

Background: The host inflammatory response can contribute to the pathogenesis of retinal degeneration.
Results: Photoreceptor proteins in degenerating retinas can activate microglial cells through Toll-like receptor 4 (Tlr4).
Conclusion: Microglial activation can be a common pathology of retinal degeneration.
Significance: Modulating microglial activation is a potential treatment strategy for human retinal degenerative diseases, including age-related macular degeneration and retinitis pigmentosa.
Keywords: Inflammation, Microglia, Retinal Degeneration, Retinal Metabolism, Toll-like Receptors (TLR), Stargardt Diseases, TLR4, Age-related Macular Degeneration, All-trans-retinal, Retinal Pigment Epithelium
Abstract
Although several genetic and biochemical factors are associated with the pathogenesis of retinal degeneration, it has yet to be determined how these different impairments can cause similar degenerative phenotypes. Here, we report microglial/macrophage activation in both a Stargardt disease and age-related macular degeneration mouse model caused by delayed clearance of all-trans-retinal from the retina, and in a retinitis pigmentosa mouse model with impaired retinal pigment epithelium (RPE) phagocytosis. Mouse microglia displayed RPE cytotoxicity and increased production of inflammatory chemokines/cytokines, Ccl2, Il1b, and Tnf, after coincubation with ligands that activate innate immunity. Notably, phagocytosis of photoreceptor proteins increased the activation of microglia/macrophages and RPE cells isolated from model mice as well as wild-type mice. The mRNA levels of Tlr2 and Tlr4, which can recognize proteins as their ligands, were elevated in mice with retinal degeneration. Bone marrow-derived macrophages from Tlr4-deficient mice did not increase Ccl2 after coincubation with photoreceptor proteins. Tlr4−/−Abca4−/−Rdh8−/− mice displayed milder retinal degenerative phenotypes than Abca4−/−Rdh8−/− mice. Additionally, inactivation of microglia/macrophages by pharmacological approaches attenuated mouse retinal degeneration. This study demonstrates an important contribution of TLR4-mediated microglial activation by endogenous photoreceptor proteins in retinal inflammation that aggravates retinal cell death. This pathway is likely to represent an underlying common pathology in degenerative retinal disorders.
Introduction
Many neurodegenerative diseases, including retinal degeneration, are associated with genetic mutations, most of which are found in completely unrelated genes. Although these diseases can result from different biological and biochemical impairments, clinical assessments show many similarities. Defining these similarities offers hope in discovering therapeutic interventions with broad applicability. Major retinal degenerative diseases include age-related macular degeneration (AMD),4 Stargardt disease, and retinitis pigmentosa (RP). AMD is the leading cause of legal blindness in industrialized countries (1), and Stargardt disease is the most common form of juvenile macular degeneration (2). RP refers to a heterogeneous group of inherited diseases that show more dominant rod photoreceptor degeneration (3). Genetic defects in diverse genes, including complement factor H (CFH) (4, 5), ATP-binding cassette transporter 4 (ABCA4) (6), and rhodopsin (7) are known to cause these disorders, but all these diseases share similarities in their pathophysiology.
The aging retina, as well as the brain, show features of low-grade chronic inflammation, recently termed para-inflammation (8, 9). This state can evolve into clinical pathologies including retinal degeneration and Alzheimer disease. Characteristic features of retinal para-inflammation are microglial activation and complement deposition. Retinal macrophages are subdivided into tissue-resident microglia in the inner retina, and peripheral macrophages that migrate to this site from local blood vessels (10). Histological studies suggest that macrophages are the predominant infiltrating cells in the retinas of patients with AMD (9, 11) and RP (12). Complement dysregulation has also been implicated in the pathogenesis of Stargardt disease (13, 14).
Excessive all-trans-retinal (atRAL) accumulation after light exposure can cause retinal toxicity because it is a reactive aldehyde. ABCA4 and retinol dehydrogenase 8 (RDH8) are the visual cycle proteins involved in clearance of atRAL from the retina (15). The Abca4−/−Rdh8−/− mouse features an AMD-like retinal phenotype that includes lipofuscin accumulation, drusen formation, and photoreceptor/RPE atrophy followed by choroidal neovascularization (16). This phenotype is exacerbated by light exposure, which causes acute retinal degeneration (17). Moreover, genetic ablation of Toll-like receptor 3 (Tlr3) in Abca4−/−Rdh8−/− mice, yielding Tlr3−/−Abca4−/−Rdh8−/− mice, attenuated the severity of this retinal degeneration (18), suggesting that immunological mechanisms participate in the pathology of atRAL-associated retinal degeneration.
Here, we investigated retinal degenerative events in two mouse models, Abca4−/−Rdh8−/− mice that exhibit light-induced and age-related retinal degeneration and RP-like retinal degeneration in c-mer proto-oncogene tyrosine kinase-deficient (Mertk−/−) mice, which are unable to conduct RPE phagocytosis (15). Subretinal accumulation of microglia/macrophages accompanied by other inflammatory changes was observed in both models. Release of dying photoreceptor cell proteins increased expression of Tlr2 and Tlr4, which stimulated the activation of microglia and RPE cells. Furthermore, Tlr4 deficiency and microglial inactivation by minocycline attenuated retinal degeneration in Abca4−/−Rdh8−/− mice. These observations indicate that microglial activation by endogenous retinal proteins arising from injured photoreceptors plays an important role in the pathogenesis of retinal degeneration.
EXPERIMENTAL PROCEDURES
Animals
Abca4−/−Rdh8−/− mice were generated as described previously, and all mice were genotyped by an established method (16, 17). Only mice with the leucine variation at amino acid 450 of RPE65 were used. Mertk−/− mice were purchased from Jackson Laboratory (Bar Harbor, ME). Tlr4−/− mice were obtained from Dr. S. Akira (Research Institute for Microbial Diseases, Osaka University, Japan). Genotyping for Tlr4 was performed with primers: TLR4-WT, 5′-CAAGATCAACCGATGGACGTGTAAAC-3′; TLR4-common, 5′-CTTGTTGCCCTTCAGTCACAGAGAC-3′; TLR4-KO, 5′-GTTGGGTCGTTTGTTCGGATCCGTC-3′. C57BL/6 or littermates of mutant mice were used as controls. Equal numbers of males and females were employed. All mice were housed in the animal facility at the School of Medicine, Case Western Reserve University, where they were maintained either under complete darkness or in a 12-h light (∼10 lux)/12-h dark cycle environment. Experimental manipulations in the dark were done under dim red light transmitted through a Kodak No. 1 safelight filter (transmittance >560 nm). All animal procedures and experiments were approved by the Case Western Reserve University Animal Care Committees and conformed to recommendations of both the American Veterinary Medical Association Panel on Euthanasia and the Association of Research for Vision and Ophthalmology.
Induction of Light Damage
Mice were dark-adapted for 48 h before their exposure to light. Light damage was induced by exposing mice to 10,000 lux of diffuse white fluorescent light (150 W spiral lamp; Commercial Electric, Cleveland, OH) for 30 min. Before such light exposure, pupils of mice were dilated with mixture of 0.5% tropicamide and 0.5% phenylephrine hydrochloride (Midorin-P®, Santen Pharmaceutical Co., Ltd., Osaka, Japan). After exposure animals were kept in the dark until evaluation.
Histological Analysis
All procedures to create sections for immunohistochemistry and light microscopy were performed by established methods (18, 19). Fluorescent intensity was measured with ImageJ (National Institutes of Health, Bethesda, MD). The following antibodies (Abs) were used for immunohistochemistry (IHC): rabbit anti-Iba1 Ab (1:400, Wako, Chuo-ku, Osaka, Japan), rat anti-mouse F4/80 Ab (1:100, AbD Serotec, Raleigh, NC), rabbit anti-glial fibrillary acidic protein Ab (GFAP; 1:400, Dako, Carpinteria, CA), mouse anti-rhodopsin 1D4 Ab (1:100, gift from Dr. Robert Molday, University of British Columbia, Vancouver, Canada), Alexa 488-conjugated peanut agglutinin (1:200, Invitrogen), mouse anti-MHC class II Ab (1:200, Abcam, Cambridge, MA), and rabbit anti-C3 Ab (1:100, Santa Cruz Biotechnology, Santa Cruz, CA).
Scanning Laser Ophthalmoscopy (SLO) Imaging and Spectral Domain Optical Coherence Tomography (SD-OCT)
HRAII (Heidelberg Engineering, Heidelberg, Germany) for SLO and SD-OCT (EnvisuTM C-Class SDOIS, Bioptigen, Research Triangle Park, NC) were employed for in vivo imaging of mouse retinas. Mice were anesthetized by intraperitoneal injection using a mixture (20 μl/g of body weight) containing ketamine (6 mg/ml) and xylazine (0.44 mg/ml) in 10 mm sodium phosphate, pH 7.2, and 100 mm NaCl. Pupils were dilated with a mixture of 0.5% tropicamide and 0.5% phenylephrine hydrochloride (Midorin-P, Santen Pharmaceutical Co., Ltd.). Five pictures acquired in the B-scan mode were used to construct each final averaged SD-OCT image. SD-OCT images were scored using our previously established scoring system (15).
Flat Mount Retina and RPE Preparation for Immunostaining
Radial incisions were made in enucleated eyecups and the vitreous was removed completely to produce flat mount retinas. RPE flat mounts were prepared by peeling the retina away from the eyecup. For immunostaining, flat mount retina or RPE was fixed with 4% paraformaldehyde for 16 h. After fixation, the sample was washed in PBST buffer (136 mm NaCl, 11.4 mm sodium phosphate, 0.1% Triton X-100, pH 7.4) for 2 h at room temperature (RT) and then placed on a coated slide (Superfrost Plus®, Fisher Scientific, Pittsburgh, PA). The mounted sample was air-dried 2 h at RT and then blocked with 5% normal goat serum in PBST for 3 h.
qRT-PCR
Retinal samples from each group were collected from 16 eye balls. Total RNA was isolated using a RiboPure Kit (Applied Biosystems, Austin, TX), and cDNA was synthesized with SuperScriptTM II Reserve Transcriptase (Invitrogen) following the manufacturer's instructions. Real-time PCR amplification was performed using iQTM SYBR® Green Supermix (Bio-Rad). Primers were designed using web tool Primer 3 and synthesized by Eurofins MWG Operon (Hunstville, AL). qRT-PCR analyses were performed with the following primers: Ccl2 (187 bp), forward 5′-GCTGACCCCAAGAAGGAATG-3′, reverse 5′-GTGCTTGAGGTGGTTGTGGA-3′; Ccr2 (227 bp), forward 5′-ATTCTCCACACCCTGTTTCG-3′, reverse 5′-ATGCAGCAGTGTGTCATTCC-3′; Ccl12 (188 bp), forward 5′-CAGTCCTCAGGTATTGGCTGGA-3′, reverse 5′-TCCTTGGGGTCAGCACAGAT-3′; Cx3cr1 (202 bp), forward 5′-CACCATTAGTCTGGGCGTCT-3′, reverse 5′-GATGCGGAAGTAGCAAAAGC-3′; Il1β (167 bp), forward 5′-CCTGCAGCTGGAGAGTGTGG-3′, reverse 5′-CCAGGAAGACAGGCTTGTGC-3′; Tnf-R (102 bp), forward 5′-GGTTCCTTTGTGGCACTTG-3′, reverse 5′-TTCTCTTGGTGACCGGGAG-3′; C3 (190 bp), forward 5′-GACCAAGTGCCAGACACAGA-3′, reverse 5′-CGGTCTGGTCCAGGTAGTGT-3′; Cfh (163 bp), forward 5′-TGGACTTCCTTGTGGACCTC-3′, reverse 5′-TGGGTCAGACCACTTTCCTC-3′; Vegfa (86 bp), forward 5′-AGCACAGCAGATGTGAATGC-3′, reverse 5′-AATGCTTTCTCCGCTCTGAA-3′; C-reactive protein (Crp, 194 bp) forward 5′-TCTGCACAAGGGCTACACTG-3′, reverse 5′-AAACATTGGGGCTGAGTGTC-3′; Pyrimidinergic receptor P2Y, G-protein coupled, 6 (P2ry6, 187 bp) forward 5′-CATTAGCTTCCAGCGCTACC-3′, reverse 5′-GCTCAGGTCGTAGCACACAG-3′; Arginase, liver (Arg1, 181 bp) forward 5′-CGCCTTTCTCAAAAGGACAG-3′, reverse 5′-ACAGACCGTGGGTTCTTCAC-3′; Transforming growth factor, β1 (Tgfb, 185 bp) forward 5′-TGAGTGGCTGTCTTTTGACG-3′, reverse 5′-GGTTCATGTCATGGATGGTG-3′; Tlr2 (241 bp) forward 5′-TGGTTCTTTTCCCAAACTGG-3′, reverse 5′-GAGAAGGGCACAGCAGACTC-3′; Tlr4 (154 bp) forward 5′-GTGGCCCTACCAAGTCTCAG-3′, reverse 5′-GACCCATGAAATTGGCACTC-3′. Relative expression of genes was normalized to the housekeeping gene Gapdh.
Enzyme-linked Immunosorbent Assay (ELISA)
Production of Ccl2, Il1β, and Tnf was quantified by ELISA kits (Ccl2; MEJ00, Il1β; MLB00C, Tnf; MTA00B) purchased from R&D Systems (Minneapolis, MN) with 50 μl of cell culture supernatants. Concentrated cell lysates were prepared with Nonidet P-40 lysis buffer containing 20 mm Tris, pH 8.0, 137 mm NaCl, and 1% Nonidet P-40. Then the protein concentration was measured with a BCA protein assay kit (Pierce).
Isolation of Primary RPE, Microglia, and Macrophages
Primary mouse RPE cells, retinal microglial cells, and bone marrow (BM)-derived macrophages were prepared from 2-week-old mice based on previously published methods (20–22) with modifications. Enucleated eyes were incubated with 2% dispase (Invitrogen) in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) for 1 h at 37 °C, and neural retinas and eyecups were separated under a surgical microscope (ILLUMIN-i, Endure Medical, Cumming, GA). The RPE layer was peeled from eyecups, passed through 70- and 40-μm nylon mesh filters (Falcon Plastics, Brookings, SD), and cultured in DMEM containing minimal essential medium non-essential amino acids (Invitrogen), penicillin/streptomycin (Invitrogen), 20 mm HEPES, pH 7.0, and 10% fetal bovine serum. To enrich microglial cells, neural retinas were homogenized and cultured in DMEM containing minimal essential medium non-essential amino acids (Invitrogen), penicillin/streptomycin (Invitrogen), 20 mm HEPES, pH 7.0, and 10% fetal bovine serum for 7 days at 37 °C. Adherent cells to the plastic surface were treated with 0.05% trypsin (Invitrogen), and less adhesive cells were collected as microglial cells. BM cells were collected from femurs and tibias and were incubated in DMEM containing 10% FBS and 30% L929-conditioned medium for 7–10 days to differentiate them into macrophages. Homogeneity of these cells was confirmed by immunocytochemistry with anti-Iba1 and anti-F4/80 Abs for microglia/macrophages, and anti-Zo-1, anti-RPE65, and anti-LRAT Abs for RPE cells. RT-PCR was conducted to check expression of RPE-specific proteins with the following primers: Rpe65 (561 bp), forward 5′-CAATTGACAAGGTCGACACAG-3′, reverse 5′-CATCTCTGGAATATGTTCAGG-3′; Lrat (334 bp), forward 5′-CARCCTAGTCAATCACCTAGAC-3′, reverse 5′-CTAGCCAGACATCATCCACAAG-3′; rhodopsin (562 bp), forward 5′-ATGAACGGCACAGAGGGCCC-3′, reverse 5′-CGCATGAACATTGCATGCCCTC-3′.
Photoreceptor Outer Segment (POS) Preparation
POS membranes were prepared from 4-week-old Abca4−/−Rdh8−/− and WT mice using a published method (23). Fifteen to 20 animals were used for each preparation. AtRAL was purchased from Sigma and lipopolysaccharide (LPS) and Pam3CSK4 were purchased from Invivogen (San Diego, CA). To remove DNA and RNA from POS, 125 units of benzonase (Sigma) was incubated with 30 μg of POS for 30 min at 37 °C. Lipase at 2.5 units (Sigma) was added and mixed with 30 μg of POS, and then incubated 60 min at 37 °C to degrade POS lipids. Endotoxin levels in prepared POS were measured using the Limulus Amebocyte Lysate assay (Pierce) according to the manufacturer's protocol.
Cell Death Assay
Activity of lactate dehydrogenase (LDH) released from dead cells into the culture supernatants was measured after a 24-h incubation at 37 °C with an LDH assay kit from BioVision (Mountain View, CA). The percentage of cytotoxicity was calculated as [(test sample − cell negative control)/(lysis control − cell negative control)] × 100.
Detection of Reactive Oxygen Species (ROS)
Production of ROS was examined by using the fluorescent ROS probe DCF-DA purchased from Sigma. After a 24-h incubation, cells were washed twice with PBS, including 137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4, 1.4 mm KH2PO4, pH 7.2, and then 0.25 μm DCF-DA was co-incubated for 30 min. After washing twice with PBS, images of the ROS signal were obtained with a Leica DMI 6000B inverted microscope. Fluorescence intensity was measured with ImageJ (National Institutes of Health) or MetaMorph (Molecular Devices, Sunnyvale, CA) software.
Minocycline Treatment
Minocycline was purchased from Sigma. Primary RPE, retinal microglia, or BM-derived macrophages were co-incubated with 30 μg/ml of minocycline at 37 °C for 24 h to determine the effects of cellular minocycline. For in vivo evaluation, mice were administrated an intraperitoneal injection of minocycline (0, 4, 50, and 100 mg/kg) daily from 1 day before to 7 days after light exposure.
Data Analysis
Data representing the mean ± S.D. for the results of at least three independent experiments were compared by the one-way analysis of variance test. A p value of <0.05 was considered statistically significant.
RESULTS
Subretinal Infiltration of Microglia/Macrophages in Light-exposed Abca4−/−Rdh8−/− Mice
Delay in atRAL clearance after light exposure in Abca4−/−Rdh8−/− mice causes acute photoreceptor apoptosis (24). To identify additional retinal degenerative symptoms associated with light-induced photoreceptor death, 4-week-old Abca4−/−Rdh8−/− mice were exposed to light at 10,000 lux for 30 min, and in vivo SLO imaging was performed. Accumulation of autofluorescent (AF) spots were detected in retinas of light-exposed Abca4−/−Rdh8−/− mice (Fig. 1A). This accumulation became evident at 3 days and peaked 7 days after light exposure (Fig. 1A, right panel). RPE/choroid flat mounts were examined to characterize the localization of these AF spots. Cellular infiltration was observed on the apical side of the RPE where the RPE and POS form junctions. AF spots and infiltrated cells appeared similar in size, and these cells were stained with Abs for Iba-1, a microglia marker and F4/80, a macrophage marker (Fig. 1B). The fact that these Iba-1- and F4/80-positive cells evidenced extensive pseudopodia, short microvilli, and lamellipodia indicates that these cells were activated forms of microglia/macrophages (25). Translocation of Iba-1-positive cells from the inner retina to the subretinal space was detected in light-exposed Abca4−/−Rdh8−/− mice (Fig. 1C). These data indicate that AF spots detected by SLO in light-exposed Abca4−/−Rdh8−/− mice are caused by infiltrating microglia/macrophages into the subretinal space.
FIGURE 1.

Light-exposed Abca4−/−Rdh8−/− mice show subretinal microglia accumulation. Retinas of Abca4−/−Rdh8−/− (dKO) and C57BL/6 WT mice at 4 weeks of age were exposed to light at 10,000 lux for 30 min. A, SLO images 7 days after light-exposed and non-light exposed mice are shown (left panels). Right panel shows time course changes in numbers of AF spots. Error bars indicate mean ± S.D. (n > 6). * indicates p < 0.05. B, RPE flat mounts of light-exposed Abca4−/−Rdh8−/− mice were stained with anti-Iba-1 (red) and anti-F4/80 (green) Abs. Nuclei were stained with 4′-6-diamidino-2-phenylindole (DAPI) (blue). Relative sizes of AF spots by SLO and infiltrating cells against the diameter of the ONH were compared (upper panels). Magnified image of a RPE flat mount (lower left) and one of an Iba-1- and F4/80-double positive cell are shown (lower middle). Numbers of Iba-1- and F4/80-positive cells were counted in RPE flat mounts (lower right). Bars indicate 30 μm. Error bars indicate mean ± S.D. (n > 50 spots; n > 3 RPE flat mounts). * indicates p < 0.05. C, immunohistochemistry with anti-Iba-1 Ab (red) and DAPI (blue) is shown. Days after light exposure are indicated. Bars indicate 50 μm. INL, inner nuclear layer.
Light Exposure Increases mRNA Expression of Inflammatory Molecules in Retinas of Abca4−/−Rdh8−/− Mice
To determine whether the increased numbers of microglia/macrophages in Abca4−/−Rdh8−/− mice correlate with increased expression of genes associated with retinal inflammation, we examined expression of proinflammatory and chemotactic cytokines using qRT-PCR. These genes included chemokine (C-C motif) ligand 2 (Ccl2/Mcp-1) (25, 26), chemokine (C-C motif) receptor 2 (Ccr2) (26), Ccl12/Mcp-5, chemokine (C-X3-C) receptor 1 (Cx3cr1/fractalkine receptor) (27), interleukin 1β (Il1b) (28), tumor necrosis factor receptor superfamily, member 1A (Tnf-R) (29–31), complement component 3 (C3) (32), complement factor H (Cfh) (4, 5), and vascular endothelial growth factor A (Vegfa) (33) (Fig. 2). Expression of Ccl2, Ccr2, Ccl12, Cx3cr1, Il1b and Tnf-R were increased by 12 h and peaked 3 days after light exposure in Abca4−/−Rdh8−/− mice. WT mice showed a transient increase of Ccr2 12 h after light exposure and a persistent, but lesser expression of Cx3cr1. Tnf-R levels were increased in light-exposed WT mice, whereas Il1b did not change in these mice. C3 expression was similar to that of chemokines and cytokines in Abca4−/−Rdh8−/− mice. Immunohistochemisty showed C3 expression of cells in the subretinal space (data not shown). Cfh expression was elevated between 12 h and 3 days after light exposure in Abca4−/−Rdh8−/− mice, and a persistent, higher expression of Cfh was also observed in WT mice. Vegfa expression was continuously up-regulated in Abca4−/−Rdh8−/− mice with the highest expression occurring 12 h after light exposure. These data indicate that light exposure in Abca4−/−Rdh8−/− mice activates the retinal immune system in a manner similar to that reported for human retinal disease.
FIGURE 2.

mRNA expression of disease-related immune components in light-exposed Abca4−/−Rdh8−/− and WT mice. RNA samples were collected from 16 retinas at each time point. Fold changes in mRNA expression relative to non-light exposed retinas of Abca4−/−Rdh8−/− (dKO) and C57BL/6 WT mice are presented. Relative expression of genes was normalized against the housekeeping gene Gapdh. Error bars indicate mean ± S.D. (n = 3). * indicates p < 0.05 versus non-light exposed Abca4−/−Rdh8−/− mice. # indicates p < 0.05 versus non-light exposed WT mice.
Inflammatory Changes in Retinas of 6-Month-old Abca4−/−Rdh8−/− Mice
Abca4−/−Rdh8−/− mice show age-related retinal degeneration and inflammation with a phenotype similar to human macular degeneration (16, 17). Mice were kept in a 12-h light (∼10 lux)/12-h dark cycle environment until they reached 6 months of age. As expected, Abca4−/−Rdh8−/− mice at 6 months of age showed retinal degeneration with infiltrates in the subretinal space, whereas 6-month-old WT mice did not display these changes (data not shown). SLO imaging also revealed increased numbers of AF spots in 6-month-old Abca4−/−Rdh8−/− mice (data not shown). To examine retinal inflammatory changes further, qRT-PCR for other inflammatory mediators was performed. Expression of C3, Il1b, Crp, Vegfa, P2ry6, Cfh, Arg1, and Tgfb in 4-week-old Abca4−/−Rdh8−/− mice, which did not yet exhibit retinal degeneration at 4 weeks of age (16), was similar to that in WT animals. Age-related increases in expression of these molecules were detected in Abca4−/−Rdh8−/− mice (Fig. 3), whereas only an increased expression of Vegfa was observed in older WT mice. These results indicate that older Abca4−/−Rdh8−/− mice kept under 12-h room light/12-h dark conditions show retinal inflammatory changes similar to those observed in light-exposed young Abca4−/−Rdh8−/− mice.
FIGURE 3.
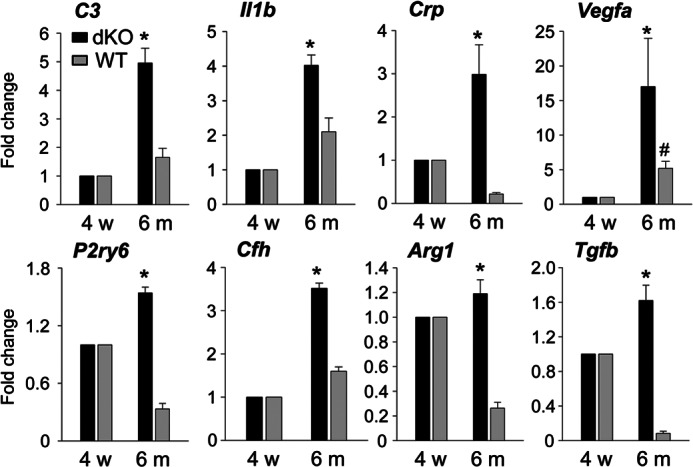
Retinal inflammation in 6-month-old Abca4−/−Rdh8−/− mice. Abca4−/−Rdh8−/− and littermate WT mice at 6 months of age were investigated. RNA samples were collected from 16 retinas from 4-week-old and 6-month-old mice. Fold-changes in expression relative to 4-week-old Abca4−/−Rdh8−/− (dKO) or littermate WT mice are presented. Relative expression of genes was normalized to the housekeeping gene Gapdh. Error bars indicate mean ± S.D. (n = 3). * indicates p < 0.05 versus 4-week-old Abca4−/−Rdh8−/− mice. # indicates p < 0.05 versus 4-week-old WT mice.
Increased Adherent Leukocytes, Glial Disruption, and a Breakdown of the Blood-Retinal Barrier in the Inner Retina
Increased expression of Vegfa in light-exposed Abca4−/−Rdh8−/− retinas (Fig. 2) prompted us to examine the integrity of retinal vessels because VEGFA is a key factor in the pathogenesis of vascular complications (34). Mammalian eyes have two blood-retinal barriers (BRBs). The outer BRB consists of a tight junction between the RPE and choroidal capillaries and the inner BRB is formed by the tight junction between neighboring capillary endothelial cells and foot processes of astrocytes and Müller glia (35).
Even though choroidal neovascularization develops in some aged Abca4−/−Rdh8−/− mice (16), no such changes were detected in Abca4−/−Rdh8−/− mice with light-induced acute retinal degeneration. This finding suggests that transient VEGF production after bright light exposure could play a role in the collapse of the inner BRB. Leukocyte adhesion to vascular endothelial cells is required for invasion of circulating leukocytes into retinal tissue and this is a marker for inner BRB breakdown (36). To examine adherent leukocytes and inner BRB integrity, staining with concanavalin A lectin (ConA) (36) to identify adherent leukocytes and GFAP, a marker of astrocyte and activated Müller glia, was performed by using flat mount retinas. FITC-conjugated ConA (FITC-ConA) was injected into the hearts of 4-week-old Abca4−/−Rdh8−/− mice 7 days after exposure to 10,000 lux for 30 min.
Whereas non-light exposed Abca4−/−Rdh8−/− mice and WT mice showed few adherent leukocytes, light-exposed Abca4−/−Rdh8−/− mice displayed a significant (p < 0.05) increase in adherent leukocytes (Fig. 4, A and D, left). In non-light exposed Abca4−/−Rdh8−/− mice and WT mice, normal astrocytes with flattened cell bodies and radiating processes and healthy Müller glia with foot processes were observed after GFAP staining. In contrast, light-exposed Abca4−/−Rdh8−/− mice showed irregular and increased GFAP expression, indicating glial disruption (Fig. 4, B and D, right). Although all retinal vessels in non-light exposed Abca4−/−Rdh8−/− mice and WT mice were enveloped with GFAP signals in merged images, light-exposed Abca4−/−Rdh8−/− mice showed scattered unwrapped GFAP staining on retinal vessels (Fig. 4C). Fundus fluorescein angiography revealed fluorescent leakage from retinal vessels, especially near the optic nerve head in light-exposed Abca4−/−Rdh8−/− mice, whereas light-exposed WT mice did not show this change (Fig. 4E). These data indicate a breakdown of the inner BRB, which enables circulating leukocytes, such as monocytes/macrophages, to invade the retina.
FIGURE 4.

Increased adherent leukocytes and glial disruption in retinas of light-exposed Abca4−/−Rdh8−/− mice. FITC-conjugated concanavalin A lectin (FITC-ConA) was injected into the hearts of 4-week-old Abca4−/−Rdh8−/− mice 7 days after light exposure at 10,000 lux for 30 min. Flat mount retinas were immediately prepared after FITC-ConA injection. A, FITC-ConA labeling of adherent leukocytes and vascular endothelial cells are presented. The small rectangle of each photograph corresponds with a higher magnified image (lower right). Bars indicate 50 μm. B, GFAP staining of astrocytes and Müller glia is presented. Higher magnified images of the small squares are shown (lower right). Bars indicate 50 μm. C, merged images of A and B with nuclear staining by DAPI are shown. Unwrapped retinal vessels by GFAP are indicated with arrows. A small square of each photograph corresponds to a higher magnified image (lower right). Bars indicate 50 μm. D, numbers of adherent leukocytes (left) and fluorescent intensity (right) calculated by ImageJ software are shown. * indicates p < 0.05. Error bars indicate mean ± S.D. (n = 5). E, fundus fluorescein angiography was performed with Abca4−/−Rdh8−/− and C57BL/6 WT mice 7 days after light exposure at 10,000 lux for 30 min. A dashed line square (left) corresponds with a higher magnified image (middle). Arrows indicate fluorescein leakage. The incidence of fluorescent leakage is presented with numbers of eyes with leakage/eyes without leakage (right).
Overproduction of Photoreceptor Debris Results in Subretinal Microglia/Macrophage Infiltration
The presence of microglia/macrophages in the subretinal space in light-exposed Abca4−/−Rdh8−/− mice (Fig. 1) led to the hypothesis that degenerating photoreceptors exceeding the clearance capacity of RPE phagocytosis initiates the events leading to macrophage infiltration. MERTK belongs to a family of receptor tyrosine kinases that includes AXL and TYRO3, and plays an indispensable role in the clearance of photoreceptor debris by RPE phagocytosis (37). Accumulation of photoreceptor debris in the subretinal space is closely associated with photoreceptor death in the Royal College of Surgeons rat with disabled Mertk (38), although the role of retinal inflammation in these animals has yet to be determined.
To test this hypothesis, we quantified the number of translocated cells in Mertk−/− mice by SLO. SLO examination revealed increased numbers of AF spots in 6-week-old Mertk−/− mice (data not shown) and a further increase at 16 weeks of age (Fig. 5A, left). SD-OCT also revealed progressive retinal degeneration in 16-week-old Mertk−/− mice (Fig. 5A, right). Neither accumulation of AF spots nor retinal degeneration was detected at 3 weeks of age. Translocation of microglia/macrophages from the inner retina where they normally reside to the outer retina including outer nuclear layers (ONL), photoreceptor layers, and subretinal space, was detected in the retinas of 6-week-old Mertk−/− mice (Fig. 5B). The accumulation of photoreceptor debris in Mertk−/− mice, retinal degenerative pathology including the formation of retinal whorls, pyknotic photoreceptor nuclei, and retinal vacuoles were also observed as previously noted (39). Increased numbers of microglia/macrophages were also detected in the subretinal space (Fig. 5C). Elevated expression of inflammatory components were detected in 8-week-old Mertk−/− mice with degenerating retinas relative to non-degenerative 3-week-old Mertk−/− mice (Fig. 5D). These observations indicate that subretinal accumulation of photoreceptor cellular debris is associated with retinal inflammation, including microglia/macrophage infiltration into the subretinal space.
FIGURE 5.

Mertk−/− mice with disabled RPE phagocytosis reveal subretinal microglia/macrophage infiltration. A, SLO (left) and SD-OCT (right) images of retinas from 3- and 16-week-old Mertk−/− mice are shown. Bars indicate 50 μm. INL, inner nuclear layer. B, retinas of 6-week-old Mertk−/− mice were stained with anti-Iba-1 and anti-F4/80 Abs. Inset shows one of Iba-1- and F4/80- double positive cells in higher magnification. Bars indicate 50 μm. C, epon-prepared retinal sections from 16-week-old Mertk−/− and WT mice are presented. Red arrows indicate microglia/macrophages. Bars indicate 20 μm. IS, inner segments; OS, outer segments. D, qRT-PCR was performed with RNA samples collected from retinas of Mertk−/− mice at 3 or 8 weeks of age. Fold-changes in expression relative to 3-week-old mice are presented. Error bars indicate mean ± S.D. (n = 3). * indicates p < 0.05.
Distinct Roles of RPE and Microglia in Activation of Retinal Inflammation and Cytotoxicity
In addition to the essential role for RPE cells in maintaining the visual system (35, 40, 41), RPE cells are also involved in immune responses of the posterior eye (42). To investigate whether RPE cells or retinal microglia are the major source of cytokines produced after light exposure in Abca4−/−Rdh8−/− mice (Fig. 2), primary RPE and retinal microglia were isolated from 2-week-old mice. Homogeneity of these isolated cells was confirmed by immunocytochemistry and RT-PCR. Isolated RPE showed hexagonal shaped tight junctions after staining with Zo-1 Ab, and RPE-specific Rpe65 and Lrat were amplified without photoreceptor-specific rhodopsin amplification (Fig. 6A, left panels). Isolated retinal microglial cells were identified by reactivity with Iba-1 and F4/80 Abs (Fig. 6A, right panels). Co-incubation of activated monocytes with RPE cells reportedly causes RPE death (43). To examine the toxic effects of microglial cells on RPE cells, RPE cell death was measured by LDH release. Production of LDH was observed when RPE cells were co-cultured with microglia, and was elevated after incubation with the TLR1/2 ligand Pam3CSK4 or lipopolysaccharide (LPS) (a TLR4 agonist) (Fig. 6B). No LDH production was observed when microglia or RPE cells were incubated alone, and a calculated cell death rate of microglia or RPE cells in the presence of 1 μm Pam3CSK4 for 24 h was 2.6 or 1.4%, respectively. Incubation of these cells with 1 μm LPS for 24 h showed their cell death rate of 3.3% for microglia and 1.0% for RPE cells.
FIGURE 6.
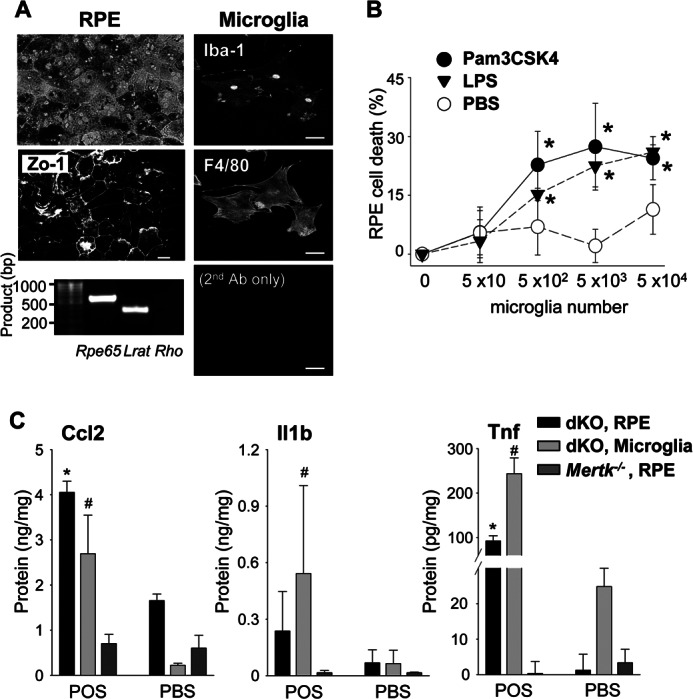
Primary cultured RPE and microglial cells produce Ccl2, Il1b, and Tnf in response to photoreceptor proteins. A, primary RPE and retinal microglial cells were isolated from 2-week-old Abca4−/−Rdh8−/− mice. Morphology of RPE cells after 7 days in culture was captured with bright-field microscopy (left upper), and immunohistochemistry with anti-Zo-1 Ab was carried out (left middle). RT-PCR showed RPE-specific Rpe65 and Lrat amplification at day 14 in the primary RPE culture (left bottom) without photoreceptor-specific rhodopsin (Rho) amplification. Cultured microglia at 7 days displayed microglia- and macrophage-specific staining with anti-Iba-1 and anti-F4/80 Abs (right). Bars indicate 10 μm. B, primary RPE cells from 2-week-old Abca4−/−Rdh8−/− mice (5 × 103) were co-incubated with different numbers of microglia cultured for 7 days with or without 1 μm Pam3CSK4 or 1 μm LPS for 24 h, and the activity of LDH released from dead cells was measured. Error bars indicate mean ± S.D. (n = 3). * indicates p < 0.05 versus PBS. C, production of Ccl2, Il1b, and Tnf was quantified by ELISA with culture supernatants of primary cultured RPE or microglia from 2-week-old Abca4−/−Rdh8−/− (dKO) or Mertk−/− mice after co-incubation with or without 6 μg/100 μl of mouse POS for 24 h. Error bars indicate mean ± S.D. (n = 9). * and # indicate p < 0.05 versus PBS.
Additionally, primary RPE and microglial cells were co-cultured with 6 μg/100 μl of photoreceptor outer segments (POS) from 4-week-old Abca4−/−Rdh8−/− mice to examine whether phagocytosis of POS could stimulate these cells to produce chemokines and cytokines, because debris from photoreceptor cells including POS debris is produced after bright light exposure in mice. RPE and microglial cells were incubated with POS, and production of Ccl2, Il1b, and Tnf in the culture supernatant was quantified by ELISA. RPE and microglial cells produced Ccl2, Il1b, and Tnf, whose production was accelerated in light-exposed Abca4−/−Rdh8−/− mice (Fig. 2) when these cells were incubated with POS (Fig. 6C). RPE cells of Mertk−/− mice lack phagocytotic ability (39) and isolated RPE cells from Mertk−/− mice did not produce Ccl2, Il1b, and Tnf when they were co-incubated with POS. Co-culture with atRAL did not result in significant changes in the production of these molecules, whereas 1 μm Pam3CSK4 and LPS increased Ccl2, Il1b, and Tnf (data not shown). Phagocytosis of non-pathogenic latex beads did not produce these molecules (data not shown). Notably production of Ccl2, Il1b, and Tnf was observed when wild-type RPE or microglial cells were co-incubated with POS from WT mice (data not shown). These findings were not due to LPS contamination as the endotoxin level in POS <0.3 EU/ml (∼0.03 ng/ml), which is less than that in commercially prepared cell culture medium (44). These data indicate that phagocytosis of POS by RPE and microglial cells is associated with the production of chemokines and cytokines under normal and disease conditions.
Endogenous Photoreceptor Proteins Contribute to the Microglial Activation via TLRs
To examine POS-induced cytokine production, microglial cells were incubated with 6 μg/100 μl of heat-treated or untreated POS. Ccl2 production from microglial cells was not increased with heat-treated POS compared with untreated POS (Fig. 7A), indicating a role for heat labile molecules. Ccl2 production was maintained when microglial cells were co-incubated with heat-treated LPS (data not shown). In contrast, incubation with POS treated with benzonase, which degrades DNA and RNA, or lipase produced similar levels of Ccl2 as non-treated POS. These data suggest that endogenous ligands for microglial activation are proteins. Perfluorooctanoic acid (PFOA) was used to solubilize POS proteins for detection of candidate proteins that cause immune reactions by proteomics analysis (45). POS proteins were solubilized by 0.5% PFOA and the insoluble fraction of 0.5% PFOA was further solubilized by 2% PFOA. POS proteins solubilized by 2% PFOA enhanced Ccl2 production, whereas proteins extracted by 0.5% PFOA did not increase Ccl2 levels (Fig. 7B). POS protein profiles extracted by 0.5 and 2% PFOA also were analyzed by LC-MS/MS. Peptides from 1168 proteins were detected in POS extracted by 0.5 and 2% PFOA but 278 proteins were identified only in POS extracted by 2% PFOA followed by 0.5% PFOA extraction (data not shown). Heat-shock proteins that can activate TLRs (46) were included in these 278 proteins.
FIGURE 7.
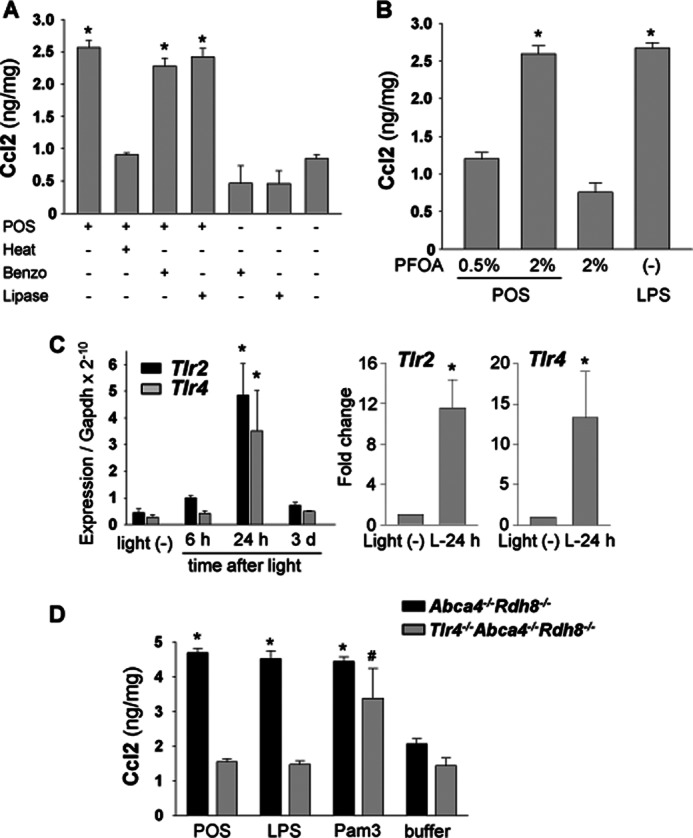
Tlr4-associated Ccl2 production by photoreceptor proteins and increase in mRNA level of retinal Tlrs after light exposure. A, production of Ccl2 was quantified by ELISA with culture supernatant of primary cultured microglia from 2-week-old Abca4−/−Rdh8−/− mice. Microglial cells were co-incubated with 6 μg/100 μl of POS from Abca4−/−Rdh8−/− mice for 24 h, which were treated with or without heat (98 °C, 10 min), benzonase (Benzo) (25 units, 37 °C, 30 min), and lipase (0.5 unit, 37 °C, 60 min). * indicates p < 0.05. B, proteins from POS were extracted by 0.5 and 2% PFOA, and co-incubated at 6 μg/100 μl with microglia for 24 h. * indicates p < 0.05. C, mRNA levels of Tlr2 and Tlr4 were measured with retinas from Abca4−/−Rdh8−/− mice by qRT-PCR. Mice were exposed to 10,000 lux light for 30 min, and retinas were harvested 6 h, 24 h, and 3 days after exposure. Right panel presents relative expression normalized to Gapdh and left panels show the fold-changes 24 h after light relative to mice without light exposure, * indicates p < 0.05 versus no-light exposed Abca4−/−Rdh8−/− mice. D, BM-derived macrophages of 4–6-week-old Tlr4−/−Abca4−/−Rdh8−/− and Abca4−/−Rdh8−/− mice were co-incubated with POS from Abca4−/−Rdh8−/− mice at 6 μg/100 μl for 24 h, and production of Ccl2 was quantified by ELISA in the culture supernatant. * and # indicate p < 0.05 versus supernatant of cells incubated with buffer only.
Recently, endogenous ligands from damaged tissues were reported to activate pattern-recognition receptors such as TLRs, and these receptors may play a role in the immune response in degenerative diseases. Among these receptors, TLR2 and TLR4 can use proteins as natural ligands (46–48). Therefore, changes in expression of Tlr2 and Tlr4 were examined in retinas of light-exposed Abca4−/−Rdh8−/− mice. Significant elevation of mRNA expression of both Tlr2 and Tlr4 was observed 24 h after light exposure when maximum photoreceptor cell apoptosis occurred (24) (Fig. 7C).
To examine further the role of TLR4 in retinal degeneration, we generated Tlr4−/−Abca4−/−Rdh8−/− mice. Because Abca4 is located on chromosome 3, crossing Abca4−/−Rdh8−/− mice with mice deficient in Tlr4 located on chromosome 4 can result in the generation of triple knock-out mice. (Tlr2 is located on chromosome 3.) BM-derived macrophages were isolated from Tlr4−/−Abca4−/−Rdh8−/− and Abca4−/−Rdh8−/− mice, and these cells (5 × 103 cells/well in a 96-well plate) were incubated with 6 μg/100 μl of POS from Abca4−/−Rdh8−/− mice. In contrast to macrophages from Abca4−/−Rdh8−/− mice, which secreted Ccl2, macrophages from Tlr4−/−Abca4−/−Rdh8−/− mice showed significantly (p < 0.05) lower Ccl2 (Fig. 7D). These data indicate that photoreceptor proteins can activate microglia/macrophages to produce chemokines by activating TLR4.
Genetic Deletion of Tlr4 Results in Milder Light-induced and Age-related Retinal Degeneration in Abca4−/−Rdh8−/− Mice
To explore more directly if TLR4 contributes to light-induced retinal degeneration, we examined the phenotype of Tlr4−/−Abca4−/−Rdh8−/− mice. Six-week-old mice were exposed to 10,000 lux for 30 min to induce light-induced retinal degeneration and ONL thickness was measured 7 days later by in vivo OCT imaging. Although both mouse strains showed retinal degeneration at this time, Tlr4−/−Abca4−/−Rdh8−/− mice had less severe degeneration with a thicker ONL and significantly less AF spots than Abca4−/−Rdh8−/− mice (Fig. 8A). Immunohistochemistry with anti-rhodopsin Ab to detect rod photoreceptors, peanut agglutinin lectin to stain the cone photoreceptor sheath, and anti-GFAP Ab revealed not only better preserved rod and cone photoreceptor cells but also weaker gliosis of glial cells in Tlr4−/−Abca4−/−Rdh8−/− mice (Fig. 8B). As compared with Abca4−/−Rdh8−/− mice, Tlr4−/−Abca4−/−Rdh8−/− mice also evidenced significantly fewer activated microglia, which were double positively stained by anti-Iba1 and anti-MHC II Abs in the subretinal space (Fig. 8C). Double positive cells were not detected in retinas of WT mice. Together, these data indicate that TLR4 contributes to light-induced retinal degeneration in this mouse model.
FIGURE 8.

Genetic elimination of Tlr4 results in milder light-induced retinal degeneration in Abca4−/−Rdh8−/− mice. Tlr4−/−Abca4−/−Rdh8−/− mice were established by cross-breeding Abca4−/−Rdh8−/− mice with Tlr4−/− mice. Abca4−/−Rdh8−/− mice from littermates were used as controls. Retinal degeneration was induced by exposing mice to 10,000 lux light for 30 min. A, ONL thickness was measured by OCT (left) and numbers of AF spots were counted by SLO (right) 7 days after light exposure. *, indicates p < 0.05. B, rod and cone photoreceptors were stained by anti-rhodopsin (Rho) Ab (red) and peanut agglutinin lectin (PNA) (green), respectively (upper and middle panels). Middle panels are magnified images of the dashed rectangles in the upper panel. Müller cells and astrocytes were stained by anti-GFAP Ab (lower panels). Bars indicate 50 μm. C, microglial activation was examined by staining with anti-MHC Class II (green) and anti-Iba1 (red) Abs. Nuclei were stained by DAPI (blue). Fewer numbers of activated microglia with double positive staining were counted in Tlr4−/−Abca4−/−Rdh8−/− (tKO) mice relative to Abca4−/−Rdh8−/− (dKO) mice. Bars indicate 50 μm. * indicates p < 0.05.
The role of TLR4 was also investigated in 6-month-old mice. Retinal sections from Tlr4−/−Abca4−/−Rdh8−/− mice had a more normal retinal architecture than Abca4−/−Rdh8−/− mice (Fig. 9A, upper panels). Additionally only 50% of Tlr4−/−Abca4−/−Rdh8−/− mice exhibited age-related degenerative retinal changes, whereas 100% Abca4−/−Rdh8−/− mice displayed such degeneration (Fig. 9B). The severity of retinal degeneration was also milder in Tlr4−/−Abca4−/−Rdh8−/− mice than in Abca4−/−Rdh8−/− mice. Fewer subretinal AF spots were present in 6-month-old Tlr4−/−Abca4−/−Rdh8−/− mice than in Abca4−/−Rdh8−/− mice (Fig. 9, A, lower panels, and C). Collectively, deficiency of Tlr4 in Abca4−/−Rdh8−/− mice was associated with milder retinal inflammation and degeneration.
FIGURE 9.
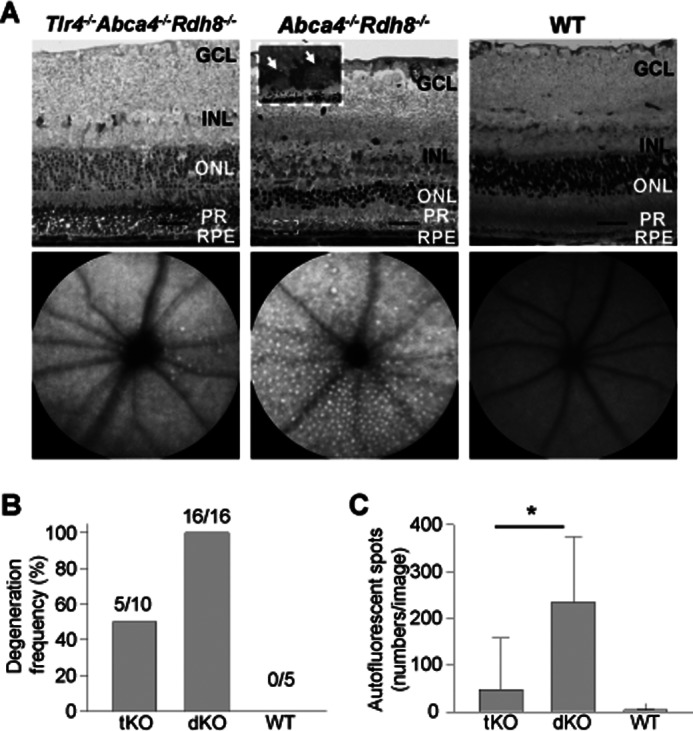
Tlr4 deficiency in Abca4−/−Rdh8−/− mice attenuates age-related retinal degeneration. A, retinal morphology (upper panels) and SLO images (lower panels) of 6-month old Tlr4−/−Abca4−/−Rdh8−/− (tKO), littermate Abca4−/−Rdh8−/− (dKO), and WT mice were examined after using a plastic embedment. A higher magnified image indicated as a dashed-line rectangle in the Abca4−/−Rdh8−/− section is presented in the inset. White arrows indicate infiltrating cells in the subretinal space. Bars indicate 20 μm. GCL, ganglion cell layer; INL, inner nuclear layer. B, the frequency of retinal degeneration was examined by OCT imaging. Although all dKO mice showed retinal degeneration, only 50% of tKO mice displayed this pathology. C, numbers of AF spots were assessed by using SLO. Numbers of AF spots were significantly decreased in tKO mice compared with dKO mice (right). * indicates p < 0.05.
Systemic Macrophage Depletion Attenuates Subretinal Infiltration of Microglia/Macrophages and Retinal Degeneration after Light Exposure
Findings of subretinal invasion of microglia/macrophages (Fig. 1), cytotoxicity of activated microglia (Fig. 6B), and a breakdown of the inner BRB (Fig. 4) in light-exposed Abca4−/−Rdh8−/− mice suggest a role of subretinal microglia/macrophages, especially those originating from circulating leukocytes, in the pathogenesis of light-induced retinal degeneration in Abca4−/−Rdh8−/− mice. To examine the effect of macrophage depletion on retinal degeneration, clodronate (Cl2MBP)-liposomes were treated. Systemic injection of Cl2MBP-liposomes is an established method for macrophage depletion used in disease models such as those for rheumatoid arthritis and neurological disorders (49). Cl2MBP is a non-toxic bisphosphonate and liposomes are not intrinsically toxic either (50). Cl2MBP-liposomes or PBS-liposomes were injected intravenously into Abca4−/−Rdh8−/− mice immediately after light exposure at 10,000 lux for 30 min because its effects can only be achieved after ingestion by activated macrophages. Effects observed after Cl2MBP-liposome administration included: 1) decreased numbers of AF spots upon SLO imaging (Fig. 10A, left panels and right upper); 2) much fewer Iba-1-positive cells in both the inner and outer retina of Cl2MBP-liposome-injected mice (Fig. 10A, right lower); 3) better preserved ONL structure, whereas vehicle injected groups showed complete loss of ONL 7 days after light (Fig. 10B); and 4) significantly reduced GFAP expression as compared with vehicle injection (Fig. 10C). These observations indicate a preventive effect of macrophage depletion on retinal inflammation and degeneration.
FIGURE 10.
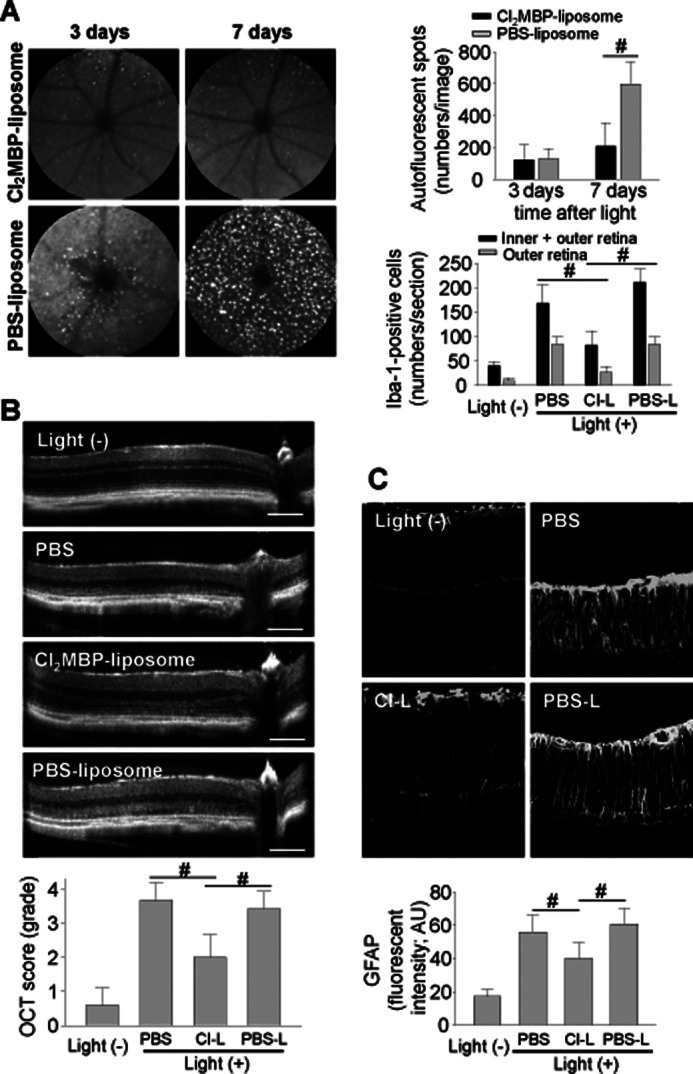
Systemic depletion of macrophages attenuates light-induced retinal degeneration and inflammation in Abca4−/−Rdh8−/− mice. Abca4−/−Rdh8−/− mice at 4 weeks of age were injected in the tail vein with Cl2MBP-liposomes (50 mg/kg) immediately after light exposure at 10,000 lux for 30 min. A, SLO images of the same animals 3 and 7 days after light exposure are shown (upper panels). Numbers of AF spots are presented (right upper panel). Error bars indicate mean ± S.D. (n > 10). Numbers of Iba-1-positive cells in the inner plus outer retina and the outer retina 7 days after light were counted (right lower panel). Cl-L, Cl2MBP-liposomes; PBS-L, PBS-liposomes. Error bars indicate mean ± S.D. (n > 6). #, indicates p < 0.05. B, SD-OCT imaging of Cl2MBP-liposomes (Cl-L), PBS-liposomes (PBS-L), and PBS injected Abca4−/−Rdh8−/− mice was carried out 7 days after light exposure (upper panels). OCT scores (15) are presented (lower panel). Bars indicate 100 μm. Error bars indicate mean ± S.D. (n > 10). # indicates p < 0.05. C, GFAP expression in Cl2MBP-liposome (Cl-L), PBS-liposome (PBS-L), and PBS injected Abca4−/−Rdh8−/− mice was examined 7 days after light exposure by staining with anti-GFAP Ab. Intensities of GFAP fluorescent signals were calculated with ImageJ software (lower panel). Bars indicate 50 μm. Error bars indicate mean ± S.D. (n > 5). # indicates p < 0.05.
Minocycline Administration Attenuates Activation of Microglia/Macrophages and Light-induced Retinal Degeneration in Abca4−/−Rdh8−/− Mice
The current study outlines the sequence of events caused by excessive accumulation of atRAL after bright light exposure, which could be a common factor in various degenerative retinal diseases (Fig. 11). Initial photoreceptor death from atRAL in Abca4−/−Rdh8−/− mice leads to overproduction of cellular debris, which can trigger infiltration of immune cells into the subretinal space. Accumulation of cellular debris in Mertk−/− mice also attracts immune cells into the subretinal space. Activation of these cells then results in a breakdown of the inner BRB, which in turn invites systemic macrophages into the retina. These observations indicate that residential and circulating macrophages contribute to secondary inflammatory retinal damage, and that reducing the numbers and/or inactivating these cells can ameliorate retinal degeneration. Indeed, systemic administration of Cl2MBP-liposomes, which effectively eliminates circulating macrophages, successfully attenuated not only subretinal microglia/macrophage infiltration but also retinal degeneration (Fig. 10).
FIGURE 11.
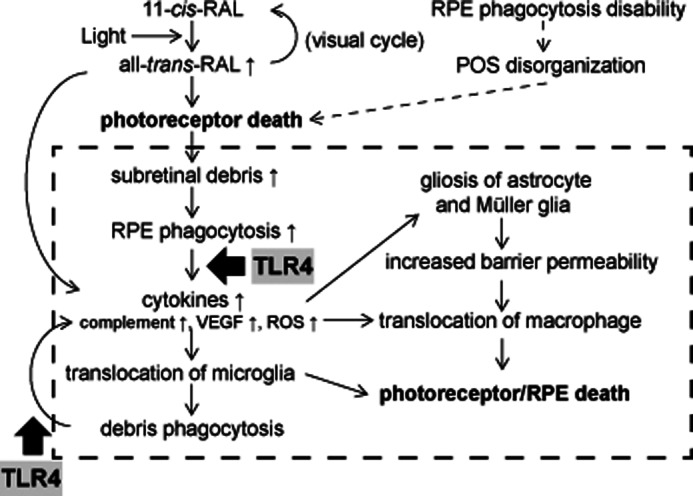
Sequence of events after light illumination of Abca4−/−Rdh8−/− mouse retinas. The visual chromophore, 11-cis-retinal, is isomerized to atRAL by light. Under normal conditions atRAL is reduced to all-trans-retinol, and used to regenerate 11-cis-retinal through the visual cycle (41). However, if excessive amounts of atRAL accumulate in photoreceptors, atRAL can cause photoreceptor death. RPE cells phagocytize debris from photoreceptors. Accelerated RPE phagocytosis of such debris results in higher production of chemokines and cytokines, which promote translocation of retinal microglia to the subretinal space. Although these microglial cells are involved in the clearance of photoreceptor debris, their phagocytosis of retinal proteins results in production of additional cytokines and chemokines that causes further inflammation. Gliosis of astrocytes and Müller glia in response to inflammation contributes to the breakdown of retinal blood barrier properties that allows monocytes/macrophages to infiltrate into the retina from the circulation. As long as overproduction of photoreceptor debris exists, this series of inflammatory reactions continues, leading to a chronic inflammatory and degenerative state. Possible common events in degenerative retinal disorders are indicated in the dotted line rectangle.
To gain further information about whether activation of microglia/macrophage affects retinal degenerative pathology and whether inactivation of these cells could be useful therapeutically, minocycline was tested for its ability to inhibit activation of microglia. Minocycline, a second generation tetracycline analog, which effectively crosses the blood-brain barrier was reported to be an inhibitor of microglial activation in the brain (51, 52) and the retina (53, 54). Inhibitory effects of minocycline on microglial activation were initially tested by using primary cultured retinal microglial cells. Because increased levels of Ccl2, Il1b, and Tnf were detected when microglia were co-cultured with POS (Fig. 6C), amounts of these proteins were quantified by ELISA in culture supernatants of retinal microglia incubated with POS in the presence of minocycline. Minocycline tended to inhibit the production of these proteins (Fig. 12A). ROS were examined in the presence or absence of 30 μg/ml of minocycline, because one of the hallmarks of activated microglia/macrophages is ROS generation (55). Co-incubation of isolated retinal microglia with 6 μg/100 μl of POS or 2 μm atRAL facilitated ROS generation, and this generation was inhibited when cells were treated with minocycline (Fig. 12B). RPE death was also prevented when microglia were co-incubated with minocycline (Fig. 12C). Similar results were obtained with BM-derived macrophages (data not shown).
FIGURE 12.
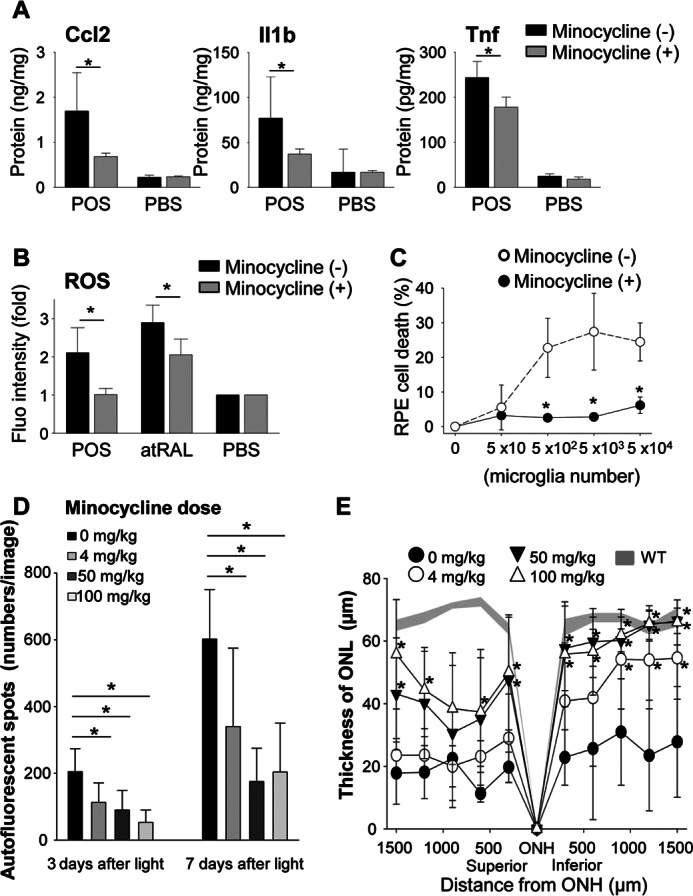
Minocycline inhibits activation of microglia in vitro and attenuates light-induced retinal degeneration in vivo. Retinal microglia (5 × 103/well in 96-well plates) isolated from 2-week-old C57BL/6 WT mice were incubated at 37 °C with or without 6 μg/100 μl of POS from 4-week-old WT mice. Cultured cells were divided into two groups without or with minocycline (30 μg/ml). A, production of Ccl2, Il1β, and Tnf was quantified by ELISA using culture supernatants of microglia cells with or without 24 h stimulation by POS. Minocycline was administrated as noted above. Error bars indicate mean ± S.D. (n = 6–9). * indicates p < 0.05. B, generation of ROS was examined by the ROS probe, DCF-DA. After 24 h incubation, cells were washed with PBS twice and 0.25 μm DCF-DA was co-incubated for 30 min. Error bars indicate mean ± S.D. (n = 3). * indicates p < 0.05. C, the influence of minocycline was tested using the RPE cell death system described in the legend to Fig. 6B. Minocycline administration prevented RPE cell death caused by microglia activation. Error bars indicate mean ± S.D. (n = 3). * indicates p < 0.05. D and E, the therapeutic effect of minocycline in vivo was tested in light-induced retinal degeneration model. Minocycline was administrated as described under ”Experimental Procedures.“ Minocycline administration attenuated AF spot accumulation (D) observed by using SLO, and retinal degeneration (E) represented by ONL thickness measured by OCT in a dose-dependent manner.
Next, in vivo effects of minocycline were examined on light-induced retinal degeneration in Abca4−/−Rdh8−/− mice. Minocycline was administered daily from 1 day before light exposure at 10,000 lux for 30 min to 7 days thereafter via intraperitoneal injection at different doses to 4-week-old Abca4−/−Rdh8−/− mice. In agreement with in vitro experiments, minocycline at all tested doses reduced the numbers of AF spots (Fig. 12D) and attenuated retinal degeneration (Fig. 12E). Of note, the 4 mg/kg dose, which is a clinical dose in humans (200 mg/day) (56), displayed significant protection (p < 0.05) from this retinal degeneration. Taken together, these observations suggest that activation of microglia/macrophages by photoreceptor proteins is a major contributor to the pathology of atRAL-associated retinal degeneration.
DISCUSSION
Microglia/Macrophage Infiltration into the Subretinal Space in Mice with Retinal Degeneration
Abca4−/−Rdh8−/− and Mertk−/− mice with retinal degeneration in this study evidenced cellular infiltration into the subretinal space (Figs. 1, 5, and 9). These subretinal cells are identified as microglia/macrophages because they are both Iba-1- and F4/80-positive by immunohistochemistry. Microglial cells are retinal resident macrophages that play a predominant role in retinal inflammation (9). Along with retinal microglia, BM-derived macrophages in circulating blood are also important contributors to tissue immune responses (10). Classification of microglia or macrophages by immunohistochemistry alone is difficult because Iba-1 is expressed in both types of cells (57). However, previous reports involving the brain have classified Iba-1-positive cells as microglia when they display a ramified shape and as macrophages when they show a round shape (58). In this study, most subretinal cells after light exposure displayed a ramified shape and thus are considered microglia (Fig. 1B), whereas round shaped Iba-1-positive cells were detected 14 days after light exposure (data not shown). Additionally, subretinal microglia/macrophages feature AF, which enables us to observe these invading cells by in vivo SLO imaging. Electron microscopic analyses of these cells demonstrated photoreceptor debris in their cell bodies, material thought to elicit the fluorescent signal (10, 59, 60). Subretinal infiltration of microglia/macrophages was also reported in dogs with retinal degeneration (61, 62).
Chemokines, Proinflammatory Cytokines, and Complement in Retinal Degeneration
Phagocytosis of retinal proteins resulted in the production of chemokines and proinflammatory cytokines including Ccl2, Il1b, and Tnf (Fig. 6C) as well as complement activation in Abca4−/−Rdh8−/− mice (Fig. 2). Retinal inflammation in these mice appears similar to that observed in human retinal disorders. Among these molecules, the role of CCL2 in retinal degeneration has been well studied in humans and mouse models. Ccl2 or its cognate Ccr2 knock-out mice were originally reported to represent human AMD-like retinal phenotypes (26). However, another study reported that Ccl2−/− or Ccr2−/− mice have macrophages in the retina, and that their AMD-like retinal phenotypes result from the normal aging process (60). A more recent study with the same mouse models reported age-related accumulation of subretinal microglia with photoreceptor and RPE cell death and impaired phagocytosis and impaired chemotactic function of monocytes. These investigators concluded that subretinal microglia in Ccl2−/− or Ccr2−/− mice are less able to migrate away from the subretinal space, and thus prolonged exposure to these microglia adversely affects photoreceptor and RPE cells (25). In addition to Ccl2, we found that expression of Ccl12 was also highly up-regulated (Ccl2, ∼180-fold; Ccl12, ∼75-fold increase relative to non-light exposed Abca4−/−Rdh8−/− mice) 3 days after light exposure (Fig. 2), and that this higher expression was associated with recruitment of microglia/macrophages into the subretinal space.
In addition to microglia/macrophages, RPE cells also produced Ccl2 when incubated with POS (Fig. 6C). This finding indicates that chemokine production from RPE cells after phagocytosis of POS plays an important role in recruiting microglia/macrophages into the subretinal space under normal conditions to maintain retinal homeostasis. When POS phagocytosis increases over maintenance levels, increased production of chemokines leads to additional recruitment of microglia/macrophages into the subretinal space. Although RPE cells are not typical immune cells, they do have important immune regulatory roles at the retinal/choroidal interface (9, 63–65). RPE cells may therefore have multiple immune-modulating functions including removal of photoreceptor cellular debris and microglial activation in vivo.
Phagocytosis of Photoreceptor Cellular Debris
Maintenance of POS involves production of POS proteins in the inner segments of photoreceptor cells and removal of shed POS disks by RPE cells. RPE cells phagocytose shed POS disks and 10% of the POS disks are replaced daily (40). We showed that light exposure at 10,000 lux for 30 min causes massive photoreceptor cell death in Abca4−/−Rdh8−/− mice within 24 h (24). We propose that this level of photoreceptor cell death exceeds the phagocytic capacity of the RPE. Primary cultured RPE cells co-cultured with POS produced Ccl2, Il1b, and Tnf (Fig. 6C), which can attract microglia/macrophages into the subretinal space and aid in the clearance of dead and degenerating photoreceptors. Notably, elevated mRNA expression levels of chemokines and their receptors started at 12 h and peaked 3 days after light exposure (Fig. 2). This time-dependent change correlates with the removal of dead cells observed by OCT (66). These observations also indicate a relationship between the production of cellular debris and the activation of subretinal immune cells. Even though subretinal translocation of microglia/macrophages is part of a defense mechanism, these cells may in turn cause secondary photoreceptor cell death through retinal inflammation by producing additional chemokines and cytokines.
Mertk−/− mice with non-phagocytic RPE cells also exhibited microglia/macrophage invasion into the subretinal space (Fig. 5), suggesting that accumulation of cellular debris in the subretinal space is an important factor in initiating subretinal translocation of microglia/macrophages. MERTK plays an important role in macrophage phagocytosis, and elimination of this protein nearly abolishes thymic apoptotic cell phagocytosis by macrophages in mice (67). However, phagocytosis by retinal microglia/macrophages in Mertk−/− mice is not well understood. A recent report indicated that the role of Axl/Mertk/Tyro3 in mouse macrophages, dendritic cells, and RPE cells are different. Whereas Mertk is indispensable for RPE phagocytosis, dendritic cells rely primarily on Axl and Tyro3 for their phagocytosis. This study also suggests that phagocytosis by tissue-specific macrophages can be regulated by a mechanism that differs from that of monocyte-derived macrophages (37). Existence of dendritic cells in the retina has been reported (68), and these cells could produce chemokines to attract microglia/macrophages in Mertk−/− mice. Further investigation is required to address the mechanism(s) of microglia/macrophage infiltration in Mertk−/− mice.
Possible Roles for TLRs in Degenerative Retinal Diseases
Involvement of dysregulated innate immunity and chronic inflammation in pathogenesis of retinal degenerative diseases including AMD has been suggested by evidence that includes increased AMD susceptibility of individuals with certain single nucleotide polymorphisms for key molecules involved in complement activation (4, 5, 69, 70). Although single nucleotide polymorphisms of TLR3 and TLR4 have been reported to correlate with AMD susceptibility (71, 72), other conflicting reports (73, 74) indicate only a weak relationship between AMD and these TLRs. Studies other than single nucleotide polymorphism analyses are needed to resolve this discrepancy. Additional evidence for the role of TLRs in retinal physiology and pathology has been reported (75–78). Possible contributions of TLRs to the pathogenesis of neurodegeneration and heart disease have also been discussed (79, 80), and endogenous ligands for these receptors have been identified (47, 81).
We previously reported a role for TLR3, the receptor for double-stranded RNA, in retinal degeneration observed in Abca4−/−Rdh8−/− mice (17). That study demonstrated important pathogenic roles for Tlr3, including: 1) changes in expression of Tlr3 and other Tlrs in retinas of mice with retinal degeneration; 2) weaker phenotypes of retinal degeneration in mice deficient in Tlr3 and Trif, the latter a downstream target of the TLR3 signal; and 3) activation of NFκB via Tlr3 by endogenous products that are produced from dying/dead photoreceptors (17). Because the endogenous products from dying/dead photoreceptors fail to activate Tlr3 after treatment with nucleases, we concluded that Tlr3 recognizes endogenous nucleotides.
In the current study, increased expression of Tlr2 and Tlr4 were observed after light exposure in Abca4−/−Rdh8−/− mice (Fig. 7C), and milder retinal degeneration was observed in Tlr4-deficient mice (Figs. 8 and 9). Of note, Tlr4-deficient macrophages did not produce chemokines in response to photoreceptor proteins (Fig. 7D). Endogenous TLR4 ligands were detected in the protein fraction, which was extracted by 2% PFOA (Fig. 7B), indicating that they are not soluble under native conditions. A possible source of these ligands under normal conditions could be small amounts of POS, which have undergone daily phagocytosis by RPE cells. This could contribute to the basal production of chemokines required to maintain the subretinal environment. Because of these features, TLR4 ligands are not likely to elicit strong immune reactions unless larger amounts of photoreceptor debris are released into the subretinal space from injured photoreceptors, phagocytosed, and digested into small pieces.
Together, these observations indicate that TLRs can recognize endogenous products as their ligands, and play an important role in pathogenesis of degenerative neural diseases, especially in the retina. Although the predominant TLR4/MD-2 ligand is the Lipid A moiety of LPS, several proteins have been shown to activate TLR4. These include danger-associated molecular pattern molecules such as heat shock proteins (Hsp 60/70), high mobility group box-1, and calcium-binding proteins (S100A8/A9) (82–84).
Contribution of Microglia/Macrophages to Retinal Degeneration and Possible Therapeutic Strategies
Invasion of immune cells, including microglia/macrophage, into the subretinal space has been observed during retinal degeneration caused by genetic impairments (12, 85), age (42, 86), and light (10, 60). However, it remains to be determined whether these cells contribute to retinal disruption (20) or to photoreceptor protection (87, 88). Conflicting data may be explained by the presence of macrophages at different states of activation including M1 and M2 macrophages (89). Results from the current study demonstrate that activation of retinal immune cells by overproduction of cellular debris due to photoreceptor death results in inner BRB breakdown, which invites systemic macrophages into the retina. These observations indicate that resident and circulating macrophages contribute to secondary retinal damage from inflammation, and that reduced numbers and/or inactivation of these cells can ameliorate retinal degeneration. Systemic administration of Cl2MBP-liposomes, which can effectively eliminate circulating macrophages, successfully attenuated not only subretinal microglia/macrophage infiltration but also retinal degeneration (Fig. 10). Minocycline, reported as a microglial inhibitor (51–54), prevented production of ROS, Ccl2, Il1b, and Tnf from retinal microglia (Fig. 12A). Furthermore, minocycline treatment in vivo significantly ameliorated light-induced retinal degeneration in Abca4−/−Rdh8−/− mice (Fig. 12, D and E). These results provide evidence for the involvement of microglia/macrophages in retinal tissue damage of mice.
In summary, excessive production of photoreceptor cellular debris by atRAL and accumulation of photoreceptor debris resulting from impaired phagocytosis trigger overproduction of chemokines and cytokines from RPE and phagocytic cells resulting in translocation of microglia and macrophages into the subretinal space. Although these infiltrated cells initially help to remove retinal debris, inadequate removal of the debris later causes destructive retinal inflammation that can result in chronic retinal degeneration. Activation of the retinal immune system due to the imbalance of production and digestion of photoreceptors could represent a pathology common to many retinal degenerative diseases. Inhibiting microglia/macrophage infiltration into the subretinal space and modulating their activation could provide a new treatment strategy for retinal disorders such as AMD, Stargardt disease, and RP.
Acknowledgments
We thank Drs. M. Golczak, Z. Dong, B. Wang, S. Howell, M.-J. Lee, and L. Perusek (Case Western Reserve University) for comments and technical support.
This work was supported, in whole or in part, by National Institutes of Health Grants EY022658, EY019031, EY019880, EY009339, EY021126, and EY11373 and grants from the Research to Prevent Blindness Foundation, Foundation Fighting Blindness, Fight for Sight, and Ohio Lions Eye Research Foundation.
- AMD
- age-related macular degeneration
- atRAL
- all-trans-retinal
- ABCA4
- ATP-binding cassette transporter 4
- AF
- autofluorescent
- BRB
- blood-retinal barrier
- CFH
- complement factor H
- GFAP
- glial fibrillary acidic protein
- Mertk
- c-mer proto-oncogene tyrosine kinase
- POS
- photoreceptor outer segments
- RDH
- retinol dehydrogenase
- RP
- retinitis pigmentosa
- RPE
- retinal pigment epithelium
- SD-OCT
- spectral-domain optic coherence tomography
- SLO
- scanning laser ophthalmoscopy
- TLR
- Toll-like receptor
- LDH
- lactate dehydrogenase
- ROS
- reactive oxygen species
- Vegfa
- vascular endothelial growth factor A
- ConA
- concanavalin A
- ONL
- outer nuclear layers
- DCF-DA
- dichlorofluorescein diacetate
- PFOA
- perfluorooctanoic acid
- BM
- bone marrow
- qRT
- quantitative RT.
REFERENCES
- 1. Friedman D. S., O'Colmain B. J., Muñoz B., Tomany S. C., McCarty C., de Jong P. T., Nemesure B., Mitchell P., Kempen J. (2004) Prevalence of age-related macular degeneration in the United States. Arch. Ophthalmol. 122, 564–572 [DOI] [PubMed] [Google Scholar]
- 2. Molday R. S., Zhang K. (2010) Defective lipid transport and biosynthesis in recessive and dominant Stargardt macular degeneration. Prog. Lipid Res. 49, 476–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berger W., Kloeckener-Gruissem B., Neidhardt J. (2010) The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 29, 335–375 [DOI] [PubMed] [Google Scholar]
- 4. Hageman G. S., Anderson D. H., Johnson L. V., Hancox L. S., Taiber A. J., Hardisty L. I., Hageman J. L., Stockman H. A., Borchardt J. D., Gehrs K. M., Smith R. J., Silvestri G., Russell S. R., Klaver C. C., Barbazetto I., Chang S., Yannuzzi L. A., Barile G. R., Merriam J. C., Smith R. T., Olsh A. K., Bergeron J., Zernant J., Merriam J. E., Gold B., Dean M., Allikmets R. (2005) A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 102, 7227–7232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Edwards A. O., Ritter R., 3rd, Abel K. J., Manning A., Panhuysen C., Farrer L. A. (2005) Complement factor H polymorphism and age-related macular degeneration. Science 308, 421–424 [DOI] [PubMed] [Google Scholar]
- 6. Allikmets R., Shroyer N. F., Singh N., Seddon J. M., Lewis R. A., Bernstein P. S., Peiffer A., Zabriskie N. A., Li Y., Hutchinson A., Dean M., Lupski J. R., Leppert M. (1997) Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 277, 1805–1807 [DOI] [PubMed] [Google Scholar]
- 7. Dryja T. P., McGee T. L., Reichel E., Hahn L. B., Cowley G. S., Yandell D. W., Sandberg M. A., Berson E. L. (1990) A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 343, 364–366 [DOI] [PubMed] [Google Scholar]
- 8. Medzhitov R. (2008) Origin and physiological roles of inflammation. Nature 454, 428–435 [DOI] [PubMed] [Google Scholar]
- 9. Xu H., Chen M., Forrester J. V. (2009) Para-inflammation in the aging retina. Prog. Retin. Eye Res. 28, 348–368 [DOI] [PubMed] [Google Scholar]
- 10. Joly S., Francke M., Ulbricht E., Beck S., Seeliger M., Hirrlinger P., Hirrlinger J., Lang K. S., Zinkernagel M., Odermatt B., Samardzija M., Reichenbach A., Grimm C., Remé C. E. (2009) Cooperative phagocytes. Resident microglia and bone marrow immigrants remove dead photoreceptors in retinal lesions. Am. J. Pathol. 174, 2310–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ding X., Patel M., Chan C. C. (2009) Molecular pathology of age-related macular degeneration. Prog. Retin. Eye Res. 28, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gupta N., Brown K. E., Milam A. H. (2003) Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp. Eye Res. 76, 463–471 [DOI] [PubMed] [Google Scholar]
- 13. Radu R. A., Hu J., Yuan Q., Welch D. L., Makshanoff J., Lloyd M., McMullen S., Travis G. H., Bok D. (2011) Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J. Biol. Chem. 286, 18593–18601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou J., Jang Y. P., Kim S. R., Sparrow J. R. (2006) Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc. Natl. Acad. Sci. U.S.A. 103, 16182–16187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maeda A., Golczak M., Chen Y., Okano K., Kohno H., Shiose S., Ishikawa K., Harte W., Palczewska G., Maeda T., Palczewski K. (2012) Primary amines protect against retinal degeneration in mouse models of retinopathies. Nat. Chem. Biol. 8, 170–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maeda A., Maeda T., Golczak M., Palczewski K. (2008) Retinopathy in mice induced by disrupted all-trans-retinal clearance. J. Biol. Chem. 283, 26684–26693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shiose S., Chen Y., Okano K., Roy S., Kohno H., Tang J., Pearlman E., Maeda T., Palczewski K., Maeda A. (2011) Toll-like receptor 3 is required for development of retinopathy caused by impaired all-trans-retinal clearance in mice. J. Biol. Chem. 286, 15543–15555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Haeseleer F., Jang G. F., Imanishi Y., Driessen C. A., Matsumura M., Nelson P. S., Palczewski K. (2002) Dual-substrate specificity short chain retinol dehydrogenases from the vertebrate retina. J. Biol. Chem. 277, 45537–45546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maeda A., Maeda T., Imanishi Y., Kuksa V., Alekseev A., Bronson J. D., Zhang H., Zhu L., Sun W., Saperstein D. A., Rieke F., Baehr W., Palczewski K. (2005) Role of photoreceptor-specific retinol dehydrogenase in the retinoid cycle in vivo. J. Biol. Chem. 280, 18822–18832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ma W., Zhao L., Fontainhas A.M., Fariss R. N., Wong W. T. (2009) Microglia in the mouse retina alter the structure and function of retinal pigmented epithelial cells. A potential cellular interaction relevant to AMD. PLoS One 4, e7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson A. C., Li X., Pearlman E. (2008) MyD88 functions as a negative regulator of TLR3/TRIF-induced corneal inflammation by inhibiting activation of c-Jun N-terminal kinase. J. Biol. Chem. 283, 3988–3996 [DOI] [PubMed] [Google Scholar]
- 22. Diemer T., Gibbs D., Williams D. S. (2008) Analysis of the rate of disk membrane digestion by cultured RPE cells. Adv. Exp. Med. Biol. 613, 321–326 [DOI] [PubMed] [Google Scholar]
- 23. Liang Y., Fotiadis D., Filipek S., Saperstein D. A., Palczewski K., Engel A. (2003) Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J. Biol. Chem. 278, 21655–21662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maeda A., Maeda T., Golczak M., Chou S., Desai A., Hoppel C. L., Matsuyama S., Palczewski K. (2009) Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J. Biol. Chem. 284, 15173–15183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen M., Forrester J. V., Xu H. (2011) Dysregulation in retinal para-inflammation and age-related retinal degeneration in CCL2 or CCR2 deficient mice. PLoS One 6, e22818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ambati J., Anand A., Fernandez S., Sakurai E., Lynn B. C., Kuziel W. A., Rollins B. J., Ambati B. K. (2003) An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat. Med. 9, 1390–1397 [DOI] [PubMed] [Google Scholar]
- 27. Tuo J., Smith B. C., Bojanowski C. M., Meleth A. D., Gery I., Csaky K. G., Chew E. Y., Chan C. C. (2004) The involvement of sequence variation and expression of CX3CR1 in the pathogenesis of age-related macular degeneration. FASEB J. 18, 1297–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lavalette S., Raoul W., Houssier M., Camelo S., Levy O., Calippe B., Jonet L., Behar-Cohen F., Chemtob S., Guillonneau X., Combadière C., Sennlaub F. (2011) Interleukin-1β inhibition prevents choroidal neovascularization and does not exacerbate photoreceptor degeneration. Am. J. Pathol. 178, 2416–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu J., Zhu D., He S., Spee C., Ryan S. J., Hinton D. R. (2011) Transcriptional regulation of bone morphogenetic protein 4 by tumor necrosis factor and its relationship with age-related macular degeneration. FASEB J. 25, 2221–2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lichtlen P., Lam T. T., Nork T. M., Streit T., Urech D. M. (2010) Relative contribution of VEGF and TNF-α in the cynomolgus laser-induced CNV model. Comparing the efficacy of bevacizumab, adalimumab, and ESBA105. Invest. Ophthalmol. Vis. Sci. 51, 4738–4745 [DOI] [PubMed] [Google Scholar]
- 31. Theodossiadis P. G., Liarakos V. S., Sfikakis P. P., Vergados I. A., Theodossiadis G. P. (2009) Intravitreal administration of the anti-tumor necrosis factor agent infliximab for neovascular age-related macular degeneration. Am. J. Ophthalmol. 147, 825–830 [DOI] [PubMed] [Google Scholar]
- 32. Anderson D. H., Mullins R. F., Hageman G. S., Johnson L. V. (2002) A role for local inflammation in the formation of drusen in the aging eye. Am. J. Ophthalmol. 134, 411–431 [DOI] [PubMed] [Google Scholar]
- 33. Witmer A. N., Vrensen G. F., Van Noorden C. J., Schlingemann R. O. (2003) Vascular endothelial growth factors and angiogenesis in eye disease. Prog. Retin. Eye Res. 22, 1–29 [DOI] [PubMed] [Google Scholar]
- 34. Cai J., Wu L., Qi X., Li Calzi S., Caballero S., Shaw L., Ruan Q., Grant M. B., Boulton M. E. (2011) PEDF regulates vascular permeability by a γ-secretase-mediated pathway. PLoS One 6, e21164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kaur C., Foulds W. S., Ling E. A. (2008) Blood-retinal barrier in hypoxic ischaemic conditions. basic concepts, clinical features and management. Prog. Retin. Eye Res. 27, 622–647 [DOI] [PubMed] [Google Scholar]
- 36. Joussen A. M., Murata T., Tsujikawa A., Kirchhof B., Bursell S. E., Adamis A. P. (2001) Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am. J. Pathol. 158, 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seitz H. M., Camenisch T. D., Lemke G., Earp H. S., Matsushima G. K. (2007) Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J. Immunol. 178, 5635–5642 [DOI] [PubMed] [Google Scholar]
- 38. Nandrot E. F., Dufour E. M. (2010) Mertk in daily retinal phagocytosis. A history in the making. Adv. Exp. Med. Biol. 664, 133–140 [DOI] [PubMed] [Google Scholar]
- 39. Duncan J. L., LaVail M. M., Yasumura D., Matthes M. T., Yang H., Trautmann N., Chappelow A. V., Feng W., Earp H. S., Matsushima G. K., Vollrath D. (2003) An RCS-like retinal dystrophy phenotype in mer knockout mice. Invest. Ophthalmol. Vis. Sci. 44, 826–838 [DOI] [PubMed] [Google Scholar]
- 40. Kevany B.M., Palczewski K. (2010) Phagocytosis of retinal rod and cone photoreceptors. Physiology 25, 8–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Palczewski K. (2010) Retinoids for treatment of retinal diseases. Trends Pharmacol. Sci. 31, 284–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nussenblatt R. B., Ferris F., 3rd. (2007) Age-related macular degeneration and the immune response. Implications for therapy. Am. J. Ophthalmol. 144, 618–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yoshida A., Elner S. G., Bian Z. M., Kindezelskii A. L., Petty H. R., Elner V. M. (2003) Activated monocytes induce human retinal pigment epithelial cell apoptosis through caspase-3 activation. Lab. Invest. 83, 1117–1129 [DOI] [PubMed] [Google Scholar]
- 44. Fishel S., Jackson P., Webster J., Faratian B. (1988) Endotoxins in culture medium for human in vitro fertilization. Fertil. Steril. 49, 108–111 [DOI] [PubMed] [Google Scholar]
- 45. Kadiyala C. S., Tomechko S. E., Miyagi M. (2010) Perfluorooctanoic acid for shotgun proteomics. PLoS One 5, e15332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kono H., Rock K. L. (2008) How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8, 279–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shichita T., Hasegawa E., Kimura A., Morita R., Sakaguchi R., Takada I., Sekiya T., Ooboshi H., Kitazono T., Yanagawa T., Ishii T., Takahashi H., Mori S., Nishibori M., Kuroda K., Akira S., Miyake K., Yoshimura A. (2012) Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat. Med. 18, 911–917 [DOI] [PubMed] [Google Scholar]
- 48. Erridge C. (2010) Endogenous ligands of TLR2 and TLR4. Agonists or assistants? J. Leukocyte Biol. 87, 989–999 [DOI] [PubMed] [Google Scholar]
- 49. van Rooijen N., van Kesteren-Hendrikx E. (2002) Clodronate liposomes. Perspectives in research and therapeutics. J. Liposome Res. 12, 81–94 [DOI] [PubMed] [Google Scholar]
- 50. Van Rooijen N., Sanders A. (1994) Liposome mediated depletion of macrophages. Mechanism of action, preparation of liposomes and applications. J. Immunol. Methods 174, 83–93 [DOI] [PubMed] [Google Scholar]
- 51. Wu D. C., Jackson-Lewis V., Vila M., Tieu K., Teismann P., Vadseth C., Choi D. K., Ischiropoulos H., Przedborski S. (2002) Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. 22, 1763–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Filipovic R., Zecevic N. (2008) Neuroprotective role of minocycline in co-cultures of human fetal neurons and microglia. Exp. Neurol. 211, 41–51 [DOI] [PubMed] [Google Scholar]
- 53. Wang A. L., Yu A. C., Lau L. T., Lee C., Wu le M., Zhu X., Tso M. O. (2005) Minocycline inhibits LPS-induced retinal microglia activation. Neurochem. Int. 47, 152–158 [DOI] [PubMed] [Google Scholar]
- 54. Zhao L., Ma W., Fariss R. N., Wong W. T. (2011) Minocycline attenuates photoreceptor degeneration in a mouse model of subretinal hemorrhage microglial. Inhibition as a potential therapeutic strategy. Am. J. Pathol. 179, 1265–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bauer M., Goldstein M., Christmann M., Becker H., Heylmann D., Kaina B. (2011) Human monocytes are severely impaired in base and DNA double-strand break repair that renders them vulnerable to oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 108, 21105–21110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen X., Ma X., Jiang Y., Pi R., Liu Y., Ma L. (2011) The prospects of minocycline in multiple sclerosis. J. Neuroimmunol. 235, 1–8 [DOI] [PubMed] [Google Scholar]
- 57. Imai Y., Ibata I., Ito D., Ohsawa K., Kohsaka S. (1996) A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem. Biophys. Res. Commun. 224, 855–862 [DOI] [PubMed] [Google Scholar]
- 58. Ji K. A., Yang M. S., Jeong H. K., Min K. J., Kang S. H., Jou I., Joe E. H. (2007) Resident microglia die and infiltrated neutrophils and monocytes become major inflammatory cells in lipopolysaccharide-injected brain. Glia 55, 1577–1588 [DOI] [PubMed] [Google Scholar]
- 59. Maeda A., Okano K., Park P. S., Lem J., Crouch R. K., Maeda T., Palczewski K. (2010) Palmitoylation stabilizes unliganded rod opsin. Proc. Natl. Acad. Sci. U.S.A. 107, 8428–8433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Luhmann U. F., Robbie S., Munro P. M., Barker S. E., Duran Y., Luong V., Fitzke F. W., Bainbridge J. W., Ali R. R., MacLaren R. E. (2009) The drusenlike phenotype in aging Ccl2-knockout mice is caused by an accelerated accumulation of swollen autofluorescent subretinal macrophages. Invest. Ophthalmol. Vis. Sci. 50, 5934–5943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Beltran W. A., Acland G. M., Aguirre G. D. (2009) Age-dependent disease expression determines remodeling of the retinal mosaic in carriers of RPGR exon ORF15 mutations. Invest. Ophthalmol. Vis. Sci. 50, 3985–3995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gu D., Beltran W. A., Pearce-Kelling S., Li Z., Acland G. M., Aguirre G. D. (2009) Steroids do not prevent photoreceptor degeneration in the light-exposed T4R rhodopsin mutant dog retina irrespective of AP-1 inhibition. Invest. Ophthalmol. Vis. Sci. 50, 3482–3494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sugita S., Seki K., Yokozawa K., Tochigi N., Furuta K., Hisaoka M., Hashimoto H., Shimoda T., Hasegawa T. (2009) Analysis of CHOP rearrangement in pleomorphic liposarcomas using fluorescence in situ hybridization. Cancer Sci. 100, 82–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Usui Y., Okunuki Y., Hattori T., Kezuka T., Keino H., Ebihara N., Sugita S., Usui M., Goto H., Takeuchi M. (2008) Functional expression of B7H1 on retinal pigment epithelial cells. Exp. Eye Res. 86, 52–59 [DOI] [PubMed] [Google Scholar]
- 65. Chen H., Liu B., Lukas T. J., Neufeld A. H. (2008) The aged retinal pigment epithelium/choroid. A potential substratum for the pathogenesis of age-related macular degeneration. PLoS One 3, e2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Maeda A., Golczak M., Maeda T., Palczewski K. (2009) Limited roles of Rdh8, Rdh12, and Abca4 in all-trans-retinal clearance in mouse retina. Invest. Ophthalmol. Vis. Sci. 50, 5435–5443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Scott R. S., McMahon E. J., Pop S. M., Reap E. A., Caricchio R., Cohen P. L., Earp H. S., Matsushima G. K. (2001) Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–211 [DOI] [PubMed] [Google Scholar]
- 68. Xu H., Dawson R., Forrester J. V., Liversidge J. (2007) Identification of novel dendritic cell populations in normal mouse retina. Invest. Ophthalmol. Vis. Sci. 48, 1701–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Haines J. L., Hauser M. A., Schmidt S., Scott W. K., Olson L. M., Gallins P., Spencer K. L., Kwan S. Y., Noureddine M., Gilbert J. R., Schnetz-Boutaud N., Agarwal A., Postel E. A., Pericak-Vance M. A. (2005) Complement factor H variant increases the risk of age-related macular degeneration. Science 308, 419–421 [DOI] [PubMed] [Google Scholar]
- 70. Yates J. R., Sepp T., Matharu B. K., Khan J. C., Thurlby D. A., Shahid H., Clayton D. G., Hayward C., Morgan J., Wright A. F., Armbrecht A. M., Dhillon B., Deary I. J., Redmond E., Bird A. C., Moore A. T., and Genetic Factors in AMD Study Group (2007) Complement C3 variant and the risk of age-related macular degeneration. N. Engl. J. Med. 357, 553–561 [DOI] [PubMed] [Google Scholar]
- 71. Yang Z., Stratton C., Francis P. J., Kleinman M. E., Tan P. L., Gibbs D., Tong Z., Chen H., Constantine R., Yang X., Chen Y., Zeng J., Davey L., Ma X., Hau V. S., Wang C., Harmon J., Buehler J., Pearson E., Patel S., Kaminoh Y., Watkins S., Luo L., Zabriskie N. A., Bernstein P. S., Cho W., Schwager A., Hinton D. R., Klein M. L., Hamon S. C., Simmons E., Yu B., Campochiaro B., Sunness J. S., Campochiaro P., Jorde L., Parmigiani G., Zack D. J., Katsanis N., Ambati J., Zhang K. (2008) Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N. Engl. J. Med. 359, 1456–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zareparsi S., Buraczynska M., Branham K. E., Shah S., Eng D., Li M., Pawar H., Yashar B. M., Moroi S. E., Lichter P. R., Petty H. R., Richards J. E., Abecasis G. R., Elner V. M., Swaroop A. (2005) Toll-like receptor 4 variant D299G is associated with susceptibility to age-related macular degeneration. Hum. Mol. Genet. 14, 1449–1455 [DOI] [PubMed] [Google Scholar]
- 73. Edwards A. O., Chen D., Fridley B. L., James K. M., Wu Y., Abecasis G., Swaroop A., Othman M., Branham K., Iyengar S. K., Sivakumaran T. A., Klein R., Klein B. E., Tosakulwong N. (2008) Toll-like receptor polymorphisms and age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 49, 1652–1659 [DOI] [PubMed] [Google Scholar]
- 74. Cho Y., Wang J. J., Chew E. Y., Ferris F. L., 3rd, Mitchell P., Chan C. C., Tuo J. (2009) Toll-like receptor polymorphisms and age-related macular degeneration. Replication in three case-control samples. Invest. Ophthalmol. Vis. Sci. 50, 5614–5618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ko M. K., Saraswathy S., Parikh J. G., Rao N. A. (2011) The role of TLR4 activation in photoreceptor mitochondrial oxidative stress. Invest. Ophthalmol. Vis. Sci. 52, 5824–5835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sen A., Chowdhury I. H., Mukhopadhyay D., Paine S. K., Mukherjee A., Mondal L. K., Chatterjee M., Bhattacharya B. (2011) Increased Toll-like receptor-2 expression on nonclassic CD16+ monocytes from patients with inflammatory stage of Eales' disease. Invest. Ophthalmol. Vis. Sci. 52, 6940–6948 [DOI] [PubMed] [Google Scholar]
- 77. Tarallo V., Hirano Y., Gelfand B. D., Dridi S., Kerur N., Kim Y., Cho W. G., Kaneko H., Fowler B. J., Bogdanovich S., Albuquerque R. J., Hauswirth W. W., Chiodo V. A., Kugel J. F., Goodrich J. A., Ponicsan S. L., Chaudhuri G., Murphy M. P., Dunaief J. L., Ambati B. K., Ogura Y., Yoo J. W., Lee D. K., Provost P., Hinton D. R., Núñez G., Baffi J. Z., Kleinman M. E., Ambati J. (2012) DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 149, 847–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kleinman M. E., Kaneko H., Cho W. G., Dridi S., Fowler B. J., Blandford A. D., Albuquerque R. J., Hirano Y., Terasaki H., Kondo M., Fujita T., Ambati B. K., Tarallo V., Gelfand B. D., Bogdanovich S., Baffi J. Z., Ambati J. (2012) Short-interfering RNAs induce retinal degeneration via TLR3 and IRF3. Mol. Ther. 20, 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lehnardt S. (2010) Innate immunity and neuroinflammation in the CNS. The role of microglia in Toll-like receptor-mediated neuronal injury. Glia 58, 253–263 [DOI] [PubMed] [Google Scholar]
- 80. Owens T. (2009) Toll-like receptors in neurodegeneration. Curr. Top. Microbiol. Immunol. 336, 105–120 [DOI] [PubMed] [Google Scholar]
- 81. West X. Z., Malinin N. L., Merkulova A. A., Tischenko M., Kerr B. A., Borden E. C., Podrez E. A., Salomon R. G., Byzova T. V. (2010) Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 467, 972–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Vogl T., Tenbrock K., Ludwig S., Leukert N., Ehrhardt C., van Zoelen M. A., Nacken W., Foell D., van der Poll T., Sorg C., Roth J. (2007) Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 13, 1042–1049 [DOI] [PubMed] [Google Scholar]
- 83. Thériault J. R., Mambula S. S., Sawamura T., Stevenson M. A., Calderwood S. K. (2005) Extracellular HSP70 binding to surface receptors present on antigen presenting cells and endothelial/epithelial cells. FEBS Lett. 579, 1951–1960 [DOI] [PubMed] [Google Scholar]
- 84. Asea A., Rehli M., Kabingu E., Boch J. A., Bare O., Auron P. E., Stevenson M. A., Calderwood S. K. (2002) Novel signal transduction pathway utilized by extracellular HSP70. Role of Toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 277, 15028–15034 [DOI] [PubMed] [Google Scholar]
- 85. Sanyal S., De Ruiter A., Hawkins R. K. (1980) Development and degeneration of retina in rds mutant mice. Light microscopy. J. Comp. Neurol. 194, 193–207 [DOI] [PubMed] [Google Scholar]
- 86. Xu H., Chen M., Manivannan A., Lois N., Forrester J. V. (2008) Age-dependent accumulation of lipofuscin in perivascular and subretinal microglia in experimental mice. Aging Cell 7, 58–68 [DOI] [PubMed] [Google Scholar]
- 87. Bruban J., Maoui A., Chalour N., An N., Jonet L., Feumi C., Tréton J., Sennlaub F., Behar-Cohen F., Mascarelli F., Dinet V. (2011) CCR2/CCL2-mediated inflammation protects photoreceptor cells from amyloid-β-induced apoptosis. Neurobiol. Dis. 42, 55–72 [DOI] [PubMed] [Google Scholar]
- 88. Sasahara M., Otani A., Oishi A., Kojima H., Yodoi Y., Kameda T., Nakamura H., Yoshimura N. (2008) Activation of bone marrow-derived microglia promotes photoreceptor survival in inherited retinal degeneration. Am. J. Pathol. 172, 1693–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Henkel J. S., Beers D. R., Zhao W., Appel S. H. (2009) Microglia in ALS. The good, the bad, and the resting. J. Neuroimmune Pharmacol. 4, 389–398 [DOI] [PubMed] [Google Scholar]