

Abstract
The histone deacetylase inhibitor belinostat is being evaluated clinically as a single agent in the treatment of Peripheral T-cell lymphomas and in combination with other anticancer agents to treat a wide range of human cancers including acute leukemias and solid tumors. To determine the pharmacokinetics of belinostat in the NCI ODWG liver dysfunction study, we developed and validated an LC-MS/MS assay for the quantitation of belinostat and five major metabolites in 0.05 mL human plasma. After protein precipitation, chromatographic separation was achieved with a Waters Acquity BEH C18 column and a linear gradient of 0.1% formic acid in acetonitrile and water. Detection with an ABI 4000Q mass spectrometer utilized both electrospray positive and negative mode ionization. The assay was linear from 30–5000 ng/mL for all six analytes and proved to be accurate (92.0–104.4%) and precise (<13.7%CV), and fulfilled FDA criteria for bioanalytical method validation. We demonstrated the suitability of this assay for measuring parent drug and five major metabolites in plasma from a patient who was administered belinostat IV at a dose of 400 mg/m2. The LC-MS/MS assay that has been developed will be an essential tool to further define the metabolism and pharmacology of belinostat in the ongoing liver organ dysfunction as well as other studies that investigate belinostat with other anticancer agents.
Keywords: HDAC, belinostat, tandem mass spectrometry, metabolites, assay, validation
1 Introduction
Research into epigenetics processes has furthered our understanding of malignant behavior and continues to provide novel targets to improve cancer therapy[1]. One of the most studied epigenetic changes is that of histone acetylation. Acetylation and deacetylation of histones plays an important role in transcriptional regulation. Lysine residues on histones and non-histone proteins are acetylated by histone acetyl-transferases (HATs) and deacetylated by histone deacetylases (HDACs), resulting in a lysine acetylation equilibrium. Acetylation of histone lysines allows transcriptional activation, and by shifting the equilibrium away from acetylation, HDACs are thought to act as transcriptional repressors. HDACs are often overexpressed in cancers and are therefore a recognized target for an anticancer pharmacological intervention. HDAC inhibitors (HDACi) can reverse this process and relax the chromatin, resulting in restoration of gene transcription. Clinical validation of this approach is the FDA approval of HDACi vorinostat and romidepsin for cutaneous T-cell lymphoma (CTCL). Belinostat (PXD-101, N-hydroxy-3-[phenylsulphamoylphenyl] acryl amide) is a novel HDACi with a structural design based on known natural HDACi[2]. It contains a zinc-chelating hydroxamic acid moiety and has similar potency as other HDACi in its class, such as vorinostat and trichostatin A. Belinostat is being studied in a variety of diseases[1,3] and is co-developed by the Cancer Therapy Evaluation Program of the National Cancer Institute (CTEP-NCI)[1].
Exploratory in vivo metabolic profiling studies in mice, rats and dogs suggested rapid and extensive metabolism, producing a variety of metabolites (see Fig. 1). Belinostat metabolites do appear to have very weak HDAC inhibitory activity, which is unlikely to be therapeutically relevant[4]. Uridine 5'-diphospho-glucuronosyltransferase 1A1 (UGT1A1) is the predominant enzyme for the overall metabolism and inactivation (via glucuronidation) of belinostat to less active metabolites[5]. UGT1A1 polymorphisms and changes in the glucuronidation efficiency e.g. because of liver dysfuncion, could have a significant effect on the toxicity and/or efficacy of belinostat.
Fig. 1.
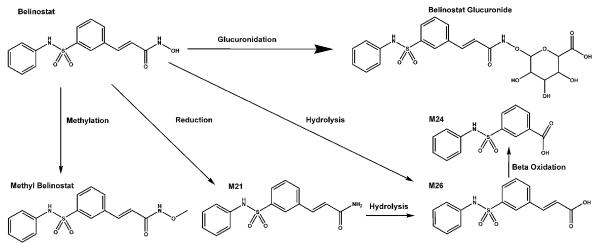
Structures and proposed metabolism of belinostat and its metabolites, belinostat glucuronide, methyl-belinostat, M21, M24, and M26.
Over 30 clinical trials are being conducted to evaluate IV and PO belinostat alone or in combination with approved chemotherapeutic agents, including 5-fluorouracil, carboplatin and paclitaxel, doxorubicin, idarubicin, bortezomib, 5-azacytidine, 13-cis-retinoic acid, and dexamethasone. Pharmacodynamic evaluations have been primarily performed in peripheral blood mononuclear cells (PBMCs) in which H-3 acetylation has been detected at various doses of belinostat and correlated with the pharmacokinetic profile.
Belinostat is presently being evaluated in a National Cancer Institute (NCI) organ dysfunction working group (ODWG) study focusing on patients with liver dysfunction (Clinicaltrials.gov identifier NCT01273155). The pharmacokinetic evaluation of belinostat and its respective metabolites across dysfunction cohorts is a particularly significant endpoint of this study. To support the ongoing clinical development of belinostat and the liver dysfunction study, in particular, we developed and validated a LC-MS/MS assay to quantitate the concentrations of belinostat and five of its metabolites in human plasma.
2 Experimental
2.1 Chemicals and reagents
Belinostat (PXD101), [13C6]-belinostat, belinostat glucuronide (TP201806), methyl belinostat (PX106507), M21 (belinostat amide, PX118624), M24 (3-(Anilinosulfonyl) benzenecarboxylic acid, 3-ASBA, TP201859), [D5]-M24 (TP203295), and M26 (belinostat acid, PXD101-6) were provided by the Topotarget (Copenhagen, Denmark), and were all declared to be >98% purity. Acetonitrile and water (all HPLC grade) were purchased from Fisher Scientific (Fairlawn, NJ, USA). Formic acid and trifluoroacetic acid was purchased from Sigma-Aldrich (St. Louis, MO, USA). Control human plasma was produced by centrifuging whole blood (Central Blood Bank, Pittsburgh, PA, USA) for 20 min at 2000 × g at room temperature. Nitrogen for evaporation of samples was purchased from Valley National Gases, Inc. (Pittsburgh, PA, USA). Nitrogen for mass spectrometrical applications was purified with a Parker Balston Nitrogen Generator (Parker Balston, Haverhill, MA, USA).
2.2 Chromatography
The LC system consisted of an Agilent (Palo Alto, CA, USA) 1200 SL autosampler and binary pump, a Waters (Milford, MA USA) Acquity UPLC BEH (1.7 μm, 50 × 2.1 mm) column kept at 40°C, and a gradient mobile phase. Mobile phase solvent A was 0.1% (v/v) formic acid in acetonitrile, and mobile phase solvent B was 0.1 % (v/v) formic acid in water. The initial mobile phase composition is 10% solvent A pumped at 0.5 mL/min for 3.8 min, and next changed to 50% solvent A. Between 3.8 and 4.0 min, the percentage of solvent A was increased to 90%, while maintaining 0.5 mL/min flow rate. Between 4.0 and 5.0 min, the percentage of solvent A was maintained at 90%. Between 5.5 and 5.6 min, the percentage of solvent A was decreased to 10%, and the flow rate was maintained 0.5 mL/min. These conditions were maintained until 7 min, followed by injection of the next sample. The total run time was 7 min.
2.3 Mass spectrometry
Mass spectrometric detection was carried out using a ABI SCIEX (San Jose, CA, USA) 4000Q hybrid linear ion trap tandem mass spectrometer with electrospray ionization in positive and negative-ion, multiple reaction monitoring (MRM) mode. The settings of the mass spectrometer in negative mode were as follows: curtain gas 30, IS voltage −4500 V, probe temperature 500°C, GS1 30, GS2 30, declustering potential −50 V, collision energy −30V, collision energy spread of −10V, and a scan rate of 700 amu/sec. The tandem positive mode scanning parameters were as follows: curtain gas 30, IS voltage 4500 V, probe temperature 500°C, GS1 30, GS2 30, declustering potential 60 V, a collision energy of 20 V, and an exit potential of 10 V. The MRM m/z transitions monitored were: positive 319.1>93.0 for belinostat; 495.3>319.1 for belinostat glucuronide; 333.1>93.0 for methyl belinostat; negative 278.1>92.0 for M24; 301.1>92.0 for M21; and 302.1>92.2 for M26; negative 281.2>97.3 for [D5]-M24; and positive 325.1>99.0 for [13C6]-belinostat. The LC system and mass spectrometer were controlled by Analyst software (version 1.4.2), and data were collected with the same software.
2.4 Preparation of calibration standards and quality control samples
Stock solutions of analytes belinostat, belinostat glucuronide, methyl belinostat, M21, M24, and M26, and internal standards [13C6]-belinostat and [D5]-M24, were prepared independently at 1 mg/mL in acetonitrile and stored at −80 °C. On the day of assay, these solutions were serially diluted (in steps of 10-fold) in acetonitrile to obtain the lower calibration working solutions. These calibration working solutions were diluted in human plasma to produce the following analyte concentrations: 30, 100, 300, 1000, 3000, 5000 ng/mL. For each calibration series, zero and blank samples were also prepared from 50 μl of control plasma.
Quality control (QC) stock solutions were prepared independently from separate weighings and stored at −80 °C. These solutions were diluted in human plasma to produce the following QC samples of either: QC Lower Limit (QCLL) 30 ng/mL, QC Low (QCL) 100 ng/mL; QC Mid (QCM) 2,500 ng/mL, and QC High (QCH) 4,000 ng/mL.
2.5 Sample preparation
Four μL of 0.1 mg/mL [D5]-M24 and 20 μL of 0.1 mg/mL [13C]-belinostat were added to 40 mL of acetonitrile with 40 μL of TFA (0.01%, v/v) to create a combination internal standard and extraction mix. 200 μL of this mix was added sequentially to each tube of 50 μL standard, QC, or sample plasma. Samples were vortexed for 1 min on a Vortex Genie-2 set at 10 (Model G-560 Scientific Industries, Bohemia, NY, USA) and then centrifuged at 13,000 × g at room temperature for 4 min. The resulting supernatants were transferred to 12 mm × 75 mm borosilicate glass tubes and evaporated to dryness under a stream of nitrogen at 37 °C. Dried residues were re-dissolved in 100 μL of acetonitrile: water: formic acid (10:90:0.1, v/v/v). The supernatants were transferred to autosampler vials, followed by injection of 3 μL into the LC-MS/MS system.
2.6 Validation procedures
2.6.1 Calibration curve and lower limit of quantitation (LLQ)
Decreasing concentrations of analytes were injected into the analytical system to determine the minimal concentration with a signal-to-noise ratio of at least 5:1. Calibration standards and blanks were prepared (see paragraph 2.4) and analyzed in triplicate to establish the calibration range with acceptable accuracy and precision. The analyte-to-internal standard ratio (response) was calculated for each sample by dividing the area of the analyte peak by the area of the internal standard peak. Standard curves were constructed individually by plotting the analyte-to-internal standard ratio versus the known concentrations in each sample. Standard curves were fitted by linear regression with weighting by 1/y2, followed by back-calculation of concentrations. The deviations of these back-calculated concentrations from the nominal concentrations were expressed as percentage of the nominal concentration.
2.6.2 Accuracy and precision
The accuracy and precision of the assay were determined by analyzing samples at the LLQ (QCLL), QCL, QCM, and QCH concentrations in 6 replicates each in 3 analytical runs, together with independently prepared, triplicate calibration curves. Accuracy was calculated at each test concentration as: (mean measured concentration / nominal concentration) × 100%.
Assay precision was calculated by ANOVA as previously described[6], by using SPSS 15.0 for Windows (SPSS Inc., Chicago, IL, USA). Back-calculated concentrations of calibration and QC samples were entered with the run number as factor. From the resulting mean squares of the within runs and mean squares of the between runs, the intra-assay and inter-assay precisions were calculated.
2.6.3 Selectivity and specificity
To investigate whether endogenous matrix constituents interfered with the assay, six individual batches of control, drug-free human plasma were processed and analyzed according to the described procedure. Responses of analytes at the LLQ concentrations were compared with the response of the blank samples. Although belinostat, belinostat glucuronide, methyl belinostat, M21, M24, and M26 are analyzed separately, cross-talk of each analyte was characterized by detection on other MRM channels.
2.6.4 Extraction recovery and ion-suppression
We determined the extraction recoveries of belinostat and its metabolites from plasma by comparing the absolute response of an extract of control plasma to which these analytes had been added after extraction, with the absolute response of an extract of plasma to which the same amounts had been added before extraction. The ion-suppression by plasma matrix components was defined as the decrease of the absolute response of an extract of control plasma to which analyte had been added after the extraction relative to the absolute response of reconstitution solvent to which the same amount of each respective analytes had been added. Experiments were performed at the four QC concentrations, in triplicate (see figure 4).
Fig. 4.

Peak areas of belinostat (A) and belinostat glucuronide (B) for a preceding mobile phase (mp) blank injection, a standard curve including blank plasma (bp), and 5 separate mobile phase blank injections displaying the decrease in carryover before a subsequent set of samples is run.
2.6.5 Stability
Long-term stability experiments were performed in plasma and in stock solution after storage at −80 °C. Stability in the stock solution was expressed as the percentage recovery of the stored solution (10 months) relative to a fresh solution. The stabilities of belinostat, belinostat glucuronide, methyl belinostat, M21, M24, and M26 in plasma at −80 °C were determined by assaying samples before and after storage. In addition, the stabilities of belinostat and metabolites in stock solution at room temperature for 4 h were determined in triplicate. All stability testing in plasma was performed in triplicate at the QCLL, QCL, QCM and QCH concentrations. The effect of 3 freeze/thaw cycles analyte concentrations on plasma was evaluated by assaying samples after they had been frozen (−80 °C) and thawed on 3 separate days and comparing the results with those of freshly prepared samples. The stabilities of belinostat and metabolites in plasma during sample preparation were evaluated by assaying samples before and after 4 h of storage at room temperature. To evaluate the stabilities of belinostat and metabolites in reconstituted samples in the autosampler, we re-injected QC samples and calibration curves approximately 72 h after the first injection and compared the concentrations derived from the second injection with those derived from the first injection. The results of the second runs were expressed as a percentage of their respective values in the first runs (see table 4).
Table 4.
Stability of belinostat and metabolites under varying conditions.
| Storage condition | Concentration (ng/mL) | Stability (%) | CV (%) | Replicates | |
|---|---|---|---|---|---|
| Belinostat | |||||
|
| |||||
| Stock solution 6 h | |||||
| Ambient temp. | 1,000 | 102.1 | 9.5 | 6 | |
| Stock solution 10 months −80 °C | |||||
| 1,000 | 100.6 | 10.4 | 6 | ||
| Plasma 4 h | |||||
| Ambient temp. | QCLL | 30 | 97.8 | 9.3 | 4 |
| QCL | 100 | 102.1 | 6.4 | 4 | |
| QCM | 2500 | 98.6 | 5.3 | 4 | |
| QCH | 4000 | 103 | 4.5 | 4 | |
| Plasma 3 freeze-thaw cycles −80 °C | |||||
| QCLL | 30 | 98.2 | 6.5 | 6 | |
| QCL | 100 | 95.0 | 5.2 | 6 | |
| QCM | 2500 | 99.6 | 4.1 | 6 | |
| QCH | 4000 | 97.9 | 3.3 | 6 | |
| Plasma 9 months −80 °C | |||||
| QCLL | 30 | 90.3 | 9.7 | 6 | |
| QCL | 100 | 86.4 | 9.3 | 6 | |
| QCM | 2500 | 88.2 | 5.2 | 6 | |
| QCH | 4000 | 91.1 | 7.6 | 6 | |
|
| |||||
| Belinostat-glucuronide | |||||
|
| |||||
| Stock solution 6 h | 6 | ||||
| Ambient temp. | 1,000 | 96.1 | 9.3 | ||
| Stock solution 10 months −80 °C | 6 | ||||
| 1,000 | 106.7 | 4.1 | |||
| Plasma 4 h | 4 | ||||
| Ambient temp. | QCLL | 30 | 98.3 | 11.1 | 4 |
| QCL | 100 | 113.8 | 10.7 | 4 | |
| QCM | 2500 | 102.9 | 9.6 | 4 | |
| QCH | 4000 | 101.6 | 9.7 | ||
| Plasma 3 freeze-thaw cycles −80 °C | 6 | ||||
| QCLL | 30 | 93.1 | 9.4 | 6 | |
| QCL | 100 | 106.3 | 14.8 | 6 | |
| QCM | 2500 | 97.4 | 10.6 | 6 | |
| QCH | 4000 | 100.1 | 8.9 | ||
| Plasma 9 months −80 °C | 6 | ||||
| QCLL | 30 | 91.5 | 11.6 | 6 | |
| QCL | 100 | 95.7 | 11.4 | 6 | |
| QCM | 2500 | 100.7 | 8.7 | 6 | |
| QCH | 4000 | 97.7 | 8.1 | 6 | |
|
| |||||
| Methyl-belinostat | |||||
|
| |||||
| Stock solution 6 h | 6 | ||||
| Ambient temp. | 1,000 | 95.6 | 9.0 | ||
| Stock solution 7 months −80 °C | 6 | ||||
| 1,000 | 104.0 | 9.4 | |||
| Plasma 4 h | 4 | ||||
| Ambient temp. | QCLL | 30 | 99.6 | 10.9 | 4 |
| QCL | 100 | 110.0 | 6.3 | 4 | |
| QCM | 2500 | 99.3 | 7.0 | 4 | |
| QCH | 4000 | 101.2 | 7.8 | ||
| Plasma 3 freeze-thaw cycles −80 °C | 6 | ||||
| QCLL | 30 | 99.3 | 11.4 | 6 | |
| QCL | 100 | 104.4 | 6.8 | 6 | |
| QCM | 2500 | 95.2 | 8.4 | 6 | |
| QCH | 4000 | 99.1 | 6.5 | ||
| Plasma 9 months −80 °C | 6 | ||||
| QCLL | 30 | 88.0 | 5.3 | 6 | |
| QCL | 100 | 93.4 | 10.8 | 6 | |
| QCM | 2500 | 100.2 | 6.0 | 6 | |
| QCH | 4000 | 96.0 | 4.8 | 6 | |
|
| |||||
| M21 | |||||
|
| |||||
| Stock solution 6 h | |||||
| Ambient temp. | 1,000 | 105.9 | 13.7 | 6 | |
| Stock solution 10 months −80 °C | |||||
| 1,000 | 104.3 | 11.6 | 6 | ||
| Plasma 4 h | |||||
| Ambient temp. | QCLL | 30 | 119.3 | 8.8 | 4 |
| QCL | 100 | 104.6 | 13.2 | 4 | |
| QCM | 2500 | 104.0 | 10.8 | 4 | |
| QCH | 4000 | 95.3 | 8.5 | 4 | |
| Plasma 3 freeze-thaw cycles −80 °C | |||||
| QCLL | 30 | 109.6 | 11.7 | 6 | |
| QCL | 100 | 106.6 | 8.4 | 6 | |
| QCM | 2500 | 106 | 8.5 | 6 | |
| QCH | 4000 | 103.2 | 11.0 | 6 | |
| Plasma 9 months −80 °C | |||||
| QCLL | 30 | 95.3 | 12.7 | 6 | |
| QCL | 100 | 86.4 | 12.5 | 6 | |
| QCM | 2500 | 87.4 | 6.5 | 6 | |
| QCH | 4000 | 91.0 | 12.4 | 6 | |
|
| |||||
| M24 | |||||
|
| |||||
| Stock solution 6 h | |||||
| Ambient temp. | 1,000 | 103.8 | 9.5 | 6 | |
| Stock solution 10 months −80 °C | |||||
| 1,000 | 104.5 | 9.7 | 6 | ||
| Plasma 4 h | |||||
| Ambient temp. | QCLL | 30 | 106.3 | 13.0 | 4 |
| QCL | 100 | 122.2 | 10.3 | 4 | |
| QCM | 2500 | 104.7 | 10.4 | 4 | |
| QCH | 4000 | 99.3 | 14.6 | 4 | |
| Plasma 3 freeze-thaw cycles −80 °C | |||||
| QCLL | 30 | 92.2 | 10.1 | 6 | |
| QCL | 100 | 115.6 | 8.5 | 6 | |
| QCM | 2500 | 101.6 | 12.6 | 6 | |
| QCH | 4000 | 97.6 | 14.9 | 6 | |
| Plasma 9 months −80 °C | |||||
| QCLL | 30 | 98.1 | 10.6 | 6 | |
| QCL | 100 | 96.5 | 10.5 | 6 | |
| QCM | 2500 | 101.3 | 9.0 | 6 | |
| QCH | 4000 | 100.7 | 12.2 | 6 | |
|
| |||||
| M26 | |||||
|
| |||||
| Stock solution 6 h | |||||
| Ambient temp. | 1,000 | 99.8 | 5.9 | 6 | |
| Stock solution 7 months −80 °C | |||||
| 1,000 | 115.8 | 2.3 | 6 | ||
| Plasma 4 h | |||||
| Ambient temp. | QCLL | 30 | 105.3 | 14.0 | 4 |
| QCL | 100 | 107.6 | 13.1 | 4 | |
| QCM | 2500 | 106.7 | 13.4 | 4 | |
| QCH | 4000 | 100.3 | 12.1 | 4 | |
| Plasma 3 freeze-thaw cycles −80 °C | |||||
| QCLL | 30 | 93.2 | 12.0 | 6 | |
| QCL | 100 | 103.8 | 14.6 | 6 | |
| QCM | 2500 | 103.1 | 10.5 | 6 | |
| QCH | 4000 | 100.8 | 12.1 | 6 | |
| Plasma 9 months −80 °C | |||||
| QCLL | 30 | 100.5 | 11.8 | 6 | |
| QCL | 100 | 96.1 | 14.3 | 6 | |
| QCM | 2500 | 100.8 | 9.1 | 6 | |
| QCH | 4000 | 95.6 | 11.6 | 6 | |
2.6.6 Parallelism
To demonstrate parallelism, the ability to dilute samples from above the upper limit of quantitation to within the validated concentration range, plasma samples containing belinostat and its metabolites above the upper limit of quantitation were diluted to within the assay range. Plasma samples (N=3) with analyte concentrations of 50.000 ng/mL were diluted 20-fold (to 2500 ng/mL) with control plasma and assayed.
2.6.7 Anti-coagulantia cross validation
To demonstrate the ability of our citrate plasma based assay to quantitate heparinized plasma samples, we quantitated our citrate QCLL, QCL, QCM, and QCH samples (N=6) against a heparinized plasma triplicate calibration curve.
2.7 Application of the assay
To document the potential applicability of the assay, we measured the pharmacokinetics of belinostat and its respective metabolites out to 24 h after administration of a 400 mg/m2 IV dose of belinostat to a patient enrolled on the ongoing organ dysfunction study of belinostat (NCT01273155). Pharmacokinetic parameters were calculated non-compartmentally using PK Solutions 2.0 (Summit Research Services, Montrose, CO http://www.summitPK.com).
3 Results and Discussion
3.1 Validation of the assay
3.1.1 Chromatography
The approximate retention times of each compound were as follows: belinostat 2.9 min, belinostat glucuronide at 2.8 min, methyl belinostat at 3.3 min, M21 at 3.1 min, M24 at 3.3 min, M26 at 3.6 min, [13C6]-belinostat at 2.9 min, and [D5]-M24 at 3.2 min.
The analytes at the retention time extremes of approximately 2.8 and 3.6 min, respectively, had corresponding capacity factors of 0.4 and 1.4, respectively, with a void time of 2.0 min. Representative chromatograms of each compound (at the LLQ), and internal standards in plasma are displayed in Fig. 2.
Fig. 2.

Representative chromatograms of: A) belinostat (m/z 319.1>93.0; 2.94 min) added to control plasma at the LLQ concentration of 30 ng/mL (top trace with an offset of 1000 counts) and control human plasma (bottom trace); B) belinostat glucuronide (495.3>319.1; 2.82 min) added to control plasma at the LLQ concentration of 30 ng/mL (top trace with an offset of 2500 counts) and control human plasma (bottom trace); C) methyl belinostat (m/z), 333.1>93.0; 3.30 min)) added to control plasma at the LLQ concentration of 30 ng/mL (top trace with an offset of 500 counts) and control human plasma (bottom trace); D) M21 (m/z 301.1>92.0; 3.10) added to control plasma at the LLQ concentration of 30 ng/mL (top trace with an offset of 200 counts) and control human plasma (bottom trace); E) M24 (m/z 273.1>92.0; 3.38 min) added to control plasma at the LLQ concentration of 30 ng/mL (top trace with an offset of 200 counts) and control human plasma (bottom trace); F) M26 (302.1>92.2; 3.62min) added to control plasma at the LLQ concentration of 30 ng/mL (top trace with an offset of 500 counts) and control human plasma (bottom trace); G) [13C6]-belinostat internal standard (m/z 325.1>99.0; 2.94 min) added to control plasma at a concentration of 200 ng/mL (top trace with an offset of 1000 counts) and control human plasma (bottom trace); H) [D5]-M24 internal standard (m/z 281.2>97.3; 3.32 min) added to control plasma at a concentration of 40 ng/mL (top trace with an offset of 2000 counts) and Scontrol human plasma (bottom trace).
3.1.2 Calibration curve and LLQ
According to the FDA guidelines for bioanalytical method validation[7], the calibration curve adequately describes the concentration versus response relationship if the observed deviation and precision are ≤20% for the LLQ and ≤15% for all other calibration concentrations. At least 4 of 6 calibration points should meet the above criteria[7].
The selected assay range of 30–5000 ng/mL fulfilled the FDA criteria for the LLQ concentration and the calibration curve. Accuracies and precisions at the different concentrations were determined from triplicate calibration curves on 3 separate days and are reported in Table 1. At most concentrations, the mean square of the within runs was greater than the mean square of the between runs, indicating that there was no significant additional variability due to the performance of the assay in different runs[6]. Representative calibration curves and corresponding correlation and regression coefficients are shown in Fig. 3.
Table 1.
Assay performance data of the calibration samples for belinostat and its metabolites in human plasma.
| Analyte | Conc. (ng/mL) | Accuracy (%) | Intra-assay precision (%) | Inter-assay precision (%) |
|---|---|---|---|---|
|
| ||||
| Belinostat | 30 | 101.4 | 13.3 | * |
| 100 | 99.3 | 7.8 | * | |
| 300 | 102.6 | 6.2 | * | |
| 1000 | 103.8 | 4.3 | 2.2 | |
| 3000 | 106.8 | 1.9 | 4.9 | |
| 5000 | 97.4 | 3.6 | 3.7 | |
|
| ||||
| Belinostat-glucuronide | 30 | 95.3 | 12.1 | * |
| 100 | 100.3 | 10.1 | * | |
| 300 | 109.3 | 2.9 | 6.0 | |
| 1000 | 107.6 | 7.8 | * | |
| 3000 | 107.0 | 5.7 | 4.0 | |
| 5000 | 96.1 | 8.3 | * | |
|
| ||||
| Methyl-belinostat | 30 | 96.2 | 11.8 | * |
| 100 | 105.8 | 9.0 | 3.2 | |
| 300 | 111.9 | 9.1 | * | |
| 1000 | 105.6 | 8.7 | 6.4 | |
| 3000 | 106.9 | 6.2 | 2.4 | |
| 5000 | 93.4 | 5.6 | * | |
|
| ||||
| M21 | 30 | 101.7 | 10.3 | * |
| 100 | 100.6 | 10.4 | * | |
| 300 | 101.7 | 6.2 | 1.1 | |
| 1000 | 99.3 | 13.6 | * | |
| 3000 | 106.7 | 10.1 | 12.5 | |
| 5000 | 104.9 | 11.8 | * | |
|
| ||||
| M24 | 30 | 104.0 | 5.2 | * |
| 100 | 93.8 | 8.8 | * | |
| 300 | 99.1 | 13.7 | * | |
| 1000 | 101.7 | 11.2 | * | |
| 3000 | 103.9 | 7.1 | * | |
| 5000 | 103.9 | 11.1 | * | |
|
| ||||
| M26 | 30 | 105.3 | 10.3 | * |
| 100 | 93.0 | 7.7 | 5.1 | |
| 300 | 96.8 | 8.5 | * | |
| 1000 | 102.6 | 5.7 | * | |
| 3000 | 106.0 | 7.4 | 5.2 | |
| 5000 | 104.0 | 9.0 | * | |
N=9; triplicate results, each in 3 separate runs, for each concentration.
The mean square of the within runs was greater than the mean square of the between runs, indicating that there was no significant additional variation due to the performance of the assay in different runs[6].
Fig. 3.

Representative calibration curves (N=3 for each concentration) used to quantitate belinostat (◇), belinostat glucuronide (□), methyl belinostat (Δ), M21 (X), M24 (+), and M26 (○) human plasma samples (response belinostat = 0.00383•conc + 0.0786; R2=0.9968, response belinostat glucuronide = 0.00.558•conc + 0.119; R2=0.9944), response methyl belinostat = 0.00398•conc + 0.0731; R2=0.9937, response M21 = 0.00265•conc + 0.0336; R2=.9944, response M24 = 0.0011•conc + 0.0204; R2=0.9942, response M26 = 0.00546•conc + 0.109; R2=0.9968). Calibration curves are depicted as response ratio versus nominal concentration (A), and as %residuals of the back-calculated relative to the nominal concentrations versus the log transformed concentration (B), the log-transformation for visual purposes.
3.1.3 Accuracy and precision
FDA guidelines specify that the accuracies for all tested concentrations should be within ±15%, and the precisions should not be > 15% CV except for the LLQ, in which case these parameters should not exceed 20%[7].
The accuracies and intra- and inter-assay precisions for the tested concentrations (QCLL, QCL, QCM, QCH) were all within the defined acceptance criteria (Table 2).
Table 2.
Assay performance data for the quantitation of QCLL, QCL, QCM and QCH belmostat and metabolite concentrations in human plasma.
| Analyte | Concentration (ng/mL) | Accuracy (%) | Intra-assay precision (%) | Inter-assay precision (%) |
|---|---|---|---|---|
| Belinostat | 30 (QCLL) | 96.8 | 5.9 | 0.9 |
| 100 (QCL) | 99.4 | 6.3 | * | |
| 2500 (QCM) | 101.0 | 4.2 | * | |
| 4000 (QCH) | 96.2 | 3.4 | * | |
|
| ||||
| Belinostat-glucuronide | 30 (QCLL) | 94.8 | 7.6 | 12.6 |
| 100 (QCL) | 101.2 | 10.2 | 3.1 | |
| 2500 (QCM) | 104.4 | 8.5 | 2.4 | |
| 4000 (QCH) | 95.3 | 7.79 | 2.9 | |
|
| ||||
| Methyl-belinostat | 30 (QCLL) | 96.7 | 9.3 | 5.4 |
| 100 (QCL) | 103.2 | 8.0 | 2.7 | |
| 2500 (QCM) | 101.3 | 8.4 | 2.5 | |
| 4000 (QCH) | 92.0 | 7.4 | 7.4 | |
|
| ||||
| M21 | 30 (QCLL) | 93.6 | 9.3 | 2.8 |
| 100 (QCL) | 97.9 | 13.7 | * | |
| 2500 (QCM) | 96.4 | 8.3 | 2.3 | |
| 4000 (QCH) | 97.7 | 9.7 | 3.0 | |
|
| ||||
| M24 | 30 (QCLL) | 99.7 | 13.6 | 6.4 |
| 100 (QCL) | 93.4 | 12.7 | 3.9 | |
| 2500 (QCM) | 95.9 | 7.8 | * | |
| 4000 (QCH) | 96.0 | 10.8 | * | |
|
| ||||
| M26 | 30 (QCLL) | 102.7 | 11.1 | 1.0 |
| 100 (QCL) | 102.5 | 9.4 | 5.0 | |
| 2500 (QCM) | 98.0 | 8.3 | 4.4 | |
| 4000 (QCH) | 97.3 | 10.3 | * | |
N=18; 6-fold results, each in 3 separate runs, for each concentration.
The mean square of the within runs was greater than the mean square of the between runs, indicating that there was no significant additional variation due to the performance of the assay in different runs[6].
3.1.4 Selectivity and specificity
According to FDA guidelines, the signal at the LLQ must be at least 5 times the signal of any co-eluting peaks[7].
Chromatograms of six individual control plasma samples contained no co-eluting peaks >20% of the analyte areas at the LLQ concentration (interference <19.9% for belinostat, <18.5% for belinostat glucuronide, <5.2 for methyl belinostat, <9.9 for M21, <16.5 for M24, and <11.7 for M26) (Fig. 2).
Cross talk calculations were performed and revealed the following results: M24 cross talks with the methyl belinostat channel at 6.6% with almost identical retention times. M21 has a 7.2% cross talk with the M26 channel, but is resolved with a 0.5 min retention time difference. M26 has an 89% cross talk with M21 but is resolved with a 0.5 min retention time difference. Belinostat glucuronide has 16% cross talk with belinostat, but with is resolved with a 0.1 min difference in retention time. Cross talk does not appear to be a major issue with any of the analytes.
3.1.5 Extraction recovery and ion-suppression
As outlined in the FDA-guidelines, there is a requirement that recovery be consistent and precise[7]. A recovery of ≥70% with a variation of 15% is generally accepted[6,7] There is no specific requirement for ion-suppression. Ultimately, the assay performance, as expressed in the precision and accuracy, is most relevant; however, a large and/or variable ion-suppression may result in lack of assay robustness.
The recoveries of belinostat and its metabolites ranged from 69.9 and 94.9%, with CVs between 36.1 and 4.7%. Ion-suppression ranged from 8.0 to −43.3% (i.e. ionization enhancement), with CVs between 2.7 and 35.4% (Table 3).
Table 3.
Recoveries of belinostat and its metabolites from human plasma and their respective ion suppressions in human plasma extract, with coefficients of variation (CV).
| Analyte | Concentration (ng/mL) | Recovery (%) | CV (%) | Ion suppression (%) | CV (%) |
|---|---|---|---|---|---|
| Belinostat | 30 (QCLL) | 69.9 | 23.5 | −19.8 | 10.4 |
| 100 (QCL) | 82.7 | 7.1 | −11.5 | 7.2 | |
| 2500 (QCM | 84.9 | 11.0 | −15.1 | 4.7 | |
| 4000 (QCH) | 85.1 | 5.3 | −4.0 | 3.6 | |
|
| |||||
| Belinostat-glucuronide | 30 (QCLL) | 80.8 | 16.2 | −12.2 | 6.9 |
| 100 (QCL) | 78.3 | 12.9 | −17.2 | 11.9 | |
| 2500 (QCM | 85.4 | 4.9 | −3.6 | 7.5 | |
| 4000 (QCH) | 85.8 | 7.0 | −3.4 | 8.1 | |
|
| |||||
| Methyl-belinostat | 30 (QCLL) | 84.8 | 14.9 | 7.8 | 11.9 |
| 100 (QCL) | 86.4 | 5.9 | 8.8 | 6.0 | |
| 2500 (QCM | 84.6 | 11.4 | −6.2 | 7.5 | |
| 4000 (QCH) | 89.5 | 4.7 | 3.5 | 2.7 | |
|
| |||||
| M21 | 30 (QCLL) | 75.0 | 20.2 | −13.1 | 21.1 |
| 100 (QCL) | 80.3 | 17.4 | 0.0 | 16.2 | |
| 2500 (QCM | 90.7 | 12.4 | 4.3 | 8.9 | |
| 4000 (QCH) | 94.9 | 9.9 | 8.0 | 8.6 | |
|
| |||||
| M24 | 30 (QCLL) | 79.3 | 22.4 | −5.0 | 35.4 |
| 100 (QCL) | 84.0 | 36.1 | −13.5 | 21.1 | |
| 2500 (QCM | 88.0 | 16.2 | −1.7 | 4.9 | |
| 4000 (QCH) | 92.8 | 7.4 | 4.9 | 6.8 | |
|
| |||||
| M26 | 30 (QCLL) | 78.4 | 10.3 | 1.3 | 15.6 |
| 100 (QCL) | 64.4 | 19.9 | −43.3 | 19.4 | |
| 2500 (QCM | 81.4 | 10.7 | 0.99 | 7.4 | |
| 4000 (QCH) | 88.3 | 9.3 | 0.83 | 7.3 | |
N=3, for each concentration.
3.1.6 Stability
Stability in biological samples is acceptable when ≥85% of the analyte is recovered. The stabilities of belinostat, belinostat glucuronide, methyl belinostat, M21, M24, and M26 stock solutions at room temperature for 6 h were 102.1%, 96.1%, 95.6%, 105.9%, 103.8%, and 99.8%, respectively (Table 4). Stabilities in stock solutions for 10 months at −80 °C were 100.6%, 106.7%, 104.0%, 104.3%, 104.5%, and 115.8% for belinostat, belinostat glucuronide, methyl belinostat, M21, M24, and M26, respectively. The stability of the analytes after 3 freeze thaw cycles (−80°C to RT) were between 92.5 and 115%. Long-term stabilities of the analytes in plasma at −80 °C were adequate with recoveries between 86.4 and 101.3%. The absolute responses of plasma extracts of belinostat at the calibration concentrations, when reconstituted and kept in the autosampler for 72 h, were 95.8 to 102.6% of the initial responses (CV 5.4–9.1%), while the response of belinostat relative to the internal standard signal ranged from 76.7 to 97.0% (CV 4.4–8.4%). For the metabolites, the absolute responses of plasma extracts after 72 h, were 88.7 to 106.45% of the initial responses (CV 7.5–15.1%), while the response of belinostat glucuronide relative to the internal standard signal ranged from 101.43 to 108.2% (CV 9.2–26.7%). Importantly, the reinjection run passed the requirements of any run set by the FDA[7].
3.1.7 Parallelism
The samples diluted from 50 μg/mL to 2500 ng/mL displayed the following accuracies for each analyte: belinostat: 107.7%, with a CV of 3.7%; belinostat glucuronide: 101.7%, with a CV of 4.2%; methyl belinostat: 103.5%, with a CV of 3.3%; M21: 99.9%, with a CV of 9.0%; M24: 96.1%, with a CV of 10.9%; M26: 111.3%, with a CV of 7.2%.
These results indicate parallelism for both the belinostat and metabolite assay.
3.1.1 Anti-coagulantia cross validation
Accuracy and precision of back-calculated concentrations at the QCLL concentrations were within 20% and within 15% at QCL, QCM, and QCH concentrations. Results are provided in Table 5.
Table 5.
Assay performance data of QCLL, QCL, QCM and QCH belinostat and metabolite concentrations in citrate plasma quantitated against a heparinized plasma calibration curve.
| Analyte | Concentration (ng/mL) | Accuracy (%) | Precision (%) |
|---|---|---|---|
| Belinostat | 30 (QCLL) | −11.4 | 11.3 |
| 100 (QCL) | −9.2 | 5.2 | |
| 2500 (QCM) | 10.5 | 2.7 | |
| 4000 (QCH) | 12.5 | 1.4 | |
|
| |||
| Belinostat-glucuronide | 30 (QCLL) | −17.7 | 6.2 |
| 100 (QCL) | −9.9 | 6.6 | |
| 2500 (QCM) | −7.3 | 5.4 | |
| 4000 (QCH) | −11.5 | 3.4 | |
|
| |||
| Methyl-belinostat | 30 (QCLL) | −14.7 | 9.7 |
| 100 (QCL) | −9.3 | 5.1 | |
| 2500 (QCM) | −4.9 | 6.5 | |
| 4000 (QCH) | −9.7 | 3.9 | |
|
| |||
| M21 | 30 (QCLL) | −1.7 | 9.1 |
| 100 (QCL) | −1.4 | 11.1 | |
| 2500 (QCM) | 2.7 | 5.4 | |
| 4000 (QCH) | 4.1 | 6.5 | |
|
| |||
| M24 | 30 (QCLL) | −2.3 | 13.7 |
| 100 (QCL) | −5.5 | 5.7 | |
| 2500 (QCM) | 10.1 | 6.5 | |
| 4000 (QCH) | 5.8 | 6.8 | |
|
| |||
| M26 | 30 (QCLL) | 8.7 | 6.9 |
| 100 (QCL) | 8.2 | 3.9 | |
| 2500 (QCM) | 10.0 | 2.4 | |
| 4000 (QCH) | 8.1 | 4.7 | |
N=6.
3.2 Development
The method development of the assay included multiple variations in column type, extraction solvents, and HPLC gradients, and strategies to minimize carry-over.
3.2.1 Extraction and sample preparation
Because of the need to include in the assay six analytes with diverse physic-chemical properties (including glucuronide metabolite), acetonitrile protein precipitation (1:4, v/v) was chosen as a suitably non-specific sample preparation. This approach also avoided saturation of the mass spectrometer signal, while sensitivity of the assay was not compromised and was proven to be sufficient for the intended application (see below). A plasma sample volume of 50 μL was applied to keep solvent use low, resulting in a practical sample preparation executable in microtubes, and fast dry-down times.
3.2.2 Chromatography
We evaluated the following five columns: Acquity BEH UPLC (50 × 2.1 mm, 1.7 μm), Synergi polar RP 80A (100 × 2.0 mm, 4 μm), Phenomenex Luna C18 100A (150 × 2.0 mm, 5 μm), Hypersil ODS (100 × 2 mm, 5 μm) and Acquity UPLC BEH Phenyl (50 × 3.0, 1.7 μm). All columns allowed for adequate separation of the 6 analytes so that any cross-talk in other MRM channels would not interfere with appropriate quantitation. However, the BEH (non-phenyl) did so in a shorter window, as opposed to the other four, which resulted in much more disparate retention times, and was therefore selected.
Between-run variability in retention times could be reduced by the use of using a column heater set at 40°C allowed for tighter and more uniform chromatographic peaks throughout each sample.
Another step taken to ensure stability and uniformity was to run the first standard curve twice and discounting the first run from the quantitation process. This, in theory, may have built up enough matrix in the system, to allow for even responses from the beginning to the end of the assay
Belinostat forms chelates with divalent metal ions such as zinc and carryover was an issue throughout development and had multiple attempts to minimize its effects. Wash methods were developed to purge the system of carryover but too high of an organic wash led to inconsistent responses most likely due to a clearing of matrix effect or non-equilibrium state in the column. The most consistent approach to minimizing carryover was to have five separate mobile phase washes (using the same method) analysed after each concurrent set of samples (e.g. after each standard curve or set of QCs). This method was adopted to the final version of the method and led to the least amount of carryover, see Fig. 4. Analysis of a zero plasma sample after the 5000 ng/mL ULQ and 5 washes resulted in an apparent concentration of ≤6 ng/mL or 0.12% carryover. Our batch organization adequately deals with the carry-over provided that unknown patient samples are analysed in the sequence they were generated. Random sequencing of patient samples (e.g. with Cmax samples preceding blanks or 24 h samples) could result in slightly biased concentration values.
3.2.3 Mass Spectrometry
Each analyte was scanned in both negative and positive ionization modes until the most sensitive mode was identified for parent drug and the respective metabolites. We determined that belinostat, methyl belinostat, and belinostat glucuronide were optimally sensitive in the positive ionization, while the M21, M24, and M26 metabolites were most sensitive in negative ionization mode.
The major problems during this phase of development were uniformity of responses from sample to sample. The same sample run multiple times may yield drastically differing responses depending on which analyte was being reviewed. Trifluoroacetic acid was added to the extraction mix to increase the solubility of belinostat and the metabolites in acetonitrile, and this appeared to stabilize the response.
The original injection volume during development was 10 μl, but as the assay was optimized, the response of the more sensitive analytes (belinostat glucuronide and belinostat) were found to saturate at 5000 ng/mL, resulting in plateauing of the calibration curve. The reduction of injection volume to 3 μl allowed for a sensitive response to the other analytes while reducing the saturation effect on belinostat and belinostat glucuronide.
3.3 Application of the assay
We applied the assay to samples obtained from a patient receiving a 400 mg/m2 dose of belinostat intravenously administered over 30 min. This patient had signed informed consent on an institutional review board approved protocol.
As seen in Fig. 5, the assay was capable of quantitating belinostat and metabolites in this patient. Non-compartmental analysis yielded the following pharmacokinetic parameters: Belinostat AUC0-inf 1,323 μg/mL•min, t½ 3.7 h; belinostat-glucuronide AUC0-inf 13,811 μg/mL•min, t½ 5.8 h; methyl belinostat AUC0-inf 443 μg/mL•min, t½ 1.4 h; M21 AUC0-inf 539 μg/mL•min, t½ 6.3 h; M24 AUC0-inf 388 μg/mL•min, t½ 1.3 h; M26 AUC0-inf 149 μg/mL•min, t½ 1.5 h.
Fig. 5.

Plasma concentrations of belinostat and metabolites in a patient after a 30 min intravenous infusion of 400 mg/m2 belinostat. Belinostat (◇), belinostat glucuronide (□), methyl belinostat (Δ), M21 (X), M24 (+), and M26 (○).
4 Conclusion
The objective of this study was to develop and validate an analytical method for the quantitation of belinostat and its metabolites in human plasma. We accomplished this using reversed phase chromatography for separation with triple quadrupole mass spectrometric MRM detection.
A previously reported assay for the quantitation of belinostat alone was validated over the range of 0.5–1000 ng/mL with a run time of 6 min, which could detect but not quantitate the glucuronide metabolite[8]. Employing UPLC chromatography allowed us to quantitate belinostat and 5 metabolites in a similar time-span, across a higher concentration range of 30–5000 ng/mL, which was adequate to evaluate the pharmacokinetic profiles of all analytes in a patient. To our knowledge, this is the first assay for simultaneously measuring all six analytes published to date, that is validated according to FDA guidelines[7].
The analytical method presented herein will be a valuable tool in quantitating belinostat and its metabolites in plasma as belinostat undergoes further clinical development either as monotherapy or in combination with other anticancer agents.
Highlights
Belinostat and 5 metabolites could be quantitated in a single run
FDA validated assay ranges from 30–5000 ng/mL using 50 μL of human plasma
Glucuronidation is the major metabolic pathway of belinostat
Acknowledgements
We thank the Merrill Egorin Writing Group of UPCI for constructive suggestions regarding the manuscript.
Funding
Support: Spectrum Pharmaceuticals Inc., Topotarget A/S, grant U01-CA099168 and contract N01-CM62209 (NCI-CTEP). This project used the UPCI Clinical Pharmacology Analytical Facility (CPAF) and was supported in part by award P30-CA47904.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5 References
- [1].Beumer JH, Tawbi H. Role of histone deacetylases and their inhibitors in cancer biology and treatment. Curr Clin Pharmacol. 2010;5:196–208. doi: 10.2174/157488410791498770. [DOI] [PubMed] [Google Scholar]
- [2].Finn PW, Bandara M, Butcher C, et al. Novel Sulphonamide Derivatives as Inhibitors of Histone Deacetylase. Helvetica Chimica Acta88. 2005:1630–1657. [Google Scholar]
- [3].Gimsing P. Belinostat: a new broad acting antineoplastic histone deacetylase inhibitor. Expert Opin Investig Drugs. 2009;18:501–508. doi: 10.1517/13543780902852560. [DOI] [PubMed] [Google Scholar]
- [4].Jones RJ, Tjornelund J, Erichsen KD, Sengelov L, de BJ. Belinostat in Combination With Carboplatin and Paclitaxel (BelCaP) for Treatment of Bladder Cancer - a Pharmacokinetic Study of Exposure to Belinostat and Its Metabolites. European Journal of Cancer. 2011;47:S506. [Google Scholar]
- [5].Wang L, Goh BC, Lwin TW, et al. Phase I pharmacokinetics and metabolic pathway of belinostat in patients with hepatocellular carcinoma [abstract] Journal of Clinical Oncology. 2010:28. Wang L, Goh BC, Lwin TW et al. [Google Scholar]
- [6].Rosing H, Man WY, Doyle E, Bult A, Beijnen JH. Bioanalytical liquid chromatographic method validation: a review of current practices. Journal of liquid chromatography & related technologies. 2000;23:329–354. [Google Scholar]
- [7].U.S.Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research. Center for Veterinary Medicine [Accessed February 1, 2007];Guidance for Industry - Bioanalytical Method Validation. http://wwwfdagov/cder/guidance/indexhtm [serial online] (2001);
- [8].Wang LZ, Chan D, Yeo W, et al. A sensitive and specific liquid chromatography-tandem mass spectrometric method for determination of belinostat in plasma from liver cancer patients. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:2409–2414. doi: 10.1016/j.jchromb.2010.07.015. [DOI] [PubMed] [Google Scholar]