

Abstract
Abscisic acid (ABA) response elements (ABREs) are a group of cis-acting DNA elements that have been identified from promoter analysis of many ABA-regulated genes in plants. We are interested in understanding the mechanism of binding specificity between ABREs and a class of bZIP transcription factors known as ABRE binding factors (ABFs). In this work, we have modeled the homodimeric structure of the bZIP domain of ABRE binding factor 1 from Arabidopsis thaliana (AtABF1) and studied its interaction with ACGT core motif-containing ABRE sequences. We have also examined the variation in the stability of the protein–DNA complex upon mutating ABRE sequences using the protein design algorithm FoldX. The high throughput free energy calculations successfully predicted the ability of ABF1 to bind to alternative core motifs like GCGT or AAGT and also rationalized the role of the flanking sequences in determining the specificity of the protein-DNA interaction.
Keywords: Abscisic acid response element, Basic leucine zipper, Protein–DNA interaction, Comparative modeling, FoldX, HADDOCK, Recognition specificity
Abbreviations: ABA, abscisic acid; ABRE, abscisic acid response element; ABF1, ABRE binding factor 1; bZIP, basic leucine zipper; SSCRE, somatostatin cAMP response element; CREB, cAMP response element-binding protein.
Highlights
▸ Abscisic acid response elements (ABREs) are bound by ABFs, a class of bZIP transcription factor. ▸ We examined the recognition of ABRE DNA sequences by ABF1 protein from A. thaliana. ▸ An initial model was constructed by comparative modeling and protein DNA docking. ▸ Relative binding affinities of the protein for mutated DNA sequences were calculated. ▸ The approach explains much of the experimental data at a low computational cost.
1. Introduction
Abscisic acid (ABA) is a vital mediator of responses in plants to various adverse environmental conditions like salinity, cold, drought, etc. Some of the ABA-mediated physiological responses are regulated by a group of basic leucine zipper (bZIP) transcription factors [1] that interact with a class of cis-acting DNA elements, collectively known as abscisic acid response elements (ABREs) [2].
Although plant bZIP proteins have similar motifs as their animal counterparts consisting of a basic DNA-binding region towards the N-terminal and a leucine zipper portion in the C-terminal, they also have some differences in the detailed structure of the motifs. The characterization and classification of plant bZIP proteins have been reviewed in detail [3–5].
Plant bZIP proteins recognizing ABRE sequences are generally thought to require the presence of an ACGT core with some degree of variability in the flanking sequences. A typical consensus sequence in case of rice was proposed as T/G/CACGTGG/TC [6]. Similar sequence requirement was also found for other species [7,8]. Experimental investigations have revealed a complex pattern of sequence recognition of various classes of arabidopsis bZIP proteins (compiled in Table 1 of Ref. [3]). We are interested here in the interaction of the ABRE binding factors (ABFs) (which belong to group A of the classification by Jakoby et al. [3]) with their various cognate and non-cognate sequences. A qualitative study of the binding affinities of the arabidopsis ABFs for the ABRE sequence and its variants has been reported by Choi et al. [9]. Similar type of studies have been reported for bZIPs from other plants [6–8]. These studies have also revealed that the ABRE binding proteins can also recognize variation within the ACGT core itself so that replacement by GCGT or AAGT results in binding by ABFs [9–12].
In spite of the evident importance of a detailed understanding of the mechanism of recognition of ABREs by members from the plant bZIP transcription factor family, no study has addressed this problem. This is presumably due to a lack of high resolution structures of plant bZIP proteins with their cognate DNA sequences. Among the plant bZIP proteins, so far only the leucine zipper portion of the HY5 transcription factor from Arabidopsis thaliana has been reported [13] along with a very recent effort of crystallization of the rice AREB8 protein together with its cognate DNA [14].
In view of the lack of experimentally obtained structures for plant bZIP DNA complexes, we sought to build the structure of ABF1–ABRE complex by comparative modeling using the crystal structure of the CREB bZIP-somatostatin cAMP response element (CRE) complex [15]. Both the proteins have comparatively similar DNA binding domain and both the DNA contain an ACGT core, suggesting that there may be similar mechanisms at work for recognition of ACGT sequences for the two classes of proteins.
To test the validity of our model and its predictive capability, we have carried out a series of mutations in the DNA sequence bound by the ABF1 and theoretically calculated the resulting free energy changes. Although for accurate evaluation of free energy changes one needs to carry out extensive molecular dynamics simulations, we instead adopted the empirical force field based high throughput method provided by the FoldX algorithm [16]. This enabled us to study a large number of mutations in a relatively short time. We also found that FoldX calculations correlated quite well with a large number of experimental results with mutated sequences thus providing a microscopic and quantitative model for interaction of the plant ABF bZIP proteins with ABRE sequences.
2. Computational details
2.1. Modeling of ABF1 homodimer
The 392 amino acid long sequence of ABRE binding factor 1 (ABF1) from A. thaliana was retrieved from UniProt (http://www.uniprot.org/). Template identification and comparative modeling were carried out by the automated modeling server SWISS-MODEL version 8.05 (http://swissmodel.expasy.org/) [17]. The homodimeric CREB bZIP (PDB id 1DH3, residues 1–55) was selected as a template for modeling the bZIP region (residues 313–367) of ABF1, the alignment having the maximum similarity score of 92.03, the minimum E-value of 1.3e−09 and a sequence identity of 32% (Fig. 1). The resulting model was energy minimized by the YASARA energy minimization server (http://www.yasar.org/minimizationserver.htm) [18].
Fig. 1.
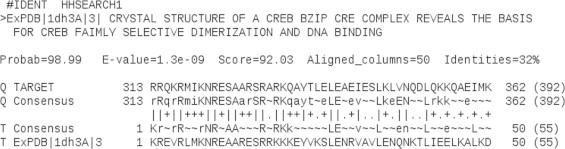
Pairwise sequence alignment between the bZIP regions of query (ABF1) and template (CREB) as obtained from HHsearch method of SWISS-MODEL template identification tool. The first 50 residues of ABF1 bZIP region (313–362) and CREB bZIP (1–50) were aligned with sequence identity 32%, E-value of 1.3e−09 with similarity score 92.03. Consensus (Q for query and T for template) is defined on the basis of profile HMMs of respective sequences where the one letter codes define the most probable amino acid residues observed at the respective positions (capital letters denote a probability of occurrence which is greater than or equal to 60%, small letters if it is greater than or equal to 40% and tilde (∼) denotes non-conserved positions).
2.2. Computational prediction of ABF1–ABRE complex
The model of ABF1–ABRE complex was built up in two successive steps. First, the modeled structure of the ABF1 homodimer was aligned with the protein portion of the CREB–CRE complex and subsequently the CREB portion was removed. The DNA portion of the ABF1–CRE complex was then mutated to ABRE sequence using the appropriate utilities in the FoldX program package. The resulting model was repaired to remove short contacts and subjected to analysis of protein–DNA interactions.
In the next step, the FoldX model was segregated into protein (ABF1) and DNA (ABRE) parts and submitted to HADDOCK for docking [19]. A restraint file was prepared from the protein–DNA interaction data (Table 1) and used as unambiguous interaction restraints. Except for the rigid body docking structure parameter which was set to 5000 as recommended for comparative models, rest of the parameters were set from defaults provided [19]. The reason was to improve the model by incorporating conformational flexibility introduced by HADDOCK while enforcing the complex to form the protein DNA interactions similar to that present in the FoldX derived model. Out of 10 clusters (each having four possible conformations), generated by HADDOCK, the lowest scoring cluster was selected. All the four conformations of this cluster were subjected to repair followed by interaction energy analysis by FoldX. The lowest energy conformation was selected as a model of ABF1–ABRE complex with which further structural and mutational studies were carried out.
Table 1.
Interaction restraintsa used in HADDOCK for docking of ABF1 and ABRE.
| Protein residue(s) | DNA base(s) |
|---|---|
| R317(A) | G17(D) |
| N321(A) | G9(B), C19(D) |
| S328(A) | C19(D) |
| R329(A) | G7(B), C6(B) |
| R721(C) | G23(D) |
| N713(C) | A3(B) |
Interaction restraints were defined by those DNA and protein residues that were found to interact by hydrogen bonding in the FoldX model.
We have also carried out docking with the same restraints in the ambiguous mode of HADDOCK, but it was observed that the docked structure failed to maintain most of the interactions observed in the model generated by FoldX (data not shown).
2.3. Structural analysis
The quality of the modeled bZIP domain of ABF1 was evaluated by QMEAN [20] and PROCHECK [21]. Interactions were predicted by using web servers like PIC [22], WHAT IF [23] and an offline resource SOCKET [24]. Probable hydrogen bonded interactions in case of ABF1–ABRE complex were defined based on the distance cutoff of <3.9 Å between donor and acceptor heavy atoms and donor–hydrogen–acceptor angle ∼120° [25]. This interaction was further categorized into two groups, namely, specific, when either donor or acceptor atom belonged to the side chain of a DNA base and non-specific, when it was from the base ring.
2.4. In silico mutational analysis
The DNA regions of the lowest energy ABF1–ABRE complex were subjected to various mutations to understand the mechanisms of interaction specificity. Mutations were carried out by FoldX as described in [26,27] except that default number of rotamers and 0.05 M ionic strength were used. Interaction energies (ΔGint) of mutant and its wild type counterpart were calculated and compared to get the change in interaction energy (ΔΔGint) which gave an idea of the relative binding affinity. We adopted the ΔΔGint cutoff proposed in [26] to define the effect of mutations on the stability of the native ABF1–ABRE structure, namely, if ΔΔGint > 1.0 Kcal/mol, mutations at the respective positions might significantly affect the stability of the complex, whereas the stability of the complex was considered to be completely disrupted if ΔΔGint > 2.0 Kcal/mol.
3. Results and discussion
3.1. Structural characteristics of the ABF1 homodimer
The comparative model of the ABF1 homodimer is a parallel coiled–coil bZIP structure. The average backbone dihedral angles for the helical region (A: 314–361, C: 706–753, Fig. 2) are −65.5 ± 4.3° (Φ) and −39.4 ± 6.8° (Ψ) which is similar to that of the HY5 (−66.5 ± 8.6° (Φ) and −40.0 ± 7° (Ψ)) homodimer [13]. Our modeled structure scored 0.78 in QMEAN [20] scoring. PROCHECK [21] analysis found 95.3% residues within the allowed region of Ramachandran plot and most of the residues fulfilling the side chain torsion angle and planarity criteria essential for optimum stability of a protein structure.
Fig. 2.
The amino acid sequence of ABF1 bZIP domain (A chain 313–367 and C chain 705–759) used for modeling.
To evaluate the interactions predicted from the modeled ABF1 bZIP homodimer we followed the classification given by Deppmann et al. [28] and compared our model with the available crystal structure of the AtbZIP protein HY5 [13] with whom it shared 33% sequence identity at the dimerization region.
As predicted by the “knob into holes” packing model [29] and similar to other classical bZIPs, the ABF1 bZIP dimer interface was found to be formed by the side chains of a, a′, d, d′ (prime indicates the other monomer) heptad residues. Rotamers and core packing angles of these residues are given in Table 2. Unlike HY5 [13], most of the Leu residues have favorable rotamer angles and adopt a perpendicular packing geometry (packing angle ∼90°).
Table 2.
Predicted knob into hole assembly and its geometry.
| ABF1 residues | Side chain torsion angles (in degree) |
Packing angle (in degree) | |
|---|---|---|---|
| χ1 | χ2 | ||
| LEU 731(C) | −67.2 | 169.9 | 97.54 |
| ILE 735(C) | −73.6 | 164.5 | 35.63 |
| LEU 738(C) | −73.2 | 168.4 | 92.96 |
| LEU 745(C) | −73.3 | 167.8 | 96.3 |
| LEU 339(A) | −68.1 | 172.8 | 93.72 |
| ILE 343(A) | −75.7 | −75.3 | 38.6 |
| LEU 346(A) | −77.7 | 165.1 | 102.24 |
| LEU 353(A) | −79.5 | 164.4 | 97.24 |
The dimeric interface was flanked by the e and g residues of the heptad repeats which normally exhibited electrostatic interactions that might be important for dimer stability and dimerization specificity. On the basis of inter-atomic distances we predicted two possible salt-bridges that obeyed the classical gi to ei+5 inter-helical ionic-interaction criteria, between Glu342 (A, gi) and Lys739 (C, ei+5) with distance 3.6 Å and between Lys347 (A, ei+5) and Glu734 (C, gi) with distance 4 Å.
Unlike the other plant bZIPs, ABF1 bZIP region contains only one asparagine (Asn) residue at the a position of the second heptad similar to other animal bZIPs like CREB, GCN4, etc. This Asn residue was found to be involved in an inter-helix Asn–Asn interaction between the positions 350(A) and 742(C) with a distance 2.9 Å.
3.2. Protein–DNA interface
The lowest energy docked structure of ABF1–ABRE complex has a similar geometry as seen in crystal structures of other bZIP DNA complexes. The ABF1 homodimer (chains A and C) recognized the ABRE duplex (chains B and D) by its N terminal domains and made seven hydrogen bonds and one hydrophobic contacts at the major groove of DNA by five amino acids, namely, Ala717(C), Arg329(A), Asn713(C), Arg721(C) and Asn321(A). Except for Ala717(C), rest of the four residues are invariant ones known for their DNA binding interactions in bZIP proteins [30]. The protein–DNA interaction profile is schematically represented in Fig. 3A.
Fig. 3.
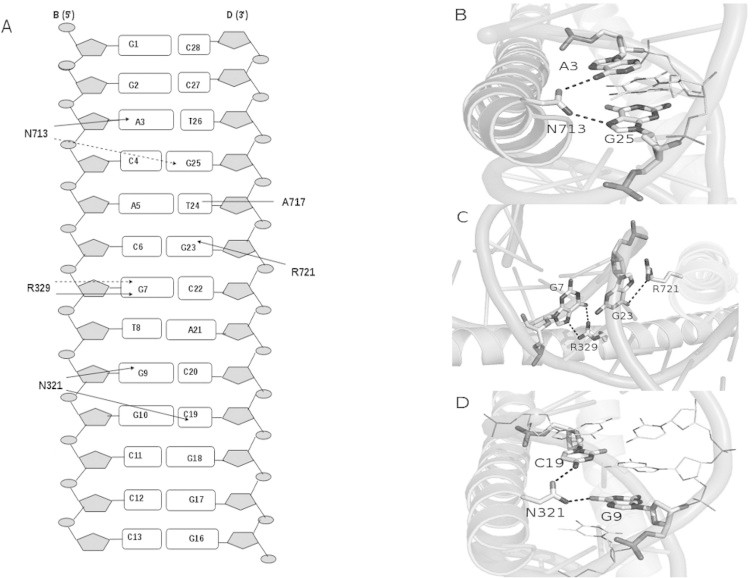
Protein–DNA interaction summary. (A) Schematic diagram representing base specific interactions predicted from the model of ABF1–ABRE complex. Non-specific hydrogen bonding is represented by dotted arrows whereas solid arrows represent specific hydrogen bonding. van der Waals contacts are shown as solid lines. (B) Hydrogen bonded interactions made by N713(C) with A3(B) and G25(D). Residues making contact are represented by sticks. (C) Hydrogen bonding network observed at the central CpG base pair step. The R329(A) is involved in a bidentate interaction with G7(B) whereas R721(C) has a hydrogen bond with G23(D). (D) Interactions made by N321(A) with G9 and C19(D).
3.3. Analysis of half-site binding
The 13 bp DNA fragment that complexed with ABF1 contains a decameric (residues 2–12) ABRE sequence, i.e., 5′GACACGTGGC3′, having a palindromic hexameric core (marked in bold) surrounded by flanking sequences. One observes from the SELEX experiment of Choi et al. [9] that although the GTG part is invariantly required, some amount of sequence variability exists in the CAC half-site for binding to the ABF1 homodimer. A possible explanation is that monomers can have asymmetric interaction with the palindromic hexameric core.
In our model, the two conserved residues asparagine (N321(A) and N713(C)) and arginine (R329(A) and R721(C)) [30] from each helix of the ABF1 homodimer were found to be involved in direct readout of the hexameric core. The N713(C) made a non-specific hydrogen bond to the N7 atom of G25(D) whereas N321(A) was involved in specific hydrogen bonding with O6 of G9(B). The other invariant residue arginine interacted with the central CpG base pair step of the hexameric core. R721(C) made a hydrogen bond to N7 of G23(D) whereas R329(A) was involved in a bifurcated hydrogen bond and contacted O6 and N7 of G7(B). A van der Waals interaction between A717(C) and T24(D) was observed only in the CAC half-site and was found to be absent in the other half. Such interaction profile indicated that despite its palindromic nature, the hexameric core was involved in an asymmetric interaction with the ABF1 homodimer. Similar asymmetry was also observed in the case of CREB SSCRE complex [15], where the R301 and R301′ residues interacted asymmetrically with the central CpG base pair step. In the flanking regions in our model, two base specific hydrogen bonding were observed. The N713(C) was found to interact with the N6 atom of 5′ flanking residue A3(B) whereas 3′ C19(D) was observed to interact with N321(A) via the donor atom N4.
3.4. Role of predicted interactions in defining specificity
In order to comprehensively evaluate the energetic contribution of DNA bases and to cover all the experimental data regarding stable ABF1–ABRE formation, the seven ABRE bases (A3(B), G25(D),T24(D), G23(D), G7(B), G9(B), and C19(D)) that made direct contact with ABF1, along with T8(B) were mutated in silico by FoldX. Effects of such mutations on the stability of protein–DNA complex were analyzed in terms of ΔΔGint (Table 3). We obtained significant correspondence with earlier experimental data regarding ABF1–ABRE interactions [6,9]. Our FoldX calculations confirmed the experimentally observed sequences CGC and CAA [9] as energetically allowed alternative combinations for the CAC half-site. We also observed a considerable degeneracy at the first cytosine residue (C4(B), in this case) of the CAC half site (Fig. 3A). FoldX calculations suggest thymine or adenine as alternatives for cytosine in this position. However, Hattori et. al. [6] suggested thymine or guanine as functional alternatives for the same position. Mutational analysis also established the energetic contribution of the two guanine residues (G7(B) and G9(B)) of the GTG half-site for stable complex formation (Table 3). In case of G7(B) any mutation completely disrupts the interaction with R329(A) explaining its experimentally observed invariant nature in ABF1–ABRE interaction [9]. For G9(B), the ΔΔGint values for all mutations except thymine are greater than the 1.0 kcal/mol cutoff. For thymine also, ΔΔGint is 0.89 kcal/mol which is very close to the cutoff, possibly making it an unlikely substitution. However, it should be noted that the present model is unable to explain the experimentally observed requirement of thymine residue (T8(B)) of the GTG half-site [6,9]. Structural analysis revealed that similar to the CREB–SSCRE complex [15], the ACGT motif of the hexameric core had alanine residues A325(A) and A717(C) in the vicinity of respective thymine bases T8(B) and T24(D). The distance between the Cβ carbon of T8(B) and A325(A) was found to be significantly larger (4.8 Å) compared to the distance observed between T24(D) and A717(C) (4.3 Å). Apparently, the larger separation between T8(B) and A325(A) in our model precluded a favorable van der Waals interaction and consequently FoldX calculations did not find any significant contribution of this base in the stability of the complex. Apart from the hexameric core, a consensus sequence for the 3′ flanking region, 3′G/TC has been reported as an essential requirement for plant bZIP DNA interaction [6,9]. Our calculations indicated in accord with experimental observations that mutations at the 3′ end flanking residue G10(B) destabilized the complex (Table 3). However, we could not explain the role of the 3′ cytosine next to G10(B) since it made no base specific direct interaction with the protein. Unlike the 3′ end, mutation of the 5′ flanking residue A3(B) was considerably more tolerated (Table 3). Mutations other than adenine to thymine at this position were energetically allowed, indicating that a cytosine or a guanine may occur at this position. In fact, SELEX experiments [9] reported sequences carrying cytosine at the 5′ flanking position to the hexameric core. Also a recent effort in crystallizing the highly similar OsAREB8–ABRE bZIP–DNA complex had cytosine at this position [14].
Table 3.
Average ΔΔintfor ABF1–ABRE complex for various mutations carried out in silicoat different positions in the ABRE sequence. The mutated position is indicated in bold.
| Type | Sequence | Change in interaction energy (ΔΔGint) kcal/mol (mutant-wildtype) |
|---|---|---|
| Wild type | G G A C A C G T G G C C C | |
| Mutated | ||
| At 5′ end | G G TC A C G T G G C C C | 1.09 ± 0.05 |
| At 5′ end | G G GC A C G T G G C C C | 0.32 ± 0.00 |
| At 5′ end | G G CC A C G T G G C C C | −0.09 ± 0.01 |
| At CACGTG | G G A T A C G T G G C C C | 0.06 ± 0.00 |
| At CACGTG | G G A GA C G T G G C C C | 1.91 ± 0.13 |
| At CACGTG | G G A AA C G T G G C C C | 0.67 ± 0.09 |
| At CACGTG | G G A C GC G T G G C C C | 0.03 ± 0.08 |
| At CACGTG | G G A C TC G T G G C C C | 1.16 ± 0.00 |
| At CACGTG | G G A C CC G T G G C C C | 1.30 ± 0.00 |
| At CACGTG | G G A C A AG T G G C C C | 0.60 ± 0.01 |
| At CACGTG | G G A C A GG T G G C C C | 1.21 ± 0.01 |
| At CACGTG | G G A C A TG T G G C C C | 2.25 ± 0.00 |
| At CACGTG | G G A C A C AT G G C C C | 2.71 ± 0.09 |
| At CACGTG | G G A C A C CT G G C C C | 2.45 ± 0.11 |
| At CACGTG | G G A C A C TT G G C C C | 1.85 ± 0.14 |
| At CACGTG | G G A C A C G CG G C C C | 0.35 ± 0.00 |
| At CACGTG | G G A C A C G GG G C C C | 0.31 ± 0.29 |
| At CACGTG | G G A C A C G AG G C C C | 0.74 ± 0.01 |
| At CACGTG | G G A C A C G T TG C C C | 0.89 ± 1.09 |
| At CACGTG | G G A C A C G T AG C C C | 1.44 ± 0.00 |
| At CACGTG | G G A C A C G T CG C C C | 1.93 ± 0.12 |
| At 3′ end | G G A C A C G T G AC C C | 1.59 ± 0.01 |
| At 3′ end | G G A C A C G T G TC C C | 1.05 ± 0.02 |
| At 3′ end | G G A C A C G T G CC C C | 2.00 ± 0.02 |
| Null mutation | G G A C C T A C A GC C C | 5.99 ± 0.49 |
4. Conclusion
Abscisic acid response elements (ABREs) are widely distributed at the upstream regions of many plant regulatory genes and need to be specifically recognized by respective transcription factors for proper gene expression. ABRE binding factor 1 (ABF1), one of the members of ABF family, is known to interact with ABRE sequences in a sequence specific manner. In order to understand its DNA recognition specificity, we have carried out comparative model building followed by extensive mutational analysis by an empirical force field based method FoldX. Our results suggest that in spite of its empirical nature, FoldX in general reproduces well the mutational data showing certain variations to be allowed within the classical hexameric core region (CACGTG) of the ABRE sequence. It is also observed from our model, in conformity with experimental data [6,9], that the flanking residues beyond the hexameric core have a role in protein DNA recognition and could affect the stability of the protein DNA complex even in the presence of a suitable hexameric core. One of the possible contributions of these flanking sequences could be the fine tuning of the discrimination of the cognate DNA sequence among a wide range of ABRE sequences for a particular transcription factor from a family.
However, it was also observed from the model that a considerable change in the sequence (Table 3, denoted as null mutation [9]) could strongly destabilize the complex. This clearly reflects that the flexibility in the recognition of cognate sequence by ABF1 is restricted.
Since other members of the ABF family share considerable sequence similarity with ABF1 [9], we expect this model to also explain the DNA recognition specificity for these sequences.
Acknowledgments
The authors would like to acknowledge the excellent initial study on this problem by Pratul Ghosh and Shibom Basu during their summer projects in A.L.’s laboratory.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Landschulz W.H., Johnson P.F., McKnight S.L. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 1988;240:1759–1764. doi: 10.1126/science.3289117. [DOI] [PubMed] [Google Scholar]
- 2.Busk P.K., Pages M. Regulation of abscisic acid-induced transcription. Plant Mol. Biol. 1996;37:425–435. doi: 10.1023/a:1006058700720. [DOI] [PubMed] [Google Scholar]
- 3.Jakoby M., Weisshaar B., Droge-Laser W., Vicente-Carbajosa J., Tiedemann J., Kroj T., Parcy F. bZIP transcription factors in Arabidopsis. Trends Plant Sci. 2002;7:106–111. doi: 10.1016/s1360-1385(01)02223-3. [DOI] [PubMed] [Google Scholar]
- 4.Nijhawan A., Jain M., Tyagi A.K., Khurana J.P. Genomic survey and gene expression analysis of the basic leucine zipper transcription factor family in rice. Plant Physiol. 2008;146:333–350. doi: 10.1104/pp.107.112821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selvaraj D., Loganathan A., Ramalingam S. Molecular characterization and phylogenetic analysis of bZIP protein in plants. J. Proteomics Bioinform. 2010;3:230–233. [Google Scholar]
- 6.Hattori T., Totsuka M., Hobo T., Kagaya Y., Yamamoto-Toyoda A. Experimentally determined sequence requirement of ACGT-containing abscisic acid response element. Plant Cell Physiol. 2002;43:136–140. doi: 10.1093/pcp/pcf014. [DOI] [PubMed] [Google Scholar]
- 7.Izawa T., Foster R., Chua N.-H. Plant bZIP protein DNA binding specificity. J. Mol. Biol. 1993;230:1131–1144. doi: 10.1006/jmbi.1993.1230. [DOI] [PubMed] [Google Scholar]
- 8.Foster R., Izawa T., Chua N.-H. Plant bZIP proteins gather at ACGT elements. FASEB J. 1994;8:192–200. doi: 10.1096/fasebj.8.2.8119490. [DOI] [PubMed] [Google Scholar]
- 9.Choi I.H., Hong H.J., Ha O.J., Kang Y.J., Kim Y.S. ABFs, a family of ABA-responsive element binding factors. J. Biol. Chem. 2000;275:1723–1730. doi: 10.1074/jbc.275.3.1723. [DOI] [PubMed] [Google Scholar]
- 10.Ezcurra I., Wycliffe P., Nehlin L., Ellerstrom M., Rask L. Transactivation of the Brassica napus napin promoter by ABI3 requires interaction of the conserved B2 and B3 domains of ABI3 with different cis-elements: B2 mediates activation through an ABRE, whereas B3 interacts with an RY/G box. Plant J. 2000;24:57–66. doi: 10.1046/j.1365-313x.2000.00857.x. [DOI] [PubMed] [Google Scholar]
- 11.Ono A., Izawa T., Chua N.-H., Shimamoto K. The rab16b promoter of rice contains two distinct abscisic acid-responsive elements. Plant Physiol. 1996;112:483–491. doi: 10.1104/pp.112.2.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hobo T., Asada M., Kowyama Y., Hattori T. ACGT-containing abscisic acid response element (ABRE) and coupling element 3 (CE3) are functionally equivalent. Plant J. 1999;19:679–689. doi: 10.1046/j.1365-313x.1999.00565.x. [DOI] [PubMed] [Google Scholar]
- 13.Yoon M.-K., Kim H.-M., Choi G., Lee J.-O., Choi B.-S. Structural basis for the conformational integrity of the Arabidopsis thaliana HY5 leucine zipper homodimer. J. Biol. Chem. 2007;282:12989–13002. doi: 10.1074/jbc.M611465200. [DOI] [PubMed] [Google Scholar]
- 14.Miyazono K., Koura T., Kubota K., Yoshida T., Fujita Y., Yamaguchi-Shinozaki K., Tanokura M. Purification, crystallization and preliminary X-ray analysis of OsAREB8 from rice, a member of the AREB/ABF family of bZIP transcription factors, in complex with its cognate DNA. Acta Crystallogr. F. 2012;68:491–494. doi: 10.1107/S1744309112009384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schumacher M.A., Goodman R.H., Brennan R.G. The structure of a CREB bZIP–somatostatin CRE complex reveals the basis for selective dimerization and divalent cation-enhanced DNA binding. J. Biol. Chem. 2000;275:35242–35247. doi: 10.1074/jbc.M007293200. [DOI] [PubMed] [Google Scholar]
- 16.Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–W388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PDBViewer: an environment for comparative protein modelling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 18.Krieger E., Joo K., Lee J., Lee J., Raman S., Thompson J., Tyka M., Baker D., Karplus K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins. 2009;77(Suppl. 9):114–122. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Dijk M., Van Dijk A.D.J., Hsu V., Boelens R., Bonvin A.M.J.J. Information-driven protein–DNA docking using HADDOCK: it is a matter of flexibility. Nucleic Acids Res. 2006;34:3317–3325. doi: 10.1093/nar/gkl412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benkert P., Künzli M., Schwede T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009;37(Web Server issue):W510–514. doi: 10.1093/nar/gkp322. <http://swissmodel.expasy.org/qmean/cgi/index.cgi> Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK – a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 22.Tina K.G., Bhadra R., Srinivasan N. PIC: protein interactions calculator. Nucleic Acids Res. 2007;35(Web Server issue):W473–W476. doi: 10.1093/nar/gkm423. <http://pic.mbu.iisc.ernet.in/> Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hekkelman M.L., Te Beek T.A., Pettifer S.R., Thorne D., Attwood T.K., Vriend G. WIWS: a protein structure bioinformatics web service collection. Nucleic Acids Res. 2010;38:W719–W723. doi: 10.1093/nar/gkq453. <http://swift.cmbi.ru.nl/servers/html/index.html> Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walshaw J., Woolfson D.N. SOCKET: a program for identifying and analysing coiled-coil motifs within protein structures. J. Mol. Biol. 2001;307:1427–1450. doi: 10.1006/jmbi.2001.4545. [DOI] [PubMed] [Google Scholar]
- 25.Torshini I.Y., Weber I.T., Harrison R.W. Geometric criteria of hydrogen bonds in proteins and identification of bifurcated hydrogen bonds. Protein Eng. Des. Sel. 2002;15:359–363. doi: 10.1093/protein/15.5.359. [DOI] [PubMed] [Google Scholar]
- 26.De Masi F., Grove C.A., Vedenko A., Alibes A., Gisselbrecht S.S., Serrano L., Bulyk M.L., Walhout A.J.M. Using a structural and logics systems approach to infer bHLH–DNA binding specificity determinants. Nucleic Acids Res. 2011;39 doi: 10.1093/nar/gkr070. 4553–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alibes A., Nadra A.D., de Masi F., Bulyk M.L., Serrano L., Stricher F. Using protein design algorithms to understand the molecular basis of disease caused by protein-DNA interactions: the Pax6 example. Nucleic Acids Res. 2010;38:7422–7431. doi: 10.1093/nar/gkq683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deppmann C.D., Acharya A., Rishi V., Wobbes B., Smeekens S., Taparowsky E.J., Vinson C. Dimerization specificity of all 67 B-ZIP motifs in Arabidopsis thaliana: a comparison to Homo sapiens B-ZIP motifs. Nucleic Acids Res. 2004;32:3435–3445. doi: 10.1093/nar/gkh653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crick F.H.C. The packing of alpha-helices: simple coiled-coils. Acta Crystallogr. 1953;6:689–697. [Google Scholar]
- 30.Keller W., Konig P., Richmond J. Crystal structure of a bZIP/DNA complex at 2.2 Å: determinants of DNA specific recognition. J. Mol. Biol. 1995;254:657–667. doi: 10.1006/jmbi.1995.0645. [DOI] [PubMed] [Google Scholar]