bromo-8-nitropyrene (70 mg, 0.21 mmol), Cs2CO3 (80 mg, 0.23 mmol), Pd(OAc)2 (4 mg, 0.01 mmol) and BINAP (80 mg, 0.12 mmol) were weighed out in a vial and flushed with argon. Anhydrous toluene (1.5 mL) was added and the flask was refilled with argon before it was sealed. The mixture was allowed to stir on a sand bath at 80 °C for 8–16 hours. It was subsequently filtered through a celite pad, concentrated under reduced pressure, and chromatographed with 8 % ethyl acetate in hexanes.
1H NMR (CDCl3): δ 8.98 (d, J = 9.6 Hz, 1H), 8.83 (d, J = 8.6 Hz, 1H), 8.69 (d, J = 8.6 Hz, 1H), 8.52 (d, J = 9.6 Hz, 1H), 8.28 (d, J = 8.2 Hz, 1H), 8.22 (d, J = 8.6 Hz, 1H), 8.14 (d, J = 8.6 Hz, 1H), 8.06 (s, 1H), 8.03 (d, J = 8.9 Hz, 1H), 7.79 (s, 1H; H-8), 7.41 (m, 2H; phenyl), 7.29 (m, 3H; phenyl), 6.44 (t, J = 6.5 Hz, 1H; H-1′), 5.60 (dd, J = 12.3, 14.0 Hz, 2H; CH2Ph), 4.58 (m, 1H; H-3′), 4.02(q, J = 3.4 Hz, 1H; H-4′), 3.80 (dq, J = 6.2, 12.3 Hz, 2H; H-5′), 2.58 (m, 1H; H-2′β), 2.42 (m, 1H; H-2′α), 0.09 (s, 3H; Si-Me), 0.094 (s, 3H; Si-Me), 0.93 (s, 9H; Me3C), 0.92 (s, 9H; Me3C), 0.11 (s, 3H; Si-Me), 0.10 (s, 3H; Si-Me). 13C NMR: δ 160.7, 155.7, 153.2, 142.0, 138.6, 136.3, 135.9, 135.6, 130.9, 128.4, 128.3, 128.0, 126.9, 125.2, 124.0, 123.1, 121.6, 121.0, 120.9, 117.2, 87.8, 83.9, 76.8, 72.0, 68.4, 62.9, 41.4, 29.99, 25.8, 18.4, 18.0, −4.65, −4.7.
HRFAB MS: 831.3748 (M+H) (Calcd. C45H55N6O6Si2: 831.3722).
2-(8-nitro-1-pyrenyl)-O6-benzyl-2′-deoxyguanosine (11)
To a stirred solution of 5 (100 mg, 0.12 mmol) in THF (3 mL) was added tetrabutylammonium fluoride (0.36 mmol, 1.0 M solution in THF). The mixture was stirred at room temperature for 1.5 h, after which the solvent was removed under reduced pressure. The crude product was purified by chromatography on silica, with 4% methanol in chloroform. The product 11 (67 mg, 95% yield) was isolated as pale yellow oil.
1H NMR (CDCl3): δ 10.11 (s, 1H, NH), 8.73-8.16 2.29 (m, 9H; H-8 and pyrene), 7.34 (m, 2H; phenyl), 7.29 (m, 3H; phenyl), 6.33 (t, J = 6.5 Hz, 1H; H-1′), 5.48 (s, 2H; CH2Ph), 5.26 (m, 1H, CH-OH), 4.86 (m, 1H, CH2-OH), 4.32 (m, 1H; H-3′), 3.83 (m, 1H; H-4′), 3.47 (m, 2H; H-5′), 2.68 (m, 1H; H-2′β), 2.29 (m, 1H; H-2′α). 13C NMR: δ 169.9, 150.6, 150.4, 137.2, 132.9, 131.9, 131.8, 128.0, 127.9, 127.4, 125.9, 125.8, 125.7, 125.6, 125.5, 125.4, 125.1, 123.1, 122.0, 119.5, 119.4., 88.5, 83.4, 71.4, 62.5, 62.4, 61.3, 55.8, 49.47, 24.5, 23.4.
HRES MS: 625.1819 (M+Na) (Calcd. C33H26N6O6Na: 625.1812).
N2-(8-nitro-1-pyrenyl)-O6-benzyl-5′-O-(4,4′dimethoxytrityl)-3′-O-[N,N′-diisopropylamino(2-cyanoethoxy)phosphinyl]-2′-deoxyguanosine (12)
The method was nearly identical to the synthesis of 8.
1H NMR (DMSO-d6): δ 10.12 (s, 1H, NH), 8.73-8.16 (m, 9H; H-8 and pyrene), 7.35 (m, 2H; phenyl), 7.28 (m, 3H, phenyl), 6.90-6.85 (m, 9H), 6.38 (d, J=6 Hz, 4H), 6.31 (t, J = 6.5 Hz, 1H; H-1′), 5.52 (s, 2H; CH2Ph), 4.32 (m, 1H; H-3′), 4.15 (m, 2H, OCH2), 3.85 (m, 1H; H-4′), 3.51 (m, 2H; H-5′), 3.46 (m, 2H), 3.35 (s, 6H), 2.86 (t, J=6 Hz, 2H), 2.68 (m, 1H; H-2′β), 2.34 (m, 1H; H-2′α), 1.05 (dd, 12H). 31P-NMR: 147.5 (diastereomeric pair).
HRMS: 1103.50 (M − H) (Calcd. C63H61N8O9P minus H: 1103.4221)
Oligonucleotide synthesis
Phosphoramidite 8 and 12 were used to synthesize modified oligonucleotides in automated synthesizer as per the manufacturer’s instructions, except a prolonged coupling time of 20 minutes was used for these monomers. Deprotection of the oligonucleotides was carried out with concentrated ammonium hydroxide for 20 h at 55 °C in the presence of 0.25 M β-mercaptoethanol. After drying to remove excess ammonia, the reaction mixture was desalted on a Sep-PAK C18 cartridge and purified by reverse-phase HPLC.
Nitroreduction and O6-benzyl deprotection
The modified oligonucleotide was suspended in a solution of 35% formamide, 35% ethanol, 10% ethyl acetate, 20% cyclohexadiene, and 0.2 mg palladium black. The reaction mixture was sonicated for one hour and then stirred at room temperature overnight. The reduced product was purified by reverse-phase HPLC on a C18 Phenomenex Ultracarb column using a gradient of 0.1 M ammonium acetate/1% acetonitrile (pH=6.9) and 80% acetonitrile/20% water over 50 min. The purified product was characterized using ESI mass spectrometry.
FIGURE 2.

Reverse-phase HPLC chromatograms (monitored at 260 nm) and UV spectra (shown in inset) of purified A. unmodified d(CGCGCG), B. d(CGCG*CG) containing dG-C8-amino-6-NP, C. d(CGCG*CG) containing dG-C8-amino-8-NP.
Acknowledgments
This work was supported by a research grant ES09127 from the National Institute of Environmental Health Sciences, NIH. CM was supported by a REU grant CHE-0354012 from NSF.
References
- 1.Rosenkranz HS, McCoy EC, Sanders DR, Butler M, Kiriazides DK, Mermelstein R. Nitropyrenes: isolation, identification, and reduction of mutagenic impurities in carbon black and toners. Science. 1980;209:1039–1043. doi: 10.1126/science.6996095. [DOI] [PubMed] [Google Scholar]
- 2.International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risk to Humans. Vol. 39. IARC; Lyon, France: 1989. Diesel and gasoline engine exhausts and some nitroarenes; pp. 1–458. [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenkranz HS, Mermelstein R. The genotoxicity, metabolism, and carcinogenicity of nitrated polycyclic hydrocarbons. J Environ Sci Health. 1985;C3:221–272. [Google Scholar]
- 4.Purohit V, Basu AK. Mutagenicity of nitroaromatic compounds. Chem Res Toxicol. 2000;13:673–692. doi: 10.1021/tx000002x. [DOI] [PubMed] [Google Scholar]
- 5.El-Bayoumy K, Hecht SS, Sackl T, Stoner GD. Tumorigenicity and metabolism of 1-nitropyrene in A/J mive. Carcinogenesis. 1984;5:1449–1452. doi: 10.1093/carcin/5.11.1449. [DOI] [PubMed] [Google Scholar]
- 6.El-Bayoumy K, Rivenson A, Johnson B, DiBello J, Little P, Hecht SS. Comparative tumorigenicity of 1-nitropyrene, 1-nitrosopyrene, and 1-aminopyrene administered by gavage to Sprague-Dawley rats. Cancer Res. 1988;48:4256–4260. [PubMed] [Google Scholar]
- 7.Vyas RR, Nolan SJ, Basu AK. Synthesis and characterization of oligodeoxynucleotides containing N-(deoxyguanosin-8-yl)-1-aminopyrene. Tetrahedron Lett. 1993;34:2247–2250. [Google Scholar]
- 8.Wolfe JP, Wagaw S, Marcoux JF, Buchwald SL. Rational development of practical catalysts for aromatic carbon-nitrogen bond formation. Acc Chem Res. 1998;31:805–818. [Google Scholar]
- 9.Hartwig JF. Carbon-heteratom bond-forming reductive eliminations of amines, ethers, and sulfides. Acc Chem Res. 1998;31:852–860. [Google Scholar]
- 10.Lakshman MK. Synthesis of biologically important nucleoside analogs by palladium-catalyzed C-N bond-formation. Curr Org Synth. 2005;2:83–112. [Google Scholar]
- 11.Elmquist CE, Stover JS, Wang Z, Rizzo CJ. Site-specific synthesis and properties of oligonucleotides containing C8-deoxyguanosine adducts of the dietary mutagen IQ. J Am Chem Soc. 2004;126:11189–11201. doi: 10.1021/ja0487022. [DOI] [PubMed] [Google Scholar]
- 12.Bonala R, Torres MC, Iden CR, Johnson F. Synthesis of the PhIP adduct of 2′-deoxyguanosine and its incorporation into oligomeric DNA. Chem Res Toxicol. 2006;19:734–738. doi: 10.1021/tx0600191. [DOI] [PubMed] [Google Scholar]
- 13.Takamura-Enya T, Enomoto S, Wakabayashi K. Palladium-catalyzed direct N-arylation of nucleosides, nucleotides, and oligonucleotides for efficient preparation of dG-N2 adducts with carcinogenic amino-/nitroarenes. J Org Chem. 2006;71:5599–5606. doi: 10.1021/jo0605243. [DOI] [PubMed] [Google Scholar]
- 14.Böge N, Gräsl S, Meier C. Synthesis and properties of oligonucleotides containing C8-deoxyguanosine arylamine adducts of borderline carcinogens. J Org Chem. 2006;71:9728–9738. doi: 10.1021/jo061803t. [DOI] [PubMed] [Google Scholar]
- 15.Gillet LCJ, Schärer OD. Preparation of C8-amine and acetylamine adducts of 2′-deoxyguanosine suitably protected for DNA synthesis. Organic Lett. 2002;4:4205–4208. doi: 10.1021/ol026474f. [DOI] [PubMed] [Google Scholar]
- 16.Chakraborti D, Colis L, Schneider R, Basu AK. Synthesis of N2 2′-deoxyguanosine adducts formed by 1-nitropyrene. Organic Lett. 2003;16:2861–2864. doi: 10.1021/ol034904b. [DOI] [PubMed] [Google Scholar]
- 17.Purohit V, Basu AK. Synthesis and characterization of oligodeoxynucleotides containing adducts formed by 1,6- and 1,8-dinitropyrene. Organic Lett. 2000;2:1871–1874. doi: 10.1021/ol000090c. [DOI] [PubMed] [Google Scholar]
- 18.Humphreys WG, Kadlubar FF, Guengerich FP. Mechanism of C8 alkylation of guanine residues by activated arylamines: evidence for initial adduct formation at the N7 position. Proc Natl Acad Sci USA. 1992;89:8278–8282. doi: 10.1073/pnas.89.17.8278. [DOI] [PMC free article] [PubMed] [Google Scholar]