

Abstract
Gene activity is regulated by transcriptional and epigenetic mechanisms. A paper in 2005 by Zhang et al.1 showed that STAT3 binds to the DNA methyl transferase, DNMT1 and their data indicated that STAT3 may cause epigenetic gene silencing by targeting DNMT1 to the PTPN6 promoter. Now, a paper by Lee et al.2 has fleshed out the mechanism. They provide evidence that acetylation of STAT3 regulates the binding of DNMT1, CpG DNA methylation and regulation of several genes, including that encoding the estrogen receptor α (ESR1) in breast cancer cells.
Keywords: STAT3, DNMT1, acetylation, DNA methylation, epigenetics, transcription, cancer
The methylation of mammalian DNA in somatic cells occurs predominantly at cytosines in CpG motifs.3 The mammalian genome, statistically CpG-deficient, contains CpG “islands” (CGI), which are CpG-rich sequences some 1,000 base pairs in length.4 The majority (70%) of protein-encoding genes are associated with such islands5 and half of CGIs contain transcriptional start sites.6 A class of orphan CGIs are also located in intragenic regions of the genome and about 40% may also have roles in transcription from alternative promoters.6,7
Only 3% of CGIs at gene promoters are methylated, whereas up to 34% of intragenic CGIs are methylated.6,7 Methylation of CpGs in CGIs is associated with transcriptional silencing (for a review, see ref. 8), although this does not occur in all cases.9 CGI methylation at specific gene loci occurs normally during embryonic development10 and aberrantly in cancers (for a review, see ref. 11). That DNA methylation at specific CpGs can initiate transcriptional silencing of the neighboring gene was shown by targeting a DNA methyl transferase (DNMT) to a specific site in the genome, containing a reporter construct. Such targeted methylation caused subsequent histone modifications and heritable silencing of the reporter gene.12 However, there is also evidence that DNA methylation follows chromatin modifications in X-chromosome inactivation and other cases and may act to cement an epigeneticically silenced state (for a review, see refs. 13 and 14).
Only some CpGs in the genome become methylated and it is likely that this is controlled in part by a balance between de-methylation, initiated by TET-proteins15 and methylation, mediated by DNMTs. DNMT1 is the methylase that maintains CpG methylation during DNA replication, whereas de novo methylation is mediated by DNMT3a, DNMT3b and their partner, DNMT3L (for a review, see ref. 16). Also pertinent is that CGIs near active promoters tend to be hypo-methylated and correlate with transcription factor binding. A seminal study showed that Sp1 sites in the mouse Aprt promoter (but not Sp1 itself) are required to maintain an unmethylated state,17,18 which could involve other factors such as VEZF1.19 Similarly, binding YY1 and Nrf1 protect CGIs from methylation20 and the binding of RNA polymerase II predicts the DNA methylation state.21 Therefore, active transcription may serve to prevent CGI methylation.
The signal transduction and activation of transcription, STAT proteins were discovered by the Stark, Kerr and Darnell laboratories in the early 1990s (for a historical perspective, see ref. 22). By 1995 all six STAT proteins had been identified, including STAT3.23-25 Soon after, STAT3 activation was shown to be regulated by tyrosine and serine phosphorylation26 and we now know that this protein is also post-translationally modified by di-methylation at K14027 and acetylation at K68528 and other sites (for a review of STAT acetylation, see ref. 29). In contrast to other STAT proteins, STAT3 is required for early development and STAT3−/− mice die between E6.5 and E7.5.30 Analyses of conditional STAT3 knockouts showed that STAT3 regulates a number of important processes, including mammary gland involution, IL-6-driven T cell proliferation, apoptosis and cell motility (for a review, see ref. 31). In humans, inborn errors in STAT3 are found in a form of hyper-IgE syndrome (HIES) and certain T cell subsets are affected in these patients.32 STAT3 is activated by a number of different stimuli, including type I and II interferons, IL-10, IL-12, IL-23 and receptor tyrosine kinases (for a review, see refs. 22, 32 and 33). Each of these stimuli results in regulation of gene expression programs and the specificity of the transcriptional programs activated is likely to be due to DNA binding of other transcriptional regulators, including STAT5,34 the interaction of STAT3 with other proteins and the epigenetic state of the chromatin. For example, STAT3 binds to the NFκB proteins RelA/p65 and p50 and STAT3 and NFκB bind some promoters in common (for a review, see ref. 33). STAT3 can also partner with other transcriptional regulators, such as c-Jun, C/EBP, β-catenin and also STAT1 and STAT6 (for a review, see ref. 35). STAT3 also binds to DNMT1 and this may cause epigenetic gene silencing by targeting DNMT1 to promoters, such as that of the PTPN6 gene (encodes the phosphatase, SHP-1).1
The article by Lee et al.2 in the Proceedings of the National Academy of Sciences, “Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation,” investigated the mechanism by which STAT3 binds DNMT1 and the consequences of such binding for CGI methylation. The authors first noticed that STAT3 K685 acetylation occurs in tissues from patients with melanoma, colon cancer and triple-negative breast cancer (TNBC), but less so in normal tissues. To investigate the role played by acetylation at this site, they expressed a STAT3 K685R mutant in the melanoma cell line, A2058. Tumors resulting from injection of the STAT3 (K685R) cells into a mouse xenograft model grew more slowly and the expression of two tumor suppressor genes suppressed by STAT3, TP53 (encodes p53) and PTPN6 were increased. In MEFs, expression of the same STAT3 mutant increased the expression of PTPN6 and CDKN2A (encodes the Cdk4/6 inhibitor, p16INK4A). They then used methylation-dependent immunoprecipitation (MeDIP) and promoter-specific PCR to determine whether CGI methylation was altered in the promoters of these genes, as well as in SOCS3. The data indicate that meC at these promoters is reduced when the STAT3 acetylation mutant is expressed. To investigate this further, the authors mutated the endogenous STAT3 gene to express STAT3 K685R in the colon cancer cell line, HCT116. Several genes known to be hypermethylated and silenced in this cell line were re-expressed. These included CDKN2A, DLEC1 and STAT1 and cytosine methylation of their promoters was reduced. Supplementary data shows that the epigenetic changes caused by the mutated STAT3 are not due to a secondary effect on phosphorylation at Y705. Since STAT3 had been shown to bind DNMT1 and recruit it to the PTPN6 promoter,1 the authors used a chromatin immunoprecipitation (ChIP) assay to determine whether DNMT1 and STAT3 also bind to the CDKN2A, STAT1 and DLEC1 promoters. The data shows that both proteins are recruited to these promoters, but not when K685 is mutated. Therefore, this study shows that STAT3 and DNMT1 bind other promoters containing STAT-binding sites, in addition to that of PTPN6.
To determine whether acetylation affects the binding of STAT3 to DNMT1, STAT3 or STAT3 K685R were overexpressed and co-immunoprecipitation experiments failed to detect the mutant in a DNMT1 complex. Expression of the p300 acetyltransferase increases STAT3 acetylation at K685 and its interaction with DNMT1. The histone deacetylase (HDAC) inhibitor, trichostatin has a similar effect. DNMT1 also co-immunoprecipitates with STAT3 from extracts of A2058 melanoma xenograft tumors, but not when the K685R mutant is expressed. Resveratol (found in red wine36) is a HDAC activator known to inhibit STAT3 acetylation and treating these mice with the drug inhibits the growth of the tumors, ablates the STAT3-DNMT1 interaction and reduces the methylation of the PTPN6, CDKN2A and SOCS3 promoters. These data are consistent with the model whereby acetylation of STAT3 enables it to interact with DNMT1 and targets it to methylate gene promoters (Fig. 1).
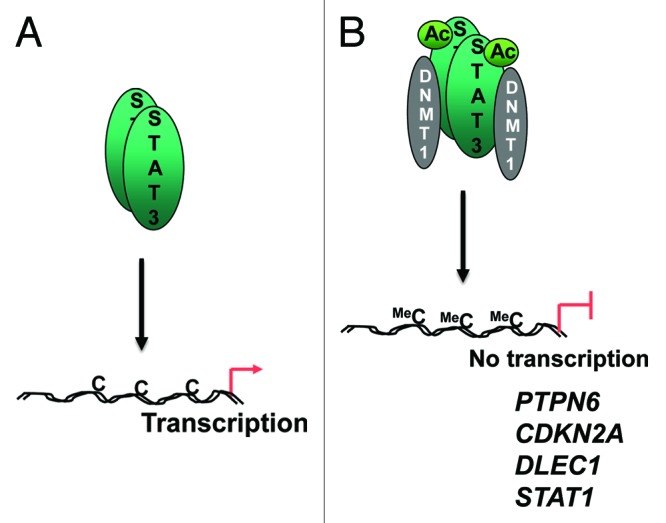
Figure 1. Interaction of STAT3 with DNMT1. (A) STAT3 binds specific gene promoters and activates transcription. (B) Acetylation of STAT3 at K685 enables it to interact with DNMT1 and targets DNMT1 to methylate CpGs in certain gene promoters. Note that the schema is drawn as a dimeric STAT3-DNMT1 complex, although the stoichiometry was not investigated in the study. Other post-translational modifications, such as phosphorylation have also been omitted.
The remaining experiments in the study investigate whether resveratol can be used to re-activate the ESR1 (ERα) gene in TNBC and melanoma cells. ERα expression is re-activated in TNBC and melanoma cell lines treated with resveratol and this correlates with a decrease in ESR1 promoter methylation. Furthermore, resveratol plus tamoxifen significantly reduces tumor formation by the TNBC cell line in the mouse xenograft model.
This is a tantalizing study, which brings out the role of STAT3 K685 acetylation in mediating its interaction with DNMT1 and the potential role of STAT3-directed DNA methylation causing an increase in promoter methylation and transcriptional repression of specific genes. STAT3 can undergo a number of different post-translational modifications, potentially creating many forms of the protein with different activities and able to interact with other proteins. It would be of interest to determine to what extent the mechanisms reported in this paper are dependent on tyrosine or serine phosphorylation, methylation or acetylation of STAT3 at other sites. Also, whether the association of acetylated STAT3 with DNMT1 requires other accessory factors. It should be noted that STAT3 has also been shown to activate DNMT1 transcription in lymphoma cells.37 The balance between DNMT1 promoter activation and DNMT1 binding by STAT3 and the precise regulation of STAT3 to control both these processes require further study.
The study uses MeDIP-PCR analyses to investigate the methylation state of specific CGIs and further, more detailed bisulfite sequencing data of at least the same CGIs are now required to ascertain which CpGs are demethylated as a consequence of expressing STAT3 K685R and the spread of demethylation in successive cell cycles. Also, we need to know where STAT3 and DNMT1 co-localize in the genome. ChIP-seq experiments would determine whether they co-localize at all or only a sub-set of STAT3-binding sites. It would also be intriguing to determine whether some of the intragenic and intronic CGIs are potentially targeted by this mechanism, in addition to known 5′ gene promoters. The studies were performed with cell lines and further studies are needed to investigate whether these mechanisms also occur in primary cells, in which cellular mechanisms are normal.
In the context of cancer, the authors show that acetylated STAT3 is highly expressed in samples from melanoma patients who do not express ERα. However, much more extensive studies now need to be done on a larger number of samples from TNBC as well as melanoma patients to determine whether the correlation holds true in all cases. Resveratol is in early clinical trials,38 which have yet to evaluate its efficacy as a single agent. Combination therapies are becoming increasingly important and the efficacy of a resveratol and tamoxifen combination clearly requires further validation.
Overall, is the study by Zhang et al. definitive? No, but it sheds light onto a path worth following.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/22436
References
- 1.Zhang Q, Wang HY, Marzec M, Raghunath PN, Nagasawa T, Wasik MA. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci U S A. 2005;102:6948–53. doi: 10.1073/pnas.0501959102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee H, Zhang P, Herrmann A, Yang C, Xin H, Wang Z, et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc Natl Acad Sci U S A. 2012;109:7765–9. doi: 10.1073/pnas.1205132109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bird AP. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980;8:1499–504. doi: 10.1093/nar/8.7.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell. 1985;40:91–9. doi: 10.1016/0092-8674(85)90312-5. [DOI] [PubMed] [Google Scholar]
- 5.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 2006;103:1412–7. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr AR, James KD, Turner DJ, et al. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 2010;6:e1001134. doi: 10.1371/journal.pgen.1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–7. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 9.Smith AE, Chronis C, Christodoulakis M, Orr SJ, Lea NC, Twine NA, et al. Epigenetics of human T cells during the G0-->G1 transition. Genome Res. 2009;19:1325–37. doi: 10.1101/gr.085530.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bogdanovicá O, Veenstra GJ. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma. 2009;118:549–65. doi: 10.1007/s00412-009-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith AE, Hurd PJ, Bannister AJ, Kouzarides T, Ford KG. Heritable gene repression through the action of a directed DNA methyltransferase at a chromosomal locus. J Biol Chem. 2008;283:9878–85. doi: 10.1074/jbc.M710393200. [DOI] [PubMed] [Google Scholar]
- 13.Payer B, Lee JT. X chromosome dosage compensation: how mammals keep the balance. Annu Rev Genet. 2008;42:733–72. doi: 10.1146/annurev.genet.42.110807.091711. [DOI] [PubMed] [Google Scholar]
- 14.Okamoto I, Heard E. Lessons from comparative analysis of X-chromosome inactivation in mammals. Chromosome Res. 2009;17:659–69. doi: 10.1007/s10577-009-9057-7. [DOI] [PubMed] [Google Scholar]
- 15.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinney SR, Pradhan S. Regulation of expression and activity of DNA (cytosine-5) methyltransferases in mammalian cells. Prog Mol Biol Transl Sci. 2011;101:311–33. doi: 10.1016/B978-0-12-387685-0.00009-3. [DOI] [PubMed] [Google Scholar]
- 17.Brandeis M, Frank D, Keshet I, Siegfried Z, Mendelsohn M, Nemes A, et al. Sp1 elements protect a CpG island from de novo methylation. Nature. 1994;371:435–8. doi: 10.1038/371435a0. [DOI] [PubMed] [Google Scholar]
- 18.Macleod D, Charlton J, Mullins J, Bird AP. Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev. 1994;8:2282–92. doi: 10.1101/gad.8.19.2282. [DOI] [PubMed] [Google Scholar]
- 19.Dickson J, Gowher H, Strogantsev R, Gaszner M, Hair A, Felsenfeld G, et al. VEZF1 elements mediate protection from DNA methylation. PLoS Genet. 2010;6:e1000804. doi: 10.1371/journal.pgen.1000804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gebhard C, Benner C, Ehrich M, Schwarzfischer L, Schilling E, Klug M, et al. General transcription factor binding at CpG islands in normal cells correlates with resistance to de novo DNA methylation in cancer cells. Cancer Res. 2010;70:1398–407. doi: 10.1158/0008-5472.CAN-09-3406. [DOI] [PubMed] [Google Scholar]
- 21.Takeshima H, Yamashita S, Shimazu T, Niwa T, Ushijima T. The presence of RNA polymerase II, active or stalled, predicts epigenetic fate of promoter CpG islands. Genome Res. 2009;19:1974–82. doi: 10.1101/gr.093310.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stark GR, Darnell JE., Jr. The JAK-STAT pathway at twenty. Immunity. 2012;36:503–14. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, et al. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994;77:63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 24.Zhong Z, Wen Z, Darnell JE., Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264:95–8. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- 25.Zhong Z, Wen Z, Darnell JE., Jr. Stat3 and Stat4: members of the family of signal transducers and activators of transcription. Proc Natl Acad Sci U S A. 1994;91:4806–10. doi: 10.1073/pnas.91.11.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wen Z, Zhong Z, Darnell JE., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–50. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 27.Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B, et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc Natl Acad Sci U S A. 2010;107:21499–504. doi: 10.1073/pnas.1016147107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Shea JJ, Kanno Y, Chen X, Levy DE. Cell signaling. Stat acetylation--a key facet of cytokine signaling? Science. 2005;307:217–8. doi: 10.1126/science.1108164. [DOI] [PubMed] [Google Scholar]
- 29.Wieczorek M, Ginter T, Brand P, Heinzel T, Krämer OH. Acetylation modulates the STAT signaling code. Cytokine Growth Factor Rev. 2012;23:293–305. doi: 10.1016/j.cytogfr.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A. 1997;94:3801–4. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akira S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene. 2000;19:2607–11. doi: 10.1038/sj.onc.1203478. [DOI] [PubMed] [Google Scholar]
- 32.Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity. 2012;36:515–28. doi: 10.1016/j.immuni.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21:11–9. doi: 10.1016/j.cytogfr.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–54. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levy DE, Darnell JE., Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 36.Kopp P. Resveratrol, a phytoestrogen found in red wine. A possible explanation for the conundrum of the ‘French paradox’? Eur J Endocrinol. 1998;138:619–20. doi: 10.1530/eje.0.1380619. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Q, Wang HY, Woetmann A, Raghunath PN, Odum N, Wasik MA. STAT3 induces transcription of the DNA methyltransferase 1 gene (DNMT1) in malignant T lymphocytes. Blood. 2006;108:1058–64. doi: 10.1182/blood-2005-08-007377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patel KR, Scott E, Brown VA, Gescher AJ, Steward WP, Brown K. Clinical trials of resveratrol. Ann N Y Acad Sci. 2011;1215:161–9. doi: 10.1111/j.1749-6632.2010.05853.x. [DOI] [PubMed] [Google Scholar]