

Abstract
CD8+ T-lymphocytes from HIV-1 infected individuals express unidentified factors that suppress viral replication by inhibiting HIV-1 gene expression. We examined the role of epigenetics in modulating the HIV-1 suppressive factors expressed by primary CD8+ T cells from subjects naturally controlling virus replication. HIV-1 suppression by CD8+ T-lymphocytes was reversed up to 40% by the addition of a histone deacetylase (HDAC) inhibitor. Noncytolytic suppression was not dependent on epigenetic changes within the target cells, as HDAC1 within the target cell was dispensable, and HIV-1 LTR histone acetylation remained unchanged in the presence of CD8+ T-lymphocytes. Histone deacetylation within CD8+ T-lymphocytes was necessary for potent HIV-1 suppression. Blocking HDACs impairs the ability of CD8+ T-lymphocytes to repress HIV-1 transcription, demonstrating that expression of a portion of the suppressive factors is regulated by epigenetics. These data provide a way to focus the search for the suppressive factors and to potentially modulate their expression.
Keywords: Histone deacetylases, CD8+ T-lymphocyte HIV-1 suppression, Virus controllers
Introduction
Understanding the antiviral immune responses that mediate potent suppression of HIV-1 infection would aid in designing vaccines and therapies. A subset of asymptomatic, antiretroviral-naïve patients have been termed controllers with the common characteristics of sustained control of virus load and HIV-1-specific immunity (Lambotte et al., 2005). Controllers are divided into several different groups based on viral load in the absence of drug therapy (Grabar et al., 2009; Walker, 2007). Virus controllers have higher viral loads than elite controllers (Walker, 2007), but maintain viral loads lower than progressing HIV-1-infected individuals (Addo et al., 2007). The immune responses of controllers are intensely studied to elucidate the types of responses that could be elicited with a vaccine to mediate control of HIV-1 infection.
CD8+ T-lymphocytes can inhibit HIV-1 replication by cytolytic and noncytolytic responses (Walker et al., 1987, 1986). The noncytolytic response is characterized by suppression of HIV-1 replication without lysis of the infected cell (Walker et al., 1991). A subset of CD8+ T-lymphocytes exhibits noncytolytic suppression without detectable cytolytic activity, distinguishing these two types of antiviral activity (Toso et al., 1995). For CCR5-tropic viruses, noncytolytic suppression can be mediated by MIP-1α, MIP-1β, and RANTES (Cocchi et al., 1995). However, the factors responsible for suppression of CXCR4-tropic viruses are still unknown. Noncytolytic suppression of CXCR4-tropic viruses can be mediated by inhibition of HIV-1 gene expression (Chen et al., 1993; Tomaras et al., 2000), with effects more specifically on transcription initiation (Overman et al., 2007). The molecules that result in suppression of HIV-1 transcription are still unknown, which has impeded the determination of the mechanism of noncytolytic suppression. Noncytolytic suppression can occur when CD8+ T-lymphocytes and HIV-1 infected cells are separated by a semi-permeable membrane, however the most potent suppression occurs with direct contact even between cells with mismatched MHC alleles (Walker and Levy, 1989). The lack of necessity for cell contact suggests that the some portion of the noncytolytic response is comprised of soluble factors (Walker and Levy, 1989). The magnitude of the noncytolytic response correlates with clinical status of HIV-1 infection; with asymptomatic individuals exerting the strongest noncytolytic suppression (Mackewicz et al., 1991) and with a preservation of the CD8 response in HIV+ subjects on early HAART (Chun et al., 2001). However, there is an unexplained variation in the presence of the noncytolytic response among HIV-infected individuals (Walker et al., 1989). Understanding what regulates this response may provide insight into how to induce a potent noncytolytic response in all HIV-1 infected individuals.
Eukaryotic chromatin consists of histone proteins and nuclear DNA (Jenuwein and Allis, 2001). The structure of chromatin can be altered to up-regulate or down-regulate the expression of genes encoded by the DNA (Jenuwein and Allis, 2001). The “histone code” contends that the structure of the chromatin is governed by post-translational modifications of the histone proteins to alter gene expression (Turner, 1993). In particular, acetylation of the lysine residues on N-terminal tails of the histones leads to less dense chromatin permissive for transcription (Hebbes et al., 1992). Furthermore, acetylated histone tails act as binding sites for “effector” molecules that influence higher-order chromatin structure and RNA polymerase recruitment (reviewed in Ruthenburg et al., 2007). Removing the acetyl groups from the histone tails compacts the chromatin and removes the binding sites of positive transcription regulators resulting in repressed transcription of the associated DNA (Hebbes et al., 1992; Turner, 2000). The acetyl groups are removed from the histone tails by a group of enzymes called histone deacetylases (HDACs) (Taunton et al., 1996). The epilepsy drug, valproic acid (VPA) is a common small molecule used to inhibit histone deacetylases (Gottlicher et al., 2001). VPA has been shown to inhibit the activity of all of the class I and II HDACs tested except HDAC10 (Catalano et al., 2007; Gottlicher et al., 2001; Gurvich et al., 2004). By increasing acetylation of histones VPA can alter the transcription profile of cells. Using various techniques and cell types, investigators have shown that histone deacetylation regulates the expression of 2–9% of human genes (Agarwal et al., 2009; Van Lint et al., 1996b). Histone acetylation also regulates HIV-1 transcription (Coull et al., 2000; Van Lint et al., 1996a). Optimal HIV-1 transcription is associated with acetylation of the histones associated with the HIV-1 long terminal repeat (LTR) (Lusic et al., 2003). Furthermore, HDAC1 is recruited to the HIV-1 LTR to repress transcription from latent provirus (Tyagi and Karn, 2007; Williams et al., 2006; Ylisastigui et al., 2004). Thus, epigenetics could modulate HIV-1 infection by altering gene expression in effector T cells or by directly influencing HIV-1 transcription in infected cells.
In this study we assessed the role of epigenetics in the noncytolytic response. We investigated the importance of histone deacetylation in the expression of antiviral factors from CD8+ T-lymphocytes, and its importance for directly inhibiting HIV-1 transcription by noncytolytic suppression. We found that a subset of the genes encoding antiviral factors from CD8+ T-lymphocytes from select virus controllers was regulated by HDACs.
Results
Inhibition of histone deacetylation during noncytolytic suppression increases HIV-1 replication
Previous studies demonstrated that noncytolytic suppression acts on HIV-1 transcription and gene expression (Chen et al., 1993; Mackewicz et al., 1995; Tomaras et al., 2000), with specific effects on transcription initiation (Overman et al., 2007). We modeled non-cytolytic suppression in vitro using JR-HVS CD8+ T-lymphocytes. JR-HVS CD8+ T-lymphocytes inhibit HIV-1 transcription initiation (Overman et al., 2007), without any cytotoxic effects (Lacey et al., 1998). Therefore, the CD8+ T-lymphocyte cell line, JR-HVS, possesses consistent noncytolytic antiviral activity making these cells a valid resource for determining the mechanism of noncytolytic suppression.
First, we examined the ability of valproic acid to inhibit histone deacetylation in CD4+ T-lymphocytes from a pool of 10 seronegative donors, an individual seronegative donor, and primary T-lymphocytes from an HIV-1+ virus controller. JR-HVS CD8+ T-lymphocytes were also cultured with valproic acid to determine if valproic acid inhibited histone deacetylation in CD8+ T-lymphocytes. Baseline levels of acetylated histone 3 (H3ac) in CD4+ T-lymphocytes are shown in Fig. 1A. Valproic acid stimulated acetylation of histones (H3ac), without altering the levels of total histone 3 (H3), in CD4+ and CD8+ T-lymphocytes (Fig. 1A). Thus, valproic acid altered the amount of H3ac specifically, and not overall expression of H3. Taken together, the effects of valproic acid on CD4+ T cells and JR-HVS CD8+ T-lymphocytes provide a model for exploration of the effect of histone acetylation on noncytolytic suppression in vitro.
Fig. 1.
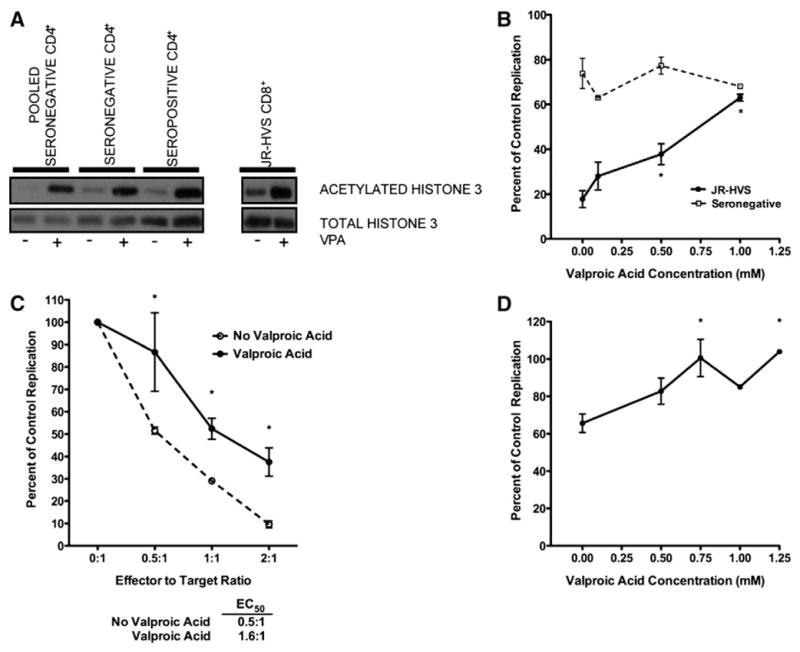
Valproic acid decreases CD8+ T-lymphocyte noncytolytic suppression. (A) Primary CD4+ T-lymphocytes or HVS-transformed CD8+ T-lymphocytes were cultured for 16 h in the presence or absence of 1.0 mM valproic acid. Acetylated and total histone 3 was analyzed by western blot. (B) Valproic acid was titrated into an autologous contact-mediated suppression assay with seronegative cells at a 1:1 E:T ratio (open squares) or a heterologous assay with JR-HVS CD8+ T-lymphocytes at a 2:1 E:T ratio (filled circles). HIV-1 replication in CD8+ T-lymphocytes co-cultures is shown as the percentage of replication when the HIV-1 infected CD4+ T-lymphocytes were cultured alone. 0.5 and 1.0 mM valproic acid significantly increased replication in the JR-HVS CD8+ T-lymphocyte co-cultures (one-way ANOVA and Tukey test, p<0.05 and p<0.001, respectively). Valproic acid had no effect on replication in the seronegative CD8+ T-lymphocyte co-cultures (one-way ANOVA and Tukey test, p=0.1177). Mean and standard error of 4 experiments are shown. (C) Heterologous contact-mediated noncytolytic suppression assays were performed at multiple effector to target ratios in the absence (open circles) or presence of 1.0 mM valproic acid (filled circles). HIV-1 replication in cultures containing CD8+ T-lymphocytes is shown as the percentage of replication when the HIV-1 infected CD4+ T-lymphocytes were cultured alone. The effector to target ratio where 50% suppression (EC50) was reached was calculated for suppression assays done in the presence and absence of valproic acid. Valproic acid significantly increased replication in CD8+ T cell co-cultures at all three effector to target ratios (two-way ANOVA and Bonferoni corrected t-tests, p=0.0002). Mean and standard error of 3 experiments are shown. (D) HIV-1 infected CD4+ T-lymphocytes were cultured alone or in a 0.4 μM transwell with JR-HVS CD8+ T cells at a 2:1 effector to target ratio. Valproic acid was added in increasing concentrations to the transwell. HIV-1 replication in CD8+ T-lymphocytes co-cultures is shown as the percentage of replication when the HIV-1 infected CD4+ T-lymphocytes were cultured alone. Significant increases in replication were observed at 0.75 and 1.25 mM valproic acid concentrations (one-way ANOVA and Tukey Test, p<0.05 for both). Mean and standard error of duplicate experiments are shown.
It is unknown if alteration of chromatin structure, through histone deacetylation, can regulate expression of the genes responsible for noncytolytic suppression of HIV-1 transcription. To examine this, we determined if inhibition of HDAC by valproic acid could reverse CD8+ T cell mediated virus suppression (Fig. 1B). Valproic acid stimulation increased virus replication in a dose-dependent manner. At 1.0 mM, replication in the CD8+ T cell co-culture significantly increased 3.7-fold to 63% of the replication control (p<0.001). In contrast, HIV replication in CD4+ lymphocytes in the presence of seronegative CD8+ T-lymphocytes was unresponsive to HDAC inhibition (Fig. 1B). These data demonstrate that alterations in chromatin, through histone deacetylation, were required for optimal and specific CD8+ T cell mediated virus suppression.
Dose-dependent CD8+ T cell mediated virus suppression requires histone deacetylation
To determine if the requirement of histone deacetylation was dependent on a specific concentrations of CD8+ effector cells, CD8+ T-lymphocytes were examined at a series of effector: target (E:T) cell ratios. HIV-1 replication in the CD8+ T cell co-cultures decreased from 51.6% at the lowest concentration (0.5:1, E:T) down to 9.6% at the highest concentration (2:1, E:T), demonstrating a CD8+ T-lymphocyte concentration-dependent response (Fig. 1C). Valproic acid stimulation significantly increased virus replication to 86.7% and 37.5% at 0.5:1 and 2:1 (E:T) ratios compared to control infection respectively (p<0.01 and p<0.05 respectively; Fig. 1C). Inhibition of histone deacetylation reduced noncytolytic suppression by an average 30% at all three E:T ratios. The addition of valproic acid in the growth media increased the EC50 more than three-fold from an E:T ratio of 0.5:1 without VPA to 1.6:1 with VPA (Fig. 1C). Thus, the inhibition of HDACs led to a consistent reduction in noncytolytic suppression irrespective of effector to target ratios.
Histone deacetylation is required for soluble noncytolytic suppression
We specifically studied only the soluble portion of virus suppression and the contribution of epigenetically regulated genes to the non-contact required portion of suppression by separating the CD8+ and CD4+ T-lymphocytes by a semi-permeable membrane (transwell cultures). Soluble factors from CD8+ T-lymphocytes suppressed HIV-1 replication in CD4+ T-lymphocytes by 34% (Fig. 1D). Similar to the effect that was observed in the direct contact co-culture system, valproic acid showed a dose-dependent reversion of soluble non-cytolytic suppression. HIV-1 replication significantly increased after stimulation with 0.75 and 1.25 mM valproic acid (p<0.05 for both; Fig. 1D). Thus, hyperacetylation inhibited both soluble and contact-mediated noncytolytic suppression of HIV-1.
Reduction of noncytolytic suppression by a histone deacetylase inhibitor is independent of the amount of HIV-1 gene expression
HDAC inhibitors have been shown to augment HTLV-1 (Mosley et al., 2006) and HIV-1 gene expression (El Kharroubi et al., 1998; Kiernan et al., 1999). A potential explanation of our results was that HIV-1 gene expression, in the presence of VPA, increases in the target cells to a level that noncytolytic suppression could not suppress. We investigated whether the reversal of noncytolytic suppression by VPA was dependent on the level of HIV-1 gene expression. Heterologous contact-mediated noncytolytic suppression assays were performed in cultures with a 20-fold range in HIV-1 gene expression, as measured by LTR-driven luciferase expression, approaching the lower limit of detection (1.6E6 to 0.08E6 relative light units). In the absence of the HDAC inhibitor, there was no difference in suppression of the infection in the CD8+ T-lymphocyte co-cultures at the different levels of HIV-1 gene expression (Fig. 2). JR-HVS CD8+ T-lymphocytes suppressed virus between 13.6% and 15.7% of the replication control at a 2:1 E:T ratio across the different amounts of HIV-1 gene expression. These results demonstrate that there is consistent noncytolytic suppression of HIV-1 infection, despite the amount of replication present. With histone deacetylation inhibited, noncytolytic suppression was alleviated in a VPA dose-dependent manner across the range of HIV-1 gene expression (Fig. 2). This reversion was equivalent across all amounts of HIV-1 gene expression. Thus, at a range of HIV-1 gene expression levels, inducing hyperacetylation of histones can reverse noncytolytic CD8+ T cell mediated suppression.
Fig. 2.
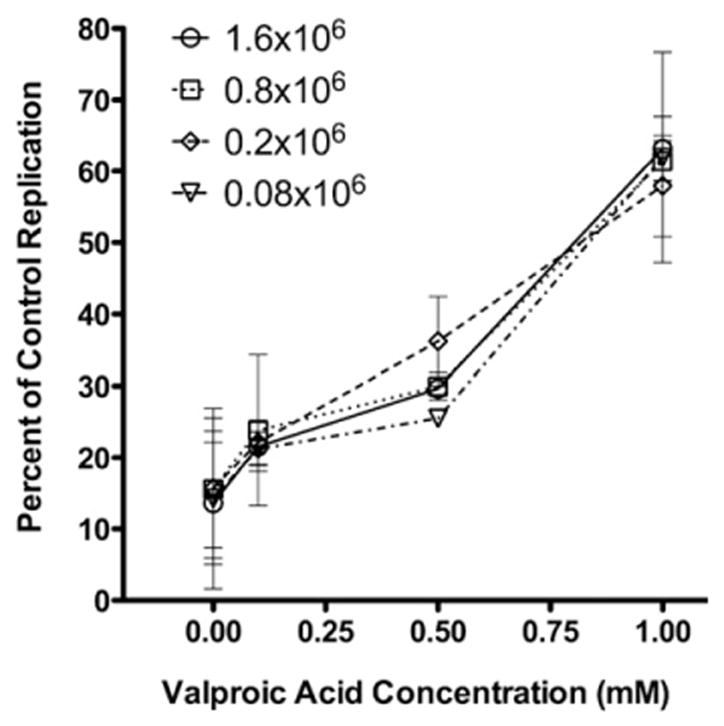
Valproic acid inhibits CD8+ T-lymphocyte noncytolytic suppression at different levels of HIV-1 gene expression. CD4+ T-lymphocytes were infected with four different amounts of HIV-1NL4-3 pseudotyped virus to generate a titration of HIV-1 replication from 1.6×106 to 0.08×106 relative light units. JR-HVS CD8+ T-lymphocytes were cultured in direct contact at a 2:1 effector to target ratio with CD4+ T-lymphocytes infected with each of the different amounts of HIV-1NL4-3 pseudotyped virus. Valproic acid was added to the growth media in increasing concentrations. HIV-1 replication in the CD8+ T-lymphocyte co-culture is shown as the percentage of replication in the infected CD4+ T cells cultured alone. Mean and standard error of duplicate experiments are shown.
Primary CD8+ T-lymphocytes from HIV-1+ virus controllers optimally suppress HIV-1 replication with a requirement for histone deacetylation
To extend the investigation to examine the impact of histone deacetylation on in vivo control of HIV-1 infection, T-lymphocytes from five HIV-1 infected subjects, naturally controlling HIV infection, were examined in autologous noncytolytic suppression assays. HIV-1 infected CD4+ T-lymphocytes from virus controllers were cultured in direct contact with autologous CD8+ T-lymphocytes at a 2:1 (E:T) ratio. CD8+ T-lymphocytes from virus controllers were able to inhibit virus replication significantly better than cells from seronegative donors (Fig. 3, p<0.001). CD8+ T-lymphocytes from HIV-1+ virus controllers suppressed virus to a mean of 20.9% of the replication control (Fig. 3). Inhibition of histone deacetylation, by valproic acid stimulation (1.0 mM), significantly reversed noncytolytic suppression mediated by CD8+ T-lymphocytes from HIV+ virus controllers (p=0.01), but had no significant effect on virus replication in the presence of CD8+ T-lymphocytes from seronegative subjects (Fig. 3, p=0.2). Valproic acid treatment reversed suppression by CD8+ T-lymphocytes from HIV-1+ virus controllers such that the amount of virus replication rose to the level of that observed in the presence of the negative control CD8+ T cell lymphocytes from seronegative donors. Inhibition of histone deacetylation significantly reduced the ability of CD8+ T-lymphocytes from HIV-1+ virus controllers to suppress HIV-1 replication (p<0.001). The absolute level of responsiveness to the histone deacetylase inhibitor was heterogeneous among the HIV-1+ virus controllers, consistent with the hypothesis that multiple mediators at varying concentrations contribute to CD8+ mediated virus suppression. However, some CD8+ T-lymphocytes from HIV-1+ virus controllers have nearly identical responsiveness to the HDAC inhibitor as the CD8+ T cell effector cell line (Figs. 1B and 3). In total, inhibition of HDACs prevented optimal noncytolytic suppression of HIV-1 by CD8+ T-lymphocytes from HIV-1+ virus controllers, which recapitulated the effects of valproic acid observed with the CD8+ T cell effector cell line.
Fig. 3.
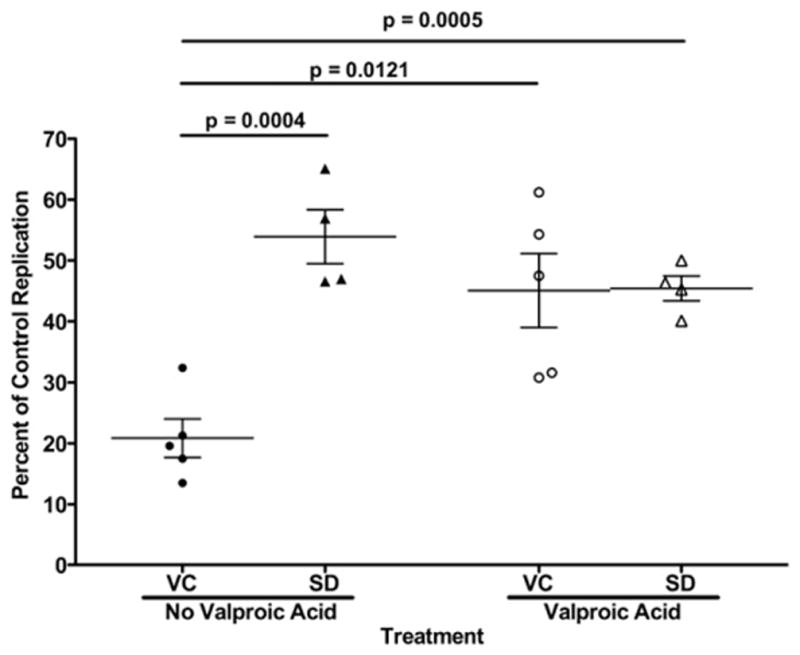
Valproic acid inhibits contact-mediated CD8+ T-lymphocyte noncytolytic suppression in virus controllers. HIV-1-infected primary CD4+ T-lymphocytes were cultured in direct contact with or without autologous CD8+ T-lymphocytes at a 2:1 effector to target ratio. CD8+ T-lymphocytes from virus controllers (VC, circles) or seronegative donors (SD, triangles) were examined. Noncytolytic suppression was examined in the absence and presence of valproic acid (filled and open symbols, respectively). Virus controller CD8 T cell co-cultures without valproic acid had significantly lower HIV-1 replication than seronegative CD8+ T cell co-cultures without and with valproic acid (unpaired t-test, p=0.0004 and p=0.0005). HIV-1 replication in the virus controller CD8+ T-lymphocyte co-cultures significantly increased with the addition of valproic acid (paired t-test, p=0.0121). There was not a significant increase in replication in the seronegative CD8+ T-lymphocyte co-cultures (paired t-test, p=0.2187). Means and standard error are represented by horizontal and vertical lines respectively.
Histone deacetylase 1 in HIV-1 infected target cells is not required for CD8+ T-lymphocyte noncytolytic suppression of HIV-1
To determine if CD8+ T-lymphocytes induce HDAC1 in HIV-1 infected CD4+ target cells to inhibit HIV-1 transcription, we used RNA interference to deplete target cells of HDAC1. We used a pool of siRNAs targeting HDAC1 to knockdown expression in infected target cells. HDAC1 siRNA transfected cells were substantially depleted of HDAC1, when compared to mock or negative control siRNA transfected cells (Fig. 4A) and HDAC1 remained knocked-down throughout the suppression assay (96 h post transfection). HDAC1-expressing and deficient cells were used in soluble noncytolytic suppression assays with CD8+ T-lymphocytes from a seronegative donor, CD8+ T-lymphocytes from an HIV-1+ virus controller (VC18), or the JR-HVS cell line. As expected, the seronegative donor CD8+ T-lymphocytes showed the least suppressive capability overall and there was no effect on HIV replication when HDAC1 was knocked-down (Fig. 4B). Also as expected, both primary CD8+ T-lymphocytes from a virus controller (VC18) and JR-HVS CD8+ T-lymphocytes substantially inhibited virus replication (Figs. 4C and D). However, there was also no change in the suppressive activity when HDAC1 was knocked-down (Figs. 4C and D). HIV-1 replication decreased with each increase in effector to target ratio to similar levels despite the differences in HDAC1 protein expression. Thus, the presence of HDAC1 in HIV-1 infected CD4+ target cells was not necessary for noncytolytic suppression of HIV-1 replication. Taken together, these data demonstrate that HDAC1 in the CD4+ target cells was not responsible for the requirement of histone deacetylation for optimal noncytolytic suppression.
Fig. 4.
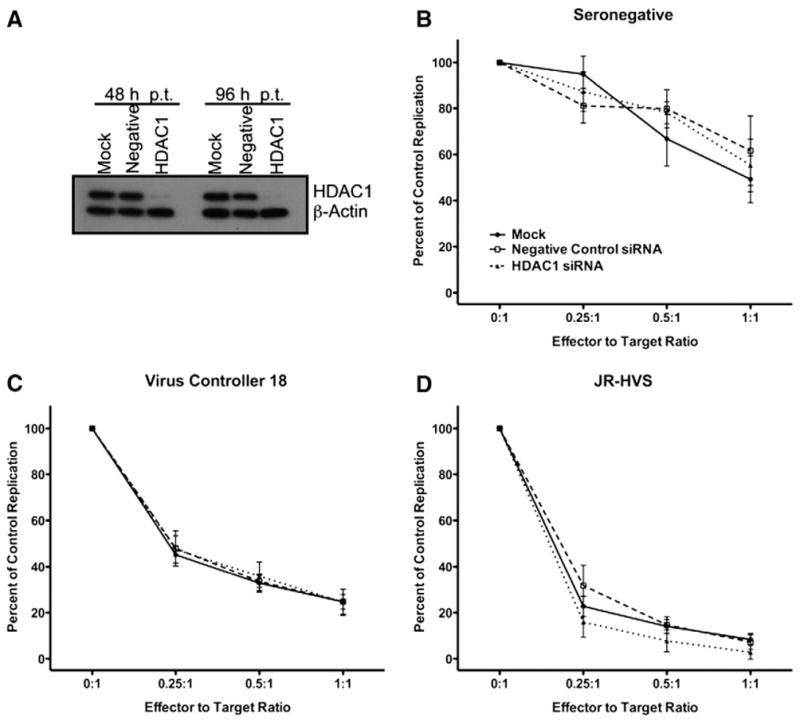
HDAC1 is not required for soluble CD8+ T-lymphocyte noncytolytic suppression. (A) siRNA-mediated knockdown of histone deacetylase 1. TZM-bl cells were transfected with no (Mock), a negative control (Negative) or HDAC1-specific (HDAC1) siRNAs. 48 and 96 h post transfection cells were lysed and HDAC1 and β-actin expression was determined by western blot. (B–D) Soluble noncytolytic suppression of HDAC1-deficient TZM-bl cells. TZM-bl cells expressing HDAC1 (mock; filled circles or Negative control; open square) or with HDAC1 knocked-down (HDAC1; filled triangle) were used as targets for soluble noncytolytic suppression assays. (B) Seronegative, (C) virus controller 18 (VC18), or (D) JR-HVS CD8+ T-lymphocytes were cultured in a transwell system with HIV-1IIIB infected TZM-bl cells at various effector to target ratios. 48 h after infection LTR-driven luciferase expression was quantified. HIV-1 replication in the culture in the presence of the CD8+ T-lymphocytes is shown as a percentage of the replication control. Mean and standard error of 3 experiments are shown.
Histone acetylation at the HIV-1 LTR in CD4+ target cells is unchanged by noncytolytic suppression
The results of the RNA interference experiments demonstrated that HDAC1 in the infected target cells was not mediating the suppression of HIV-1 transcription during noncytolytic suppression. However, recent data suggests that other HDACs may regulate HIV-1 gene expression (Archin et al., 2009), and thus could deacetylate histones in the absence of HDAC1. Thus, we examined changes in the acetylation state of the histones within the HIV-1 LTR during noncytolytic suppression. A large-scale noncytolytic suppression assay was performed, and native chromatin was isolated from these cultures. JR-HVS CD8+ T-lymphocytes strongly inhibited HIV-1 replication, but seronegative CD8+ T-lymphocytes did not (Fig. 5A). However, there was no significant decrease in acetylated histones associated with the HIV-1 LTR during noncytolytic suppression, despite potent suppression of HIV-1 replication (Fig. 5B). In the absence of CD8+ T-lymphocytes, 1.5% of the HIV-1 LTR was associated with H3ac in the absence of any CD8+ T-lymphocytes. This was very similar to the 1.4% of the LTR associated with acetylated histone 3 in infected cells cultured with seronegative CD8+ T-lymphocytes and the 1.6% in infected cells cultured with JR-HVS CD8+ T-lymphocytes. As a control for changes in histone acetylation at the promoter of host genes, association of GAPDH DNA with H3ac was determined. There was no difference in enrichment of H3ac with GAPDH DNA in any of the three conditions (Fig. 5B). Alterations of the acetylation state of histones at the viral promoter did not mediate noncytolytic suppression; and could not explain the requirement for histone deacetylases for noncytolytic suppression.
Fig. 5.
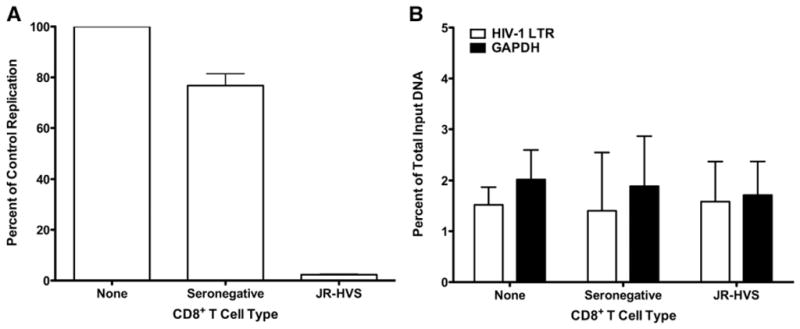
CD8+ T-lymphocyte noncytolytic suppression does not alter the acetylation of histone 3 at the HIV-1 LTR. (A) CD4+ T-lymphocytes were infected with replication-competent HIV-1IIIB and cultured alone (filled bars) or in the presence of seronegative (open bars) or JR-HVS (gray bars) CD8+ T-lymphocytes. HIV-1 replication was determined by virion-associated reverse transcriptase activity in the culture supernatant 6 days post-infection. Percent of the replication control was calculated with the same method as luciferase assays, Mean and standard error of 3 experiments are shown. (B) Native chromatin was purified from the noncytolytic suppression assays in (A). Acetylated histone 3 associated with the HIV-1 LTR (open bars) or a housekeeping gene (GAPDH; filled bars) was detected by quantitative native chromatin immunoprecipitation. The copies of DNA coimmunoprecipitated with acetylated histone 3 is shown as a percentage of the total DNA used for the immunoprecipitation input. Mean and standard error of 3 experiments are shown.
Inhibition of histone deacetylases in primary CD8+ T-lymphocytes from HIV-1+ subjects decreases their noncytolytic suppressive activity
Since the requirement for histone deacetylation in suppression of HIV-1 replication was not localized to the infected target cells, we determined whether histone deacetylation directly regulated the suppressive capacity of the CD8+ T-lymphocytes. We found that CD8+ effector lymphocytes and cell lines were resilient to attempts to knockdown HDAC, so we utilized another effective strategy of interrogating the contribution of HDAC regulation in CD8+ effector cells using valproic acid treatment. JR-HVS CD8+ T-lymphocytes were incubated with valproic acid for 16 hours and subsequently used as effector cells in a noncytolytic suppression assay. Noncytolytic suppression decreased incrementally with increasing concentrations of valproic acid pretreatment; with suppression being almost completely eliminated with 2 mM valproic acid treatment (Fig. 6A). Therefore, the shorter exposure time, 16 h instead of 72 h, appeared to warrant a higher concentration of valproic acid to exhibit the most potent effects. Suppressive activity mediated by CD8+ T-lymphocytes from either the HIV-1+ virus controller (VC6) or JR-HVS CD8+ T-lymphocytes declined in a dose-dependent manner when the CD8+ T-lymphocytes were pretreated with valproic acid (Fig. 6A).
Fig. 6.
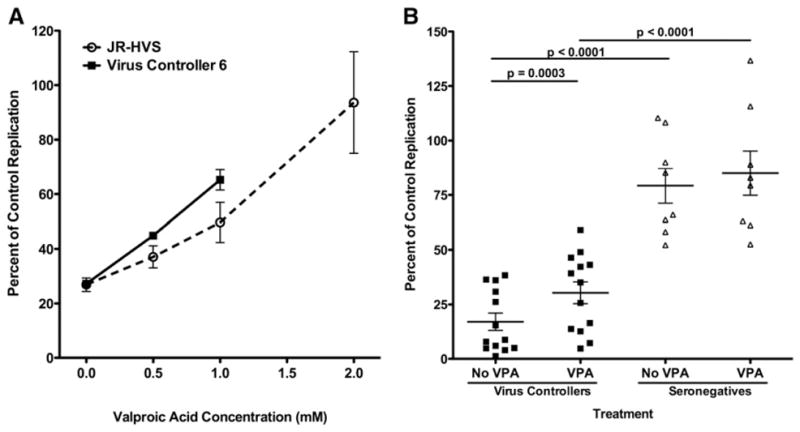
Incubation of CD8+ T-lymphocytes in valproic acid inhibits their noncytolytic suppressive activity. (A) Virus controller 6 and JR-HVS CD8+ T-lymphocytes were incubated in different concentrations of valproic acid for 16 h and subsequently cultured with HIV-1NL4-3 pseudotyped virus-infected CD4+ T-lymphocytes at a 2:1 effector to target ratio. (B) Incubation of primary CD8+ T-lymphocytes from virus controllers in valproic acid has varied effects on their suppressive activity. Panels of CD8+ T-lymphocytes from virus controllers (squares) or seronegative donors (triangles) were treated with 2.0 mM valproic acid (VPA) as in (A). The pretreated CD8+ T-lymphocytes were cultured with heterologous HIV-1-infected CD4+ T-lymphocytes at a 0.5:1 effector to target ratio. HIV-1 replication in the CD8+ T-lymphocyte co-culture is shown as the percentage of the replication control. HIV-1 replication was significantly lower in the presence of virus controller CD8+ T-lymphocytes compared to seronegative CD8+ T-lymphocytes (unpaired t-test, p<0.0001). Valproic acid significantly increased replication in the virus controller CD8+ T-lymphocyte co-cultures (paired t-test, p=0.0003). There was not a significant increase in replication in the co-cultures containing valproic acid-treated seronegative CD8+ T-lymphocytes (paired t-test, p=0.2076). Means and standard error are represented by horizontal and vertical lines respectively.
To determine how common the dependence on histone deacetylation is for CD8+ T-lymphocytes from HIV-1+ virus controllers, we went on to further examine CD8+ T-lymphocytes from 13 different subjects. Inhibiting histone deacetylation by pretreatment with valproic acid significantly decreased noncytolytic suppression across the panel of CD8+ T-lymphocytes from 13 virus controllers to varying degrees (p=0.0003, Fig. 6B). In contrast, valproic acid treatment did not significantly alter HIV-1 replication in the presence of seronegative CD8+ T-lymphocyte from eight different subjects (<6% change, p=0.2) (Fig. 6B). For one virus controller, CD8+ T-lymphocytes substantially suppressed virus replication to 8.8% of the replication control, but was increased to 43.1% of the replication control by valproic acid treatment (Fig. 6B). Virus replication in cultures containing valproic acid-treated CD8+ T-lymphocytes from a second virus controller increased up to 35.1% of the replication control (Fig. 6B). In the case of another virus controller, virus replication increased from 4% to 25% with valproic acid-treated CD8+ T-lymphocytes. Inhibition of HDACs in the CD8+ T-lymphocytes reduced suppression by more than 20% in 5 of the 13 virus controllers tested. Valproic acid pretreatment of CD8+ T-lymphocytes from VC3 reversed suppression by 11%, identical to the change seen when valproic acid was present during co-culture (Figs. 3 and 6B). Thus, valproic acid pretreatment of CD8+ T-lymphocytes from both virus controller 3 and 6 with valproic acid produced similar results to those seen when valproic acid was added during the co-culture. None of the virus controllers suppressed HIV-1 better after valproic acid pretreat-ment. Inhibition of HDACs in primary CD8+ T-lymphocytes from HIV-1+ virus controllers and the CD8+ effector cell line impaired their suppressive responses. In summary, this study demonstrates that chromatin modifications through histone deacetylation within effector CD8+ T-lymphocytes can be required for optimal CD8+ T-lymphocyte noncytolytic suppression of HIV-1.
Discussion and conclusions
Understanding the regulation of the CD8+ T cell mediated noncytolytic response has been an elusive goal since its discovery more than 20 years ago. We detail here the first evidence that the mediators of the CD8+ noncytolytic response can be regulated by histone deacetylation. Our data suggests that histone hyperacetylation induces expression of negative regulators of the antiviral factors, which leads to diminished noncytolytic suppression. Studies of the cytolytic response have shown that HDAC inhibitors augment the cytolytic response by CD8+ T-lymphocytes (Agarwal et al., 2009). Exposing CD8+ T-lymphocytes to a HDAC inhibitor during antigen stimulation increased specific killing, and secretion of gamma interferon, MIP-1 alpha and MIP-1 beta (Agarwal et al., 2009). Conversely, our data suggests that histone hyperacetylation decreases noncytolytic suppression of HIV-1. These data further distinguish the cytolytic and noncytolytic responses in CD8+ T-lymphocytes, and indicate distinct networks of genes that mediate each response. The varied responses seen with virus controller CD8+ T-lymphocytes suggests that the network of genes may be heterogeneous and that the totality of genes that can comprise the noncytolytic response may not be common among all HIV-1 infected individuals. Therefore, inhibiting HDACs may lead to different degrees of impact on virus suppression among individuals. The heterogeneous nature of the noncytolytic response has been speculated since some HIV-infected individuals exhibit contact-mediated suppression, but not soluble noncytolytic suppression (Walker and Levy, 1989). The presence of multiple mediators was also shown by size chromatography of CD8+ T-lymphocyte supernatants. In these experiments partial suppressive capacity was found in fractions of different sizes, and the sum of the suppression by each fraction totaled the suppression of the unfractionated supernatant (Geiben-Lynn et al., 2001). We propose that the noncytolytic response is a combination of several factors, with at least a subset of these factors being directly or indirectly regulated by changes in chromatin structure.
The mechanism of HIV-1 transcriptional inhibition is still unclear. HIV-1 proviral latency is maintained by histone deacetylases that inhibit transcription from the HIV-1 LTR (Tyagi and Karn, 2007; Williams et al., 2006; Ylisastigui et al., 2004). We show here that noncytolytic suppression does not block HIV-1 transcription initiation with similar mechanisms. HDAC1 is thought to be a mediator of HIV-1 proviral latency, but was dispensable for noncytolytic suppression of HIV-1 infected TZM-bl cells. Mosoian and colleagues have demonstrated that mediators of noncytolytic suppression differ in macrophages and T cells (Mosoian et al., 2006); thus, the requirement of HDAC1 in other cell types warrants further examination. HDAC1 decreases the acetylation of histones, an effect that we did not observe at the HIV-1 LTR in the presence of noncytolytic suppression of T-lymphocytes. Therefore, noncytolytic suppression inhibits HIV-1 transcription in acute infection in a manner different from what has been proposed for proviral latency.
In our study we saw impairment of the noncytolytic response at concentrations of valproic acid previously reported to be found in the blood of treated subjects (Gottlicher et al., 2001; Jennings and Romanelli, 1999). At these concentrations, valproic acid has no effect on apoptosis or the cell cycle (Gottlicher et al., 2001). Mosley et al. have found that valproic acid becomes toxic to CD8+ T-lymphocytes, impairing their cytolytic activity at levels 2.5 times the highest concentrations used in our study (Mosley et al., 2006). Similarly, we found Trichostatin A to be toxic at nanomolar concentrations to T-lymphocytes in our hands, which led us to use valproic acid in this study. However, Trichostatin A has been shown to enhance T-lymphocyte differentiation into memory T-lymphocytes capable of lysis of target cells (Agarwal et al., 2009; Northrop et al., 2008). Clearly, the amount and type of HDAC inhibitor has to be considered carefully when designing in vitro studies; and CD8+ T cell responses need to be examined more closely in HIV-1 infected patients treated with HDAC inhibitors.
The multifactorial, heterogeneous nature of the antiviral response makes antibody blocking of single molecules unproductive, and demonstrates the necessity for multigene approaches to identify the mediators and mechanisms of noncytolytic suppression. Microarray studies have shown that histone deacetylases regulate the expression of 8.7% of the genes analyzed in human CD8+ T-lymphocytes (Agarwal et al., 2009). A portion of the noncytolytic suppressive factors are likely contained within this subset of genes, since histone hyperacetylation down-modulates noncytolytic suppression. Microarray analysis of CD8+ T-lymphocytes with and without noncytolytic suppression has been complicated by determining relevant differences in CD8+ T-lymphocytes from different donors with different genetic backgrounds. Histone deacetylase inhibitors can be used to down-modulate noncytolytic suppression, allowing comparative genetic analyses of the same cells with and without noncytolytic suppressive activity. Future studies should aim to identify potential suppressive factors by analyzing the changes in mRNA or protein levels of secreted molecules from untreated or valproic acid-treated CD8+ T-lymphocytes. The availability of multiplex bead-based assays and quantitative PCR has made it increasingly easier to define expression profiles of specific cells. These technologies coupled with histone deacetylase inhibitors provide a more focused approach for identifying some of the novel antiviral molecules secreted by CD8+ T-lymphocytes.
Materials and methods
Study participants
HIV-1+ subjects controlling virus replication (virus controllers) with CD4+ T cell counts greater than 385 cells/μL and a viral load less than 5000 copies/mL were recruited from the Duke Adult and Pediatric Infectious Disease Clinics. Informed consent was obtained from all study participants according to a Duke University IRB approved protocol. Seronegative PBMCs were isolated from blood donors from the Red Cross.
Cell culture
PBMCs from seronegative donors or virus controllers were activated for 3 days with anti-CD3 and anti-CD28 in RPMI (20% FBS, 1% Penicillin–Streptomycin, 20 U/mL IL-2). CD4+-enriched cells were obtained from activated PBMCs by negative selection using anti-CD8 immunomagnetic beads (Invitrogen). CD8+-enriched cells were obtained similarly with anti-CD4 immunomagnetic beads (Invitrogen) or with the CD8+ T Cell Isolation kit and an autoMACS Cell Separator per the manufacturer’s procedure (Miltenyi Biotechnology). CD4+ and CD8+ T-lymphocyte purity was routinely 94% and 93%, respectively. The HVS-transformed CD8+ T-lymphocyte cell line from an asymptomatic HIV-1 infected subject, JR-HVS (Lacey et al., 1998), was cultured in AIM-V (20% FBS, 40 U/mL IL-2). TZM-bl cells were cultured in DMEM (10% FBS, 1% Penicillin–Streptomycin).
Acute HIV-1 CD8+ T-lymphocyte suppression assays
In autologous assays, CD4+-enriched cells were infected with a single cycle HIV-1NL4-3 pseudotyped luciferase reporter virus for 2 h in a 50 mL conical tube (Overman et al., 2007). This pseudotyped virus contains the NL4-3 genome encoding luciferase in the place of nef. Envelope production by this virus is abrogated by a frameshift mutation, limiting virus replication to one cycle. (Connor et al., 1995). HIV-1 infected CD4+ cells were plated with autologous CD8+ T cells or cultured alone. PBS or valproic acid (Calbiochem) dissolved in PBS was added to HIV-1 infected CD4+-enriched cells growing alone (HIV replication control) or together with autologous CD8+ T cells (CD8+ T cell co-culture). At 72 h post-infection, the cells were lysed with Luciferase Cell Culture Lysis reagent (Promega) and one freeze–thaw cycle. Luciferase expression was quantified with the Luciferase Assay System (Promega) on a Centro LB luminometer (Berthold). The level of HIV replication in the co-cultures containing CD8+ T cells and infected CD4+ T cells was expressed as the percent of the HIV replication in CD4+ T-lymphocytes alone: [relative light units (luciferase) in the co-culture of CD8+ and infected CD4+ T-lymphocytes (CD8+ T cell co-culture)]/[relative light units (luciferase) in infected CD4+ T cells alone (replication control)]×100%. CD8+ T cell co-cultures containing valproic acid were compared to replication control wells with the same treatment to control for effects of VPA on HIV-1 replication.
Heterologous assays were performed similarly. CD4+-enriched cells were obtained from activated PBMCs from a pool of 10 seronegative donors. Infections and valproic acid treatment were performed as described above. HVS-transformed CD8+ T cells from an asymptomatic HIV-infected individual known to demonstrate potent noncytolytic suppression were added to the culture to assess the mechanisms of noncytolytic suppression. To distinguish soluble suppression, infected CD4+ T cells were plated in the lower chamber of a 96-well 0.4 μM Pore Transwell system (Corning) and CD8+ T cells were added to the upper chamber.
Valproic acid pretreatment assay
CD8+ T cells were cultured in media containing PBS or valproic acid dissolved in PBS for 16 h. The CD8+ T cells were subsequently washed, counted, and plated in a contact-mediated noncytolytic suppression assay as stated above. Viability of the CD8+ T cells after pretreatment was above 90% as determined by Guava VIACount assays (Millipore) per the manufacturer’s protocol at the conclusion of the assay.
Western blot
Cells were lysed in RIPA buffer (0.1% SDS, 0.1% NP-40, 0.5% Sodium deoxycholate) and protein concentration was determined with the QuickStart Bradford Assay (Bio-Rad). Thirty μg of TZM-bl, 30 μg of CD4+ T-lymphocytes or 60 μg of CD8+ T-lymphocytes protein was fractionated by 7.5% SDS-PAGE. Protein was transferred to PVDF membrane and blocked with 5% BLOTTO. Membranes were probed with a 1:5000 dilution of primary antibody and a 1:10,000 dilution of anti-rabbit antibody (GE Healthcare). Total histone 3 was detected with anti-histone 3 clone ab8898 (Abcam). Acetylated histone 3 was detected with antibody clone 06-599 (Millipore). Blots were detected with autoradiography using an ECL Plus Kit (GE Healthcare).
Soluble noncytolytic suppression of HDAC1-deficient cells
siRNA-mediated knockdown of HDAC1
TZM-bl cells were plated at 80,000 cells per well in a 12-well plate in DMEM 24 h before transfection. DharmaFect1 (Dharmacon) was used to transfect siRNAs per the manufacturer’s protocol. To knockdown HDAC1 expression, SMARTpool HDAC1 siRNAs (Dharmacon) were used. Negative Control siRNA 1 (Ambion) was used a negative control. Nuclease-free water was used in the place of siRNAs for mock transfections. TZM-bl cells were transfected with a final concentration of 200 nM of siRNA. Efficient knockdown was assessed by western blot using anti-HDAC1 clone 7028 (Abcam) and anti-β-actin clone 8227 (Abcam).
Soluble noncytolytic suppression assay
Forty-eight hours post transfection, TZM-bl cells were trypsinized and washed once with complete growth media. TZM-bl cells were plated at 16,000 cells per well in the lower chamber of a 96-well 0.4 μM Pore Transwell system (Corning) and were infected with the CXCR4-tropic virus, HIV-1IIIB, at an MOI of 0.5. Activated primary or JR-HVS CD8+ T-lymphocytes were plated in the upper chamber of the transwell system at various effector to target ratios at the time of infection of the target cells. LTR-driven luciferase expression was measured using the BriteLite Plus system (Perkin Elmer) at 48 h post-infection (96 h post transfection). Viability (>92%) of the TZM-bl cells was determined using the Guava VIACount assay (Millipore) per the manufacturer’s protocol at the conclusion of the assay. The remaining transfected TZM-bl cells were analyzed for HDAC1 expression by Western blot at the initiation and at the completion of the assay.
Quantitative native chromatin immunoprecipitation of acetylated histone 3 (H3ac)
Native chromatin purification
Large-scale contact-mediated noncytolytic suppression assays were performed with HIV-1IIIB at a multiplicity of infection of 0.0075. Virion-associated reverse transcriptase activity in the culture supernatant was used to determine virus replication at 3 and 6 days post-infection as described elsewhere (Miller et al., 2009). Native chromatin was purified as previously described (Blower et al., 2002) with the following modifications: Cells from contact-mediated noncytolytic suppression assays were pelleted and resuspended in Chromatin Isolation Buffer (CIB) supplemented with Complete Mini Protease inhibitors (Roche) and NP-40 to 0.5%. Cells were broken on ice with 45 strokes of a Dounce homogenizer. Nuclei were washed once with CIB and resuspended in CIB supplemented with 1 mM CaCl2. Micrococcal nuclease was used to digest the chromatin. Nuclei were pelleted at 1000×g for 6 min and incubated on ice for 2 h in 500 mM NaCl in PBS. Insoluble chromatin was pelleted at 20,000×g for 15 min, the supernatant was retained as the soluble native chromatin.
Chromatin immunoprecipitation (ChIP)
ChIP was performed as previously described (Mravinac et al., 2009) except 55 μg of chromatin was used. Four μg of anti-acetylated Histone 3 [clone 06-599 (Millipore)] was used for immunoprecipitation. A mock immunoprecipitation containing no antibody was performed as a negative control. HIV-1 LTR DNA was detected with real-time PCR using Power SYBR Green PCR master mix (Applied Biosystems) and the following HIV-1-specific primers: HIV-1 5′ LTR forward: 5′-GCCTGGGAGCTCTCTGGCTA-3′; HIV-1 5′ LTR reverse: 5′-CAACA-GACGGGCACACACTACTT-3′. As a control, GAPDH DNA was detected with GAPDH promoter primers that have been detailed elsewhere (Spilianakis et al., 2003). ChIP results were analyzed as percent of the total input used for immunoprecipitation. Percent of the total input was calculated using the following formula: .
Acknowledgments
We thank Drs. Beth Sullivan, David Margolis, Kent J. Weinhold, and Mariano Garica-Blanco for helpful discussions about this work. We thank the Duke CFAR Clinical Core Investigators: Drs. Sunita Patil, Jason Stout, Gary Cox, David Holland, Charles Hicks, Suzanna Naggie, Mehri McKeller, Dev Anderson, Nathan Thielman, Ann Mosher, Vivian Chu, Steve Taylor and Ross McKinney and the patients and staff of the Adult and Pediatric Infectious Disease Clinics. The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: TZM-bl from Dr. John C. Kappes, Dr. Xiaoyun Wu and Tranzyme Inc. This work was supported by grants RO1-AI-052779 (G.D.T.), F31-AI-078715 (K.O.S.), Molecular Virology and Clinical Cores of the Duke University Center for AIDS Research and P30-AI-64518 (G.D.T., C.K.C.) awarded by National Institute of Allergy and Infectious Disease.
Contributor Information
Kevin O. Saunders, Email: Kevin.saunders@duke.edu.
Stephanie A. Freel, Email: Stephanie.freel@duke.edu.
R. Glenn Overman, Email: Glenn.overman@duke.edu.
Coleen K. Cunningham, Email: Coleen.cunningham@duke.edu.
Georgia D. Tomaras, Email: gdt@duke.edu.
References
- Addo MM, Draenert R, Rathod A, Verrill CL, Davis BT, Gandhi RT, Robbins GK, Basgoz NO, Stone DR, Cohen DE, Johnston MN, Flynn T, Wurcel AG, Rosenberg ES, Altfeld M, Walker BD. Fully differentiated HIV-1 specific CD8+ T effector cells are more frequently detectable in controlled than in progressive HIV-1 infection. PLoS ONE. 2007;2 (3):e321. doi: 10.1371/journal.pone.0000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P, Raghavan A, Nandiwada SL, Curtsinger JM, Bohjanen PR, Mueller DL, Mescher MF. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J Immunol. 2009;183(3):1695–1704. doi: 10.4049/jimmunol.0900592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archin NM, Keedy KS, Espeseth A, Dang H, Hazuda DJ, Margolis DM. Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. AIDS. 2009;23 (14):1799–1806. doi: 10.1097/QAD.0b013e32832ec1dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower MD, Sullivan BA, Karpen GH. Conserved organization of centromeric chromatin in fiies and humans. Dev Cell. 2002;2 (3):319–330. doi: 10.1016/s1534-5807(02)00135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano MG, Poli R, Pugliese M, Fortunati N, Boccuzzi G. Valproic acid enhances tubulin acetylation and apoptotic activity of paclitaxel on anaplastic thyroid cancer cell lines. Endocr Relat Cancer. 2007;14 (3):839–845. doi: 10.1677/ERC-07-0096. [DOI] [PubMed] [Google Scholar]
- Chen CH, Weinhold KJ, Bartlett JA, Bolognesi DP, Greenberg ML. CD8+ T lymphocyte-mediated inhibition of HIV-1 long terminal repeat transcription: a novel antiviral mechanism. AIDS Res Hum Retroviruses. 1993;9 (11):1079–1086. doi: 10.1089/aid.1993.9.1079. [DOI] [PubMed] [Google Scholar]
- Chun TW, Justement JS, Moir S, Hallahan CW, Ehler LA, Liu S, McLaughlin M, Dybul M, Mican JM, Fauci AS. Suppression of HIV replication in the resting CD4+ T cell reservoir by autologous CD8+ T cells: implications for the development of therapeutic strategies. Proc Natl Acad Sci U S A. 2001;98(1):253–258. doi: 10.1073/pnas.98.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1, and MIP-1 as the Major HIV-Suppressive Factors Produced by CD8+ T Cells. Science. 1995;270 (5243):1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206 (2):935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- Coull JJ, Romerio F, Sun JM, Volker JL, Galvin KM, Davie JR, Shi Y, Hansen U, Margolis DM. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol. 2000;74(15):6790–6799. doi: 10.1128/jvi.74.15.6790-6799.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kharroubi A, Piras G, Zensen R, Martin MA. Transcriptional activation of the integrated chromatin-associated human immunodeficiency virus type 1 promoter. Mol Cell Biol. 1998;18(5):2535–2544. doi: 10.1128/mcb.18.5.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiben-Lynn R, Kursar M, Brown NV, Kerr EL, Luster AD, Walker BD. Noncytolytic inhibition of X4 virus by bulk CD8(+) cells from human immunodeficiency virus type 1 (HIV-1)-infected persons and HIV-1-specific cytotoxic T lymphocytes is not mediated by beta-chemokines. J Virol. 2001;75(17):8306–8316. doi: 10.1128/JVI.75.17.8306-8316.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20(24):6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabar S, Selinger-Leneman H, Abgrall S, Pialoux G, Weiss L, Costagliola D. Prevalence and comparative characteristics of long-term nonprogressors and HIV controller patients in the French Hospital Database on HIV. AIDS. 2009;23 (9):1163–1169. doi: 10.1097/QAD.0b013e32832b44c8. [DOI] [PubMed] [Google Scholar]
- Gurvich N, Tsygankova OM, Meinkoth JL, Klein PS. Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res. 2004;64(3):1079–1086. doi: 10.1158/0008-5472.can-03-0799. [DOI] [PubMed] [Google Scholar]
- Hebbes TR, Thorne AW, Clayton AL, Crane-Robinson C. Histone acetylation and globin gene switching. Nucleic Acids Res. 1992;20(5):1017–1022. doi: 10.1093/nar/20.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings HR, Romanelli F. The use of valproic acid in HIV-positive patients. Ann Pharmacother. 1999;33(10):1113–1116. doi: 10.1345/aph.19014. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293 (5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Kiernan RE, Vanhulle C, Schiltz L, Adam E, Xiao H, Maudoux F, Calomme C, Burny A, Nakatani Y, Jeang KT, Benkirane M, Van Lint C. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999;18(21):6106–6118. doi: 10.1093/emboj/18.21.6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey SF, Weinhold KJ, Chen CH, McDanal C, Oei C, Greenberg ML. Herpesvirus saimiri transformation of HIV type 1 suppressive CD8+ lymphocytes from an HIV type 1-infected asymptomatic individual. AIDS Res Hum Retroviruses. 1998;14 (6):521–531. doi: 10.1089/aid.1998.14.521. [DOI] [PubMed] [Google Scholar]
- Lambotte O, Boufassa F, Madec Y, Nguyen A, Goujard C, Meyer L, Rouzioux C, Venet A, Delfraissy JF. HIV controllers: a homogeneous group of HIV-1-infected patients with spontaneous control of viral replication. Clin Infect Dis. 2005;41(7):1053–1056. doi: 10.1086/433188. [DOI] [PubMed] [Google Scholar]
- Lusic M, Marcello A, Cereseto A, Giacca M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003;22(24):6550–6561. doi: 10.1093/emboj/cdg631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackewicz CE, Ortega HW, Levy JA. CD8+ cell anti-HIV activity correlates with the clinical state of the infected individual. J Clin Invest. 1991;87(4):1462–1466. doi: 10.1172/JCI115153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackewicz CE, Blackbourn DJ, Levy JA. CD8+ T cells suppress human immunodeficiency virus replication by inhibiting viral transcription. Proc Natl Acad Sci U S A. 1995;92(6):2308–2312. doi: 10.1073/pnas.92.6.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller HB, Saunders KO, Tomaras GD, Garcia-Blanco MA. Tat-SF1 is not required for Tat transactivation but does regulate the relative levels of unspliced and spliced HIV-1 RNAs. PLoS ONE. 2009;4 (5):e5710. doi: 10.1371/journal.pone.0005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosley AJ, Meekings KN, McCarthy C, Shepherd D, Cerundolo V, Mazitschek R, Tanaka Y, Taylor GP, Bangham CR. Histone deacetylase inhibitors increase virus gene expression but decrease CD8+ cell antiviral function in HTLV-1 infection. Blood. 2006;108 (12):3801–3807. doi: 10.1182/blood-2006-03-013235. [DOI] [PubMed] [Google Scholar]
- Mosoian A, Teixeira A, High AA, Christian RE, Hunt DF, Shabanowitz J, Liu X, Klotman M. Novel function of prothymosin alpha as a potent inhibitor of human immunodeficiency virus type 1 gene expression in primary macrophages. J Virol. 2006;80(18):9200–9206. doi: 10.1128/JVI.00589-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mravinac B, Sullivan LL, Reeves JW, Yan CM, Kopf KS, Farr CJ, Schueler MG, Sullivan BA. Histone modifications within the human X centromere region. PLoS ONE. 2009;4 (8):e6602. doi: 10.1371/journal.pone.0006602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northrop JK, Wells AD, Shen H. Cutting edge: chromatin remodeling as a molecular basis for the enhanced functionality of memory CD8 T cells. J Immunol. 2008;181(2):865–868. doi: 10.4049/jimmunol.181.2.865. [DOI] [PubMed] [Google Scholar]
- Overman RG, Llorens AL, Greenberg ML, Garcia-Blanco MA, Tomaras GD. Initiation of human immunodeficiency virus type 1 (HIV-1) transcription is inhibited by noncytolytic CD8 suppression. Open Virol J. 2007;1:1–7. doi: 10.2174/1874357900701010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8(12):983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilianakis C, Kretsovali A, Agalioti T, Makatounakis T, Thanos D, Papamatheakis J. CIITA regulates transcription onset viaSer5-phosphorylation of RNA Pol II. EMBO J. 2003;22(19):5125–5136. doi: 10.1093/emboj/cdg496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272 (5260):408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- Tomaras GD, Lacey SF, McDanal CB, Ferrari G, Weinhold KJ, Greenberg ML. CD8+ T cell-mediated suppressive activity inhibits HIV-1 after virus entry with kinetics indicating effects on virus gene expression. Proc Natl Acad Sci U S A. 2000;97(7):3503–3508. doi: 10.1073/pnas.070521097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toso JF, Chen CH, Mohr JR, Piglia L, Oei C, Ferrari G, Greenberg ML, Weinhold KJ. Oligoclonal CD8 lymphocytes from persons with asymptomatic human immunodeficiency virus (HIV) type 1 infection inhibit HIV-1 replication. J Infect Dis. 1995;172(4):964–973. doi: 10.1093/infdis/172.4.964. [DOI] [PubMed] [Google Scholar]
- Turner BM. Decoding the nucleosome. Cell. 1993;75 (1):5–8. [PubMed] [Google Scholar]
- Turner BM. Histone acetylation and an epigenetic code. BioEssays. 2000;22 (9):836–845. doi: 10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Tyagi M, Karn J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007;26(24):4985–4995. doi: 10.1038/sj.emboj.7601928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996a;15(5):1112–1120. [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Verdin E. The expression of a smallfraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 1996b;5(4–5):245–253. [PMC free article] [PubMed] [Google Scholar]
- Walker BD. Elite control of HIV Infection: implications for vaccines and treatment. Top HIV Med. 2007;15(4):134–136. [PubMed] [Google Scholar]
- Walker CM, Levy JA. A diffusible lymphokine produced by CD8+ T lymphocytes suppresses HIV replication. Immunology. 1989;66 (4):628–630. [PMC free article] [PubMed] [Google Scholar]
- Walker CM, Moody DJ, Stites DP, Levy JA. CD8+ lymphocytes can control HIV infection in vitro by suppressing virus replication. Science. 1986;234 (4783):1563–1566. doi: 10.1126/science.2431484. [DOI] [PubMed] [Google Scholar]
- Walker BD, Chakrabarti S, Moss B, Paradis TJ, Flynn T, Durno AG, Blumberg RS, Kaplan JC, Hirsch MS, Schooley RT. HIV-specific cytotoxic T lymphocytes in seropositive individuals. Nature. 1987;328 (6128):345–348. doi: 10.1038/328345a0. [DOI] [PubMed] [Google Scholar]
- Walker CM, Moody DJ, Stites DP, Levy JA. CD8+ T lymphocyte control of HIV replication in cultured CD4+ cells varies among infected individuals. Cell Immunol. 1989;119(2):470–475. doi: 10.1016/0008-8749(89)90259-1. [DOI] [PubMed] [Google Scholar]
- Walker CM, Erickson AL, Hsueh FC, Levy JA. Inhibition of human immunodeficiency virus replication in acutely infected CD4+ cells by CD8+ cells involves a noncytotoxic mechanism. J Virol. 1991;65(11):5921–5927. doi: 10.1128/jvi.65.11.5921-5927.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006;25(1):139–149. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylisastigui L, Archin NM, Lehrman G, Bosch RJ, Margolis DM. Coaxing HIV-1 from resting CD4 T cells: histone deacetylase inhibition allows latent viral expression. AIDS. 2004;18 (8):1101–1108. doi: 10.1097/00002030-200405210-00003. [DOI] [PubMed] [Google Scholar]