

Abstract
Background
Intracellular signaling responsible for gastrin-releasing peptide (GRP) receptor-mediated neovascularization is not clearly understood. We sought to determine the cellular mechanisms involved in the GRP receptor regulation of vascular endothelial growth factor (VEGF) release in neuroblastoma cells.
Materials and Methods
BE(2)-C cells were treated with bombesin (BBS), the amphibian equivalent of GRP, Phorbolmyristate acetate (PMA) a PKC agonist, or GF109293X (GFX), and analyses were performed for VEGF secretion, phosphorylated protein kinase B (AKT), extracellular signal-regulated kinases (ERK) and protein kinase D (PKD) expression.
Results
BBS rapidly increased VEGF secretion at 30 min. Pre-treatment with PMA alone produced similar results; this effect was synergistic with the addition of GRP. Conversely, GFX blocked PMA-stimulated increase in VEGF secretion. Immunofluorescent staining for VEGF was correlated to BBS, PMA and GFX.
Conclusions
PKC is critically responsible for rapid VEGF secretion by GRP receptor signaling in neuroblastoma cells. Inhibition of VEGF significantly reduced GRP-mediated cell proliferation, suggesting its crucial role in neuroblastoma tumorigenesis.
Keywords: PKC, BBS, GRP, VEGF, neuroblastoma
Neuroblastoma is the most common extra-cranial solid tumor of infants and children, and accounts for 15% of all pediatric cancer-related deaths (1–3). Despite recent advances in multimodality treatment protocols for patients with neuroblastoma, the overall five-year survival rate for ‘high-risk’ neuroblastoma remains less than 40%. Angiogenesis has been implicated in aggressive tumor behavior for neuroblastoma and antiangiogenic therapies have been tested in laboratories with variable antitumor effects. Thus, a better understanding of the signaling cascades involved in angiogenesis in more aggressive forms of neuroblastoma is necessary for improving approaches to identify and to treat the ‘high-risk’ category of patients with neuroblastoma.
Gastrin-releasing peptide (GRP) is a known mitogen for various types of cancer, including breast, lung and prostate cancer (4). Previous studies have demonstrated that GRP and its receptor (GRPR) are up-regulated in undifferentiated neuroblastoma, and hence could serve as critical tumor markers and useful intracellular therapeutic targets for tumors that express high levels of GRP/GRPR(5). GRP is both an autocrine and paracrine growth factor stimulating tumor cell growth, metastasis, as well as vascular endothelial cell migration necessary for angiogenesis (5). Additionally, bombesin (BBS), the amphibian analogue of GRP, has been shown to increase expression of vascular endothelial growth factor (VEGF)and to enhance vascular density in a tumor xenograft mouse model (6).
VEGF is a crucial mediator of angiogenesis, and is intimately involved in the recruitment, proliferation, and maintenance of immature blood vessels necessary for tumor growth and metastasis. In addition to its angiogenic role, VEGF is also involved in autocrine signaling in neuroblastoma and other solid tumor types through a variety of intracellular pathways (7,8). Increased VEGF levels correlate with ‘high-risk’ neuroblastoma histologically and clinically, likely secondary to multiple signals including GRP(6,9). Previous studies have reported the effectiveness of anti-VEGF therapies such as bevacizumab, currently used to treat colorectal, breast and lung cancer, in reducing tumor growth and angiogenesis both alone and in combination with other agents (10,11). VEGF is a promising therapeutic target in undifferentiated neuroblastoma. Anti-VEGF treatment is currently in phase II trials against neuroblastoma. Both GRP and VEGF have been established as key inducers that can promote cellular growth, metastasis and angiogenesis in undifferentiated neuroblastoma; however, the exact relationship between these two growth factors is largely unknown. Therefore, the purpose of this study was to determine the mechanism of GRP-induced angiogenesis, particularly through VEGF signaling, and to elucidate the specific pathways involved in induction and regulation of BBS-induced VEGF release in neuroblastoma cells.
Materials and Methods
Reagents
BBS peptide was purchased from Bachem (Torrance, CA, USA). Phorbolmyristate acetate (PMA), a protein kinase C (PKC) agonist, was from Sigma-Aldrich (St. Louis, MO, USA). GF109293X (GFX), a PKC antagonist, was purchased from Biomol Research Laboratories, Inc. (Plymouth Meeting, PA, USA). Anti-human VEGF antibody and VEGF neutralizing antibody were from R&D Systems, Inc. (Minneapolis, MN, USA). MEK inhibitor (U0126), PI3K inhibitor(LY294002), and antibodies for total protein kinase B (AKT), phospho-AKT (ser473), total extracellular signal-regulated kinases (ERK) 1/2, phospho-ERK1/2, phospho-protein kinase D (PKD)were purchased from Cell Signaling Technology(Beverley, MA, USA).
Cell culture
Human neuroblastoma cell lines, SK-N-SH and BE(2)-C, were purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were maintained in RPMI-1640 with L-glutamine (Cellgro Mediatech, Inc., Herndon, VA, USA) supplemented with 10% fetal bovine serum (Sigma-Aldrich). The cells were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2. The cells were seeded onto 6-well plates for 24 h, then serum-starved overnight prior to treatment with BBS, GFX or PMA.
Western blot analysis
Whole-cell lysates were collected using Cell Lysis Buffer (Cell Signaling) supplemented with fresh phenylmethylsulfonyl fluoride (PMSF) at 1 mmol/l. Protein samples (20–30 μg) were run on a Tris-Bis gel (Life Tech., Grand Island, NY, USA) and transferred onto polyvinylidenedifluoride membranes. Membranes were blocked with 5% milk in TBST (120 mmol/l Tris–HCl, pH 7.4, 150 mmol/l NaCl, and 0.05% Tween 20) for 1 h at room temperature. Proteins were detected by incubating with rabbit or mouse anti-human antibodies overnight at 4°C or three h at room temperature. Membranes were then washed three times with TBST and incubated for 1 h with secondary antibody conjugated to horseradish peroxidase (HRP). Blots were developed using Western Lightning Plus-ECL substrate (Perkin Elmer, Inc., Norwalk, CT, USA). β-Actin served as a loading control.
RT-PCR
Total RNA was isolated using RNAqueous™ kit (Ambion, Austin, TX, USA) according to the manufacturer’s instructions. Isolated RNA was used to synthesize cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Austin, TX, USA). Primers designed to amplify human VEGF fragment (NM_003376) were: forward primer 5′-AGGAGGAGGGCAGAATCATCAC-3′; reverse primer 5′-ATGTCCACCAGGGTCTCGATTG-3′. Glyceraldehyde 3-phosphate dehydrogenase-specific oligonucleotide primers used were previously published(12). The level of VEGF mRNA was measure by quantitative real-time PCR using SsoFast™ EvaGreen Supermix with CFX96 Real-Time PCR System (Bio-Rad, Hercules, CA, USA).
Cell viability assay
Cells were seeded onto 96-well plates at a density of 1×104 cells per well of 96-well plate in RPMI culture medium with 10% FBS and the cell number was assessed using Cell Counting Kit-8 (Dojindo Molecular Technologies, Rockville, MD, USA) for cell viability. Cells were treated with BBS or BBS plus anti-human VEGF-neutralizing antibody, and cell viability was assessed in comparison to the control (untreated cells).
Immunofluorescent staining
BE(2)-C cells were plated on glass coverslips, incubated overnight and serum-starved for 24 h. Cells were treated with GRP (100 nM) for 30 min. The treated cells were washed once with PBS and fixed in 4% paraformaldehyde in PBS for 15 min at room temperature, then washed with PBS three times. Samples blocked in 1% bovine serum albumin(BSA)/PBS buffer for 30 min, and incubated for 60 min in 1:100 dilution of anti-VEGF antibody. The cells were washed three times with PBS and incubated with 1:500 secondary antibody Alexa Fluor 568 goat anti-rabbit (Life Tech.), then stained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min. The cells were rinsed three times with PBS, and the coverslips were mounted onto the slide glasses. The mounted cells were viewed with a fluorescence microscope using a X40 objective lens.
ELISA
The supernatant of cultured cells was collected at 0.5, 1, 2, 8 and 24 h time points, and subsequently frozen at −70° C. For assay, samples were thawed and centrifuged, and then VEGF levels were measured using a human VEGF ELISA kit according to the manufacturer’s instructions (R&D Systems, Inc.).
Statistical analysis
For in vitro experiments, conditions were compared using Student’s paired t test. One-way analysis of variance (ANOVA) was performed for multiple comparisons. For all experiments, p<0.05 was considered significant.
Results
BBS induced rapid VEGF secretion
We sought to determine the specific molecular signaling pathway responsible for BBS-induced VEGF secretion. BBS treatment for 30 min resulted in a significant increase in VEGF secretion when compared to control BE(2)-C cells (Figure 1A). An increase in VEGF secretion into the medium was noted as early as only 30 min after BBS stimulation; this increase persisted throughout the time course. In addition, increases in intracellular levels of VEGF were visualized by immunofluorescence in the cytoplasm of BE(2)-C cells (Figure 1B), suggesting a rapid post-transcriptional mobilization of VEGF in response to BBS stimulation. Moreover, we did not observe increases in VEGF mRNA at any of the time points after BBS treatment (Figure 1C), further supporting the notion that BBS-induced early VEGF release is not due to regulation of VEGF gene transcription. Taken together, these results indicate that BBS induces rapid VEGF secretion through a post-transcriptional mechanism in neuroblastoma cells. Similar results were observed in another human neuroblastoma cell line, SK-N-SH (data not shown).
Figure 1. Bombesin (BBS)-induced rapidvascular endothelial growth factor secretion (VEGF).
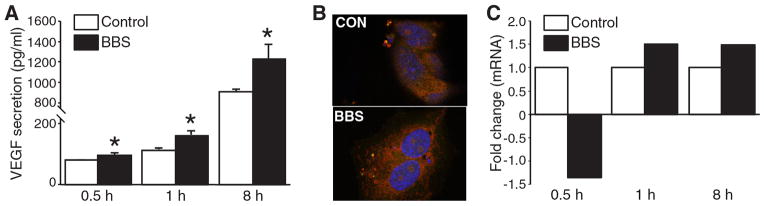
A: BE(2)-C cells were seeded onto 6-well plates for 24 h, then serum-starved overnight before treatment with BBSat100 nM. Supernatant was collected over time and VEGF enzyme-linked immunosorbent assay (ELISA) was performed (mean ±SEM of n=3; *p<0.05 vs. untreated control). B: A representative image viewed with a fluorescence microscope using a X40 objective demonstrating VEGF protein expression at 30 min after BBS (100 nM). C: After cells were treated with BBS (100 nM), quantitative real-time PCR was performed to determine VEGF mRNA levels. There was no significant difference after BBS treatment.
PKC-dependent rapid VEGF secretion
BBS has been reported to stimulate intracellular signaling pathways that can act through the activation of PKC(13). Thus, we hypothesized that PKC may play a role in the rapid secretion of VEGF after BBS stimulation. To examine this, we next treated BE(2)-C cells with PMA, a PKC agonist, and found that both BBS and PMA alone produced a rapid VEGF secretion (Figure 2A). Interestingly, they both induced similar amounts of VEGF secretion after stimulation. Moreover, when BBS and PMA were used together in combination, there was a synergistic increase in VEGF secretion. Importantly, we next determined that BBS induction of VEGF secretion was attenuated by treatment with GFX, a PKC inhibitor. Since a number of other signaling pathways have been implicated in the regulation of angiogenesis, we next assessed the effects of BBS, PMA, and GFX on activation of PI3K/AKT, ERK1/2 and PKD pathways, respectively. As expected, BBS and PMA increased levels of p-AKT and p-ERK1/2, respectively (Figure 2B). However, GFX in combination with BBS attenuated the expression of p-PKD, a downstream effector of PKC, without changing the p-AKT level (Figure 2B), further indicating that inhibitory effects of GFX on rapid VEGF secretion is independent of the phosphatidylinositol 3-kinase (PI3K)/AKT pathway. Following treatment with BBS, BBS with PMA, or BBS with GFX, BE(2)-C cells were observed using indirect immunofluorescence. We found that the addition of GFX resulted in altered cytoplasmic distribution of VEGF when compared with cells treated with BBS, or BBS with PMA alone (Figure 2C), suggesting a potential role for PKC in VEGF mRNA stabilization and/or protein production.
Figure 2. Protein kinase C (PKC)-dependent rapid vascular endothelial growth factor (VEGF) secretion.

A: BE(2)-C cells were serum-starved overnight before treatment with bombesin (BBS; 100 nM), PKC agonist phorbolmyristate acetate(PMA; 10 nM) and/or PKC antagonist GF109293X (GFX; 20 nM). Supernatant was collected 30 min later, and VEGF enzyme-linked immunosorbent assay (ELISA) performed. (mean±SEM of n=3; *p<0.05 vs. no treatment;**p<0.05 vs. BBS alone). B: BBS increased phosphorylated protein kinase B (p-AKT) (ser473) expression, while PMA increased phosphorylated extracellular signal-regulated kinases (p-ERK1/2) levels. GFX attenuated phosphorylated protein kinase D (p-PKD) expression without altering p-AKT or p-ERK by western blotting. C: A representative image viewed with a fluorescence microscope using a ×40 objective demonstrating cytoplasmic VEGF localization with30 min treatment with BBS or PMA when compared to BBS + GFX.4′,6-diamidino-2-phenylindole (DAPI) indicates nuclei staining.
BBS-induced rapid VEGF secretion is independent of PI3K/AKT and ERK pathways
We hypothesized that BBS-mediated VEGF secretion is regulated by a mechanism independent of the PI3K/AKT signaling pathway. The PI3K/AKT has been previously shown to be a key mediator of VEGF secretion that typically occurs 24 to 48 h after stimulation (14). However, in this study, we found that PI3K inhibition using LY294002 did not alter BBS-induced VEGF secretion at 30 min (Figure 3A). Additionally, we evaluated the role of the ERK pathway on BBS-induced VEGF secretion, as MEK/ERK pathways are well-described downstream cell signaling pathways of PKC, and are known to promote cell proliferation(15). Surprisingly, MEK inhibition using U0126 also failed to alter BBS-induced VEGF secretion in human neuroblastoma cells. We subsequently confirmed the specificity of LY294002 and U0126 compounds by demonstrating decreases in p-AKT and p-ERK1/2 levels without affecting the expressions of total AKT and ERK1/2 (Figure 3B).
Figure 3. Phosphatidylinositol 3-kinase (PI3K)/phosphorylated protein kinase B(p-AKT) and extracellular signal-regulated kinase (ERK)-independent rapid vascular endothelial growth factor (VEGF) secretion.
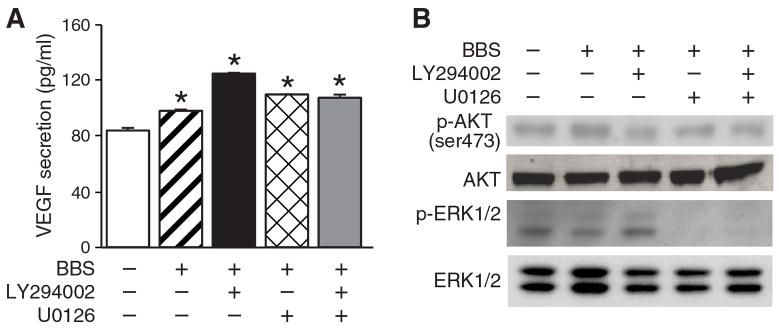
A: BE(2)-C cells were serum-starved overnight before treatment with bombesin (BBS;100 nM), LY294002 (20 μM) or U0126 (10 μM). Supernatant was collected 30 min later and VEGF enzyme-linked immunosorbent assay (ELISA) performed (mean ±SEM of n=3; *p<0.05 vs. control). B: BBS treatment increased p-AKT (ser473); this was reversed with the use of LY294002. U0126 reduced p-ERK1/2 expression by western blotting.
EGF antibody abrogated BBS-induced cell growth
GRP acts as a mitogen to stimulate cell proliferation in a variety of cancers, including neuroblastoma(5). In order to better understand the exact role of the BBS-PKC-VEGF pathway inneuroblastoma cell proliferation, we determined BE(2)-C cell proliferation over 24 h after BBS stimulation with or without anti-VEGF neutralizing antibody. As expected, BBS treatment alone significantly increased BE(2)-C cell proliferation at 24 h after treatment when compared to untreated cells. However, this growth-stimulatory effect of BBS was abolished in the presence of VEGF antibody (Figure 4). Thus, the proliferative effects of GRP appear to be intimately linked to the ability of GRP to stimulate VEGF secretion.
Figure 4. Vascular endothelial growth factor (VEGF) antibody inhibition of bombesin (BBS)-induced cell growth.
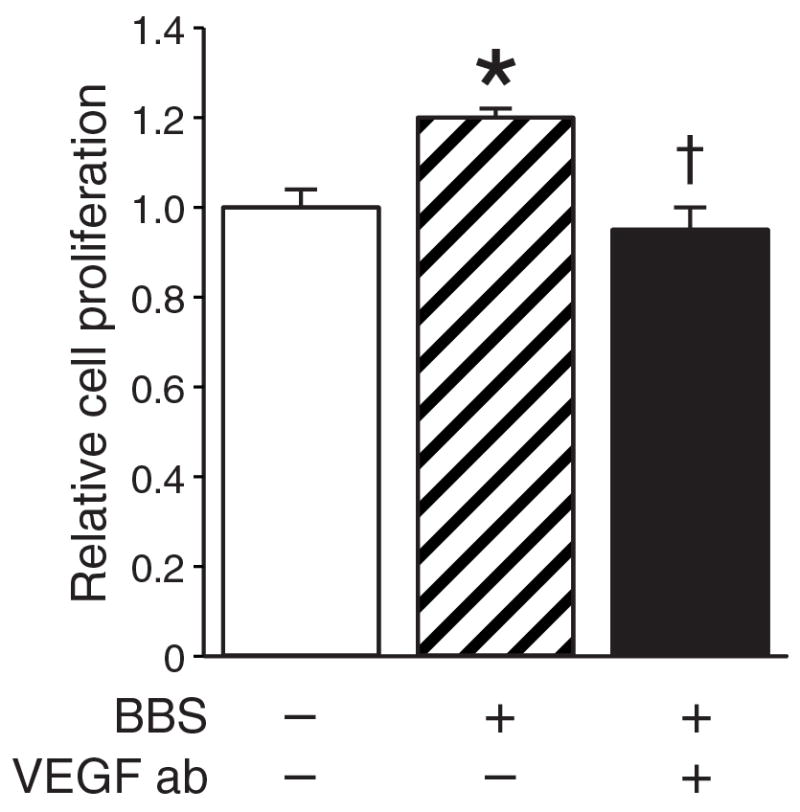
BE(2)-C cells were seeded onto 96-well plates(1 × 104 cells/well) and treated with BBS or BBS plus anti-VEGF neutralizing antibody (2 ng/ml). Cell viability was assessed using Cell Counting Kit-8 in comparison to control cells (mean ±SEM of n=3; *p<0.05 vs. control; †p<0.05 vs. BBS).
Discussion
Cellular signaling pathways are critical to tumorigenesis. Pro-angiogenic pathways are of particular interest, as they are universally required for cell growth and metastasis of all tumors. In this study, we showed that BBS stimulates rapid VEGF secretion in vitro through a PKC-dependent signaling mechanism. Inhibition of PKC pathway using GFX blocked BBS-induced VEGF secretion. However, common downstream targets of PKC, such as MEK/ERK pathways were not activated by BBS treatment, suggesting a novel alternate pathway of BBS-mediated rapid VEGF secretion in human neuroblastoma cells. Moreover, BBS-induced BE(2)-C cell proliferation was blocked in the presence of anti-VEGF neutralizing antibody. Thus, there is a critical relationship that exists between VEGF secretion and BBS-induced tumorigenesis in neuroblastoma.
PKC family member isoforms are classically divided based upon methods of activation. Conventional isoforms (α, βI, βII, γ) are activated by diacylglycerol (DAG) and Ca2+, novel isoforms (ε, η, θ) are activated by DAG alone, and atypical isoforms (ζ, λ, τ) are regulated neither by DAG nor Ca2+(16). Activation of GRPR leads to GRPR and Gq protein dissociation and subsequent stimulation of phospholipase C (PLC) (17). PLC, in turn, catalyzes the creation of two proteins, inositol 1,4,5-triphosphate (IP3) and DAG from their precursor, phosphatidylinositol 4,5-bisphosphate (PIP2). IP3 binds to the endoplasmic reticulum and triggers the rapid release of intracellular Ca2+ stores, which activates PKC and triggers DNA synthesis and mitogenesis. DAG also directly activates a variety of PKC isoforms that are involved in the regulation of cell proliferation. While PKC has been intensively studied, the specific mechanisms by which it interacts with downstream targets remain unclear. One PKC-mediated effect that has been well established is the activation of the MEK/ERK pathway, which is involved in cell proliferation and transformation. Experiments using a MEK inhibitor, U0126, suggested that downstream of this pathway is not involved in rapid VEGF secretion, and thus, PKC utilizes an alternate mechanism of inducing rapid VEGF secretion in human neuroblastoma cells.
One potential mechanism of rapid VEGF secretion may involve post-transcriptional mRNA stabilization. Shih and colleagues reported that PMA increases the half-life of VEGF mRNA in glioblastoma cells (18). They reported that PKC-α and PKC-δ reached maximum activity at 15 and 30 min, respectively, following PMA treatment, while PKC-ζ slowly increased, reaching its peak at 4 h. Anti-sense oligonucleotides targeting PKC-α and PKC-ζ specifically reduced PMA-induced VEGF mRNA in a dose-dependent manner. Our study demonstrated increases in VEGF secretion at 30 min in response to BBS and to PMA stimulation, an effect that is in turn attenuated significantly by GFX treatment. The early timing of rapid secretion observed makes PKC-α and PKC-δ likely candidates for early PKC-mediated VEGF secretion. However, it is beyond the scope of this study to discern which specific isoform is responsible for this mechanism. While our study demonstrated no detectable change in VEGF mRNA levels at 30 min, Shih et al.(18)also did not observe appreciable differences in mRNA levels until one hour post-induction. This further correlates with our own findings of the response to BBS treatment in our study.
Both transcriptional and post-transcriptional pathways of VEGF up-regulation occur in cells placed under hypoxic stress, but this dual mechanism of VEGF expression has yet to be described in neuroblastoma in response to BBS/GRP treatment. Our previous work demonstrated the role of BBS in VEGF transcription through its transcription factor, hypoxia-inducible factor-1α and the PI3K/AKT pathway, yet its relationship to PKC and early post-transcriptional VEGF secretion has yet to be described. As shown here, VEGF plays a major role in BBS-induced cell proliferation and thus, may be involved in the development and metastasis of high-risk neuroblastoma. While we have identified the involvement of PKC signaling pathway in early VEGF secretion, most likely acting through post-transcriptional modification, confirmation of this mechanism and identification of the particular PKC isoforms involved are still needed to form a complete picture of this significant signaling cascade. The identification of this early PKC-mediated pathway will allow for additional investigation into downstream regulators of VEGF secretion, and may ultimately provide insight into novel adjuvant therapies for advanced stage neuroblastoma.
Acknowledgments
We thank Karen Martin for assistance with the manuscript preparation. This work was supported by grants R01 DK61470 from the National Institutes of Health and Rally Foundation for Cancer Research.
References
- 1.Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Onc. 1999;17:2264–2279. doi: 10.1200/JCO.1999.17.7.2264. [DOI] [PubMed] [Google Scholar]
- 2.Brodeur GM. Neuroblastoma: Biological insights into a clinical enigma. Nat Rev Cancer. 2003;3:203–216. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- 3.Schwab M, Westermann F, Hero B, Berthold F. Neuroblastoma: biology and molecular and chromosomal pathology. Lancet Oncol. 2003;4:472–480. doi: 10.1016/s1470-2045(03)01166-5. [DOI] [PubMed] [Google Scholar]
- 4.Patel O, Shulkes A, Baldwin GS. Gastrin-releasing peptide and cancer. Biochim Biophys Acta. 2006;1766:23–41. doi: 10.1016/j.bbcan.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Kim S, Hu W, Kelly DR, Hellmich MR, Evers BM, Chung DH. Gastrin-releasing peptide is a growth factor for human neuroblastomas. Ann Surg. 2002;235:621–629. doi: 10.1097/00000658-200205000-00003. discussion 629–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang J, Ishola TA, Baregamian N, Mourot JM, Rychahou PG, Evers BM, Chung DH. Bombesin induces angiogenesis and neuroblastoma growth. Cancer Lett. 2007;253:273–281. doi: 10.1016/j.canlet.2007.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gee MF, Tsuchida R, Eichler-Jonsson C, Das B, Baruchel S, Malkin D. Vascular endothelial growth factor acts in an autocrine manner in rhabdomyosarcoma cell lines and can be inhibited with all-trans-retinoic acid. Oncogene. 2005;24:8025–8037. doi: 10.1038/sj.onc.1208939. [DOI] [PubMed] [Google Scholar]
- 8.Sims TL, Williams RF, Ng CY, Rosati SF, Spence Y, Davidoff AM. Bevacizumab suppresses neuroblastoma progression in the setting of minimal disease. Surgery. 2008;144:269–275. doi: 10.1016/j.surg.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukuzawa M, Sugiura H, Koshinaga T, Ikeda T, Hagiwara N, Sawada T. Expression of vascular endothelial growth factor and its receptor FLK-1 in human neuroblastoma using insitu hybridization. J Pediatr Surg. 2002;37:1747–1750. doi: 10.1053/jpsu.2002.36712. [DOI] [PubMed] [Google Scholar]
- 10.Dickson PV, Hamner JB, Sims TL, Fraga CH, Ng CY, Rajasekeran S, Hagedorn NL, McCarville MB, Stewart CF, Davidoff AM. Bevacizumab-induced transient remodeling of the vasculature in neuroblastoma xenografts results in improved delivery and efficacy of systemically administered chemotherapy. Cin Cancer Res. 2007;13:3942–3950. doi: 10.1158/1078-0432.CCR-07-0278. [DOI] [PubMed] [Google Scholar]
- 11.Rowe DH, Huang J, Li J, Manley C, O’Toole KM, Stolar CJ, Yamashiro DJ, Kandel JJ. Suppression of primary tumor growth in a mouse model of human neuroblastoma. J Pediatr Surg. 2000;35:977–981. doi: 10.1053/jpsu.2000.6946. [DOI] [PubMed] [Google Scholar]
- 12.Chatzistamou I, Schally AV, Sun B, Armatis P, Szepeshazi K. Inhibition of growth of OV-1063 human epithelial ovarian cancers and c-Jun and c-Fos oncogene expression by bombesin antagonists. Brit J Cancer. 2000;83:906–913. doi: 10.1054/bjoc.2000.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hellmich MR, Ives KL, Udupi V, Soloff MS, Greeley GH, Jr, Christensen BN, Townsend CM., Jr Multiple protein kinase pathways are involved in gastrin-releasing peptide receptor-regulated secretion. J Biol Chem. 1999;274:23901–23909. doi: 10.1074/jbc.274.34.23901. [DOI] [PubMed] [Google Scholar]
- 14.Kang J, Rychahou PG, Ishola TA, Mourot JM, Evers BM, Chung DH. N-MYC is a novel regulator of PI3K-mediated VEGF expression in neuroblastoma. Oncogene. 2008;27:3999–4007. doi: 10.1038/onc.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ueda Y, Hirai S, Osada S, Suzuki A, Mizuno K, Ohno S. Protein kinase C activates the MEK-ERK pathway in a manner independent of RAS and dependent on RAF. J Biol Chem. 1996;271:23512–23519. doi: 10.1074/jbc.271.38.23512. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Qu Y, Zu P, Han S, Gao G, Xu Q, Fang L. Increased isoform-specific membrane translocation of conventional and novel protein kinase C in human neuroblastoma SH-SY5Y cells following prolonged hypoxia. Brain Res. 2006;1093:25–32. doi: 10.1016/j.brainres.2006.03.110. [DOI] [PubMed] [Google Scholar]
- 17.Rozengurt E. Signal transduction pathways in the mitogenic response to G protein-coupled neuropeptide receptor agonists. J Cell Physiol. 1998;177:507–517. doi: 10.1002/(SICI)1097-4652(199812)177:4<507::AID-JCP2>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 18.Shih SC, Mullen A, Abrams K, Mukhopadhyay D, Claffey KP. Role of protein kinase C isoforms in phorbol ester-induced vascular endothelial growth factor expression in human glioblastoma cells. J Biol Chem. 1999;274:15407–15414. doi: 10.1074/jbc.274.22.15407. [DOI] [PubMed] [Google Scholar]