

Abstract
Molecular domestications of transposable elements have occurred repeatedly during the evolution of eukaryotes. Vertebrates, especially mammals, possess numerous single copy domesticated genes (DGs) that have originated from the intronless multicopy transposable elements. However, the origin and evolution of the retroelement-derived DGs (RDDGs) that originated from Metaviridae has been only partially elucidated, due to absence of genome data or to limited analysis of a single family of DGs. We traced the genesis and regulatory wiring of the Metaviridae-derived DGs through phylogenomic analysis, using whole-genome information from more than 90 chordate genomes. Phylogenomic analysis of these DGs in chordate genomes provided direct evidence that major diversification has occurred in the ancestor of placental mammals. Mammalian RDDGs have been shown to originate in several steps by independent domestication events and to diversify later by gene duplications. Analysis of syntenic loci has shown that diverse RDDGs and their chromosomal positions were fully established in the ancestor of placental mammals. By analysis of active Metaviridae lineages in amniotes, we have demonstrated that RDDGs originated from retroelement remains. The chromosomal gene movements of RDDGs were highly dynamic only in the ancestor of placental mammals. During the domestication process, de novo acquisition of regulatory regions is shown to be a prerequisite for the survival of the DGs. The origin and evolution of de novo acquired promoters and untranslated regions in diverse mammalian RDDGs have been explained by comparative analysis of orthologous gene loci. The origin of placental mammal-specific innovations and adaptations, such as placenta and newly evolved brain functions, was most probably connected to the regulatory wiring of DGs and their rapid fixation in the ancestor of placental mammals.
Keywords: molecular domestication, retroelement, phylogenomics, placentals, neofunctionalization, regulatory evolution
Introduction
Transposable elements (TEs) constitute a major component of eukaryotic genomes and have profound effects on the structure, function, and evolution of their host genomes. Because TEs can transpose at high frequency, they act as insertional mutagens and are powerful endogenous mutators. The mobility and amplification of TEs constitutes a major source of genomic variation, by virtue of their insertion or by triggering a variety of small- and large-scale chromosomal rearrangements. In consequence, they can have a major impact on the host phenotype (Kidwell and Lisch 2001; Kazazian 2004; Biémont and Vieira 2006; Jurka et al. 2007). Evidence is growing that TEs sometimes contribute positively to the function and evolution of genes and genomes. Genome-scale analyses have confirmed that domesticated or exapted TE-derived sequences have contributed diverse and abundant regulatory and protein-coding sequences to the host genomes (Brosius 1999; Volff 2006; Feschotte and Pritham 2007; Feschotte 2008; Sinzelle et al. 2009).
Vertebrates, especially mammals, possess numerous single copy domesticated genes (DGs) that have originated from intronless multicopy retroelements (Mi et al. 2000; Llorens and Marin 2001; Lynch and Tristem 2003; Gorinšek et al. 2004, 2005; de Parseval and Heidmann 2005; Brandt, Schrauth, et al. 2005; Brandt, Veith, et al. 2005; Kordiš 2005, 2009, 2011; Zdobnov et al. 2005; Campillos et al. 2006; Volff 2006), DNA transposons (Volff 2006; Feschotte and Pritham 2007; Sinzelle et al. 2009; Kordiš 2011), or from their remains. Domestication may require additional mutations that modify expression of the gene and the specificity of interaction of the recruited protein with nucleotide sequences or other proteins (Volff 2006). During the domestication process, de novo acquisition of the regulatory regions is a prerequisite for the survival of DGs. Molecular domestication of transposases, integrases, reverse transcriptases, and envelope proteins has occurred repeatedly during the evolution of diverse major eukaryote lineages and, during neofunctionalization, some of the newly obtained functions becoming essential for survival of the organism (Miller et al. 1999; Volff 2006). In the past decade, substantial evidence has accumulated for TEs being a dynamic reservoir for new cellular functions (Nekrutenko and Li 2001; Mariño-Ramírez et al. 2005; Medstrand et al. 2005; Britten 2006; Gotea and Makalowski 2006; Thornburg et al. 2006). Although the functions of the majority of DGs are still unknown (Campillos et al. 2006; Volff 2006; Feschotte and Pritham 2007; Sinzelle et al. 2009), some may protect against infections, some are necessary for reproduction, whereas others enable the replication of chromosomes and the control of cell proliferation and apoptosis (Volff 2006).
During evolution, many cellular protein-coding genes have been formed from genes carried by long terminal repeat (LTR) retroelements (retroviruses and LTR retrotransposons). LTR retroelements have contributed different types of coding regions to the gene repertoire of their host, including gag, envelope, integrase, and protease genes (Campillos et al. 2006; Volff 2006). LTR retrotransposons are classified into vertebrate retroviruses (Retroviridae), Metaviridae (Ty3/gypsy), Pseudoviridae (Ty1/copia), BEL (Semotivirus), and DIRS1 groups. Metaviridae populate many eukaryotic genomes. In the vast majority of Metaviridae clades, the pol domain has been found in the order protease-reverse transcriptase-ribonuclease H-integrase. Metaviridae have been classified into three genera on the basis of the presence (Errantivirus) or absence (Metavirus) of an envelope gene (env), and the presence of a chromodomain (Chromovirus). Phylogenetic analyses of the Metaviridae have shown the presence of at least 10 monophyletic clades, named Chromovirus, CsRn1, Mdg3, Osvaldo (including Gmr1 and Cigr2 families), Athila, Mag, Gypsy, Mdg1, Cer, and Tor clade. The majority of Metaviridae clades have distributions restricted to a particular taxonomic group, the only Metaviridae clade with Eukaryota-wide distribution are chromoviruses (Gorinšek et al. 2004; Kordiš 2005).
Numerous Metaviridae derived genes have been discovered in the human genome and classified into five distinct families: SASPase (ASPRV1), Sushi (=Mart), SCAN, Paraneoplastic (PNMA), and ARC (Brandt, Schrauth, et al. 2005; Campillos et al. 2006; Emerson and Thomas 2011). Large amounts of data concerning the mammalian retroelement-derived DGs (RDDGs) have been generated, such as gene structures, chromosome locations, potential biological functions, potential interacting partners, preliminary developmental expression analysis of a Mart family, and a first insight into their origin and evolution (Llorens and Marin 2001; Lynch and Tristem 2003; Gorinšek et al. 2004; Brandt, Schrauth, et al. 2005; Brandt, Veith, et al. 2005; Zdobnov et al. 2005; Campillos et al. 2006).
However, the evolutionary history and dynamics of RDDGs have been only partially explored, due to absence of genome data or to the limited analysis of a single family of RDDGs (Llorens and Marin 2001; Lynch and Tristem 2003; Gorinšek et al. 2004; Brandt, Schrauth, et al. 2005; Brandt, Veith, et al. 2005; Zdobnov et al. 2005; Campillos et al. 2006). RDDGs have, until now, been studied only in the superorder Boreoeutheria (Laurasiatheria plus Euarchontoglires), so we know nothing about them in the placental superorders Xenarthra and Afrotheria. Phylogenetic analysis of gag genes suggests at least five independent gag domestication events in mammals (Campillos et al. 2006). Because of the limited taxon sampling and lack of genome data for the key taxa, the exact timing of these domestication events could not be inferred (Campillos et al. 2006). Similarly, it has not been possible to reconstruct the evolutionary history of the Mart/sushi gene family, to identify precisely the retrotransposon ancestor(s) of the Mart genes, or to determine how many times neofunctionalization has occurred, that is, whether the Mart family is mono- or polyphyletic (Brandt, Schrauth, et al. 2005; Brandt, Veith, et al. 2005). Little is still known about the PNMA-related genes (Schüller et al. 2005; Takaji et al. 2009), the integrase-derived genes (Almeida et al. 2007; Marco and Marin 2009; Bao et al. 2010; Marín 2010), and retroelement protease-derived genes (Matsui et al. 2011).
Our aim was to gain a comprehensive insight into the origin, distribution, diversity, and evolution of the RDDGs in chordates. We traced the genesis and expansion of the RDDGs through comparative genomic and phylogenomic analyses, using publicly available whole-genome information from more than 90 chordate genomes. An extensive phylogenomic analysis of all the RDDGs in chordate, and especially mammalian, genomes has provided crucial information as to where and when Metaviridae gag, retroelement protease, and integrase domains were transformed into RDDGs. From analysis of diverse RDDGs in numerous chordate and, especially, mammalian genomes and, in the light of the novel placental phylogeny (Churakov et al. 2009; Nishihara et al. 2009), we have been able to elucidate the timing of the domestication events, clarify their origins and evolution, and provide new insights into their regulatory and functional diversification.
Results and Discussion
Phylogenomic Analysis of RDDGs
Although DGs are relatively well represented in the NCBI databases, the data are limited to the superorder Boreoeutheria. All publicly available genomic, transcriptomic, and proteomic databases have been searched for new gag-, integrase- and protease-derived DGs. In the numerous annotated genome databases, some DGs have not yet been identified, some are not correctly annotated, and some have been incorrectly assembled.
Phylogenomic analysis was the crucial part of this study because it provided a large amount of information for any novel RDDG (supplementary table S1, Supplementary Material online), including genome sequence, gene structure (fig. 1), chromosome location, protein sequence, coding and non-coding regions, as well as regulatory regions. By using phylogenomic analysis (Eisen 1998; Delsuc et al. 2005), we obtained and characterized RDDGs from all currently available mammalian (>50 different species available at NCBI) genomes, key tetrapod (amphibians and reptiles), and the remaining chordate genomes. In total, more than 90 chordate genomes were analyzed (supplementary file S1, Supplementary Material online). The rich collection of mammalian genomes belonging to all three major mammalian lineages—Eutheria (placentals), Metatheria (marsupials), and Prototheria (monotremes)—was a major advantage in studying the origin and evolution of DGs. Genomes of three placental superorders (Afrotheria, Xenarthra, and Boreoeutheria) are well represented in genome databases. In addition to the mammalian genomes, all other available vertebrate and chordate genomes were analyzed to find the transition point from TEs to DGs. By phylogenomic analysis of all available DGs in mammalian, vertebrate, and chordate genomes, unequivocal data about their origins (when and in which taxonomic group they originated) has been obtained, together with numerous gene-related information (exon/intron structure, genome location, chromosome position, etc.). Crucial information about the transition from TEs into DGs has been obtained in this study. Even more importantly, the gene structures of DGs have provided direct evidence for the extensive intron gain in placental mammals (Kordiš 2011).
Fig. 1.

Gene structures of human RDDGs. (A) Color-coded protein domains of Metaviridae retroelements and additional domains that are present in human RDDG genes. (B) Exon–intron organization of human RDDG genes. Exons are shown as boxes and introns as connecting lines. Gray regions of exons denote the protein-coding sequences, whereas UTRs are represented as orange boxes. Protein domains that are present in RDDG genes are shown schematically below the protein-coding sequences. (a) PNMA family; (b) sushi/Chromovirus family; (c) integrase-derived RDDG genes; (d) NYNRIN gene; (e) retroelement protease-derived ASPRV1 gene; and (f) ARC gene. Scale bar represents 500 bp. Distances between exons are drawn to scale except in the cases of extremely long introns. RVP, protease; RT, reverse transcriptase; INT, integrase.
RDDGs in Afrotheria and Xenarthra
We have analyzed in detail the distribution of RDDGs in all three placental superorders Boreoeutheria, Xenarthra, and Afrotheria (supplementary table S1, Supplementary Material online). RDDGs have previously been found only in a few boreoeutherian genomes and none have been reported from the genomes of the Xenarthra and Afrotheria (Llorens and Marin 2001; Lynch and Tristem 2003; Gorinšek et al. 2004; Brandt, Schrauth, et al. 2005; Brandt, Veith, et al. 2005; Zdobnov et al. 2005; Campillos et al. 2006). However, large numbers of RDDGs were found by an analysis of four afrotherian and two xenarthran genomes (supplementary table S1, Supplementary Material online). These genes provided new insights into the origin, distribution, diversity, and evolution of RDDGs.
Previous evolutionary analyses of DGs in mammals were based on comparison of only a small sample of sequences (Brandt, Schrauth, et al. 2005; Campillos et al. 2006). For this reason, the origin of numerous orthologous genes cannot be adequately inferred by using only Boreoeutheria representatives. We have demonstrated that most RDDGs are present in all three placental superorders, indicating that they were already diversified in the ancestor of placentals. These particular findings are crucial, because they provide an important insight into the origin and evolution of the RDDGs in mammals.
DGs in Monotremes and Marsupials Provide an Insight into the Ancestors of Placental Mammal-Specific DGs
The analysis of RDDGs in marsupial and monotreme genomes has shown a surprisingly limited distribution of the small number of protease-, integrase- and gag-derived genes. In the platypus genome—the only Monotremata genome available—only three DGs are present (ARC, ASPRV1, and GIN1 genes) (supplementary table S1, Supplementary Material online). The reason for such a paucity of DGs in the platypus genome is that the majority originated later, after the split of Monotremata and other mammals. In marsupials, the DGs are also rare, since only 10 DGs are present in their genomes. These genes are GIN1/2, SCAND3, KRBA2, NYNRIN, ASPRV1, ARC, PEG10, SIRH12, and PNMA progenitor (M-PNMA) (supplementary table S1, Supplementary Material online).
Paucity of DGs in Sauropsids, Amphibians, Basal Vertebrate Classes, and Chordates
The distribution of DGs has been analyzed in chordates, vertebrates and, in particular, in tetrapods and sauropsids. A surprisingly small number of DGs was demonstrated in these genomes. In sauropsids and amphibians, only a few DGs are present, such as the ARC and GIN1/2 genes (Kordiš 2009) (supplementary table S1, Supplementary Material online). In the genomes of basal vertebrate classes and the remaining chordates, only GIN1/2 genes are present (Bao et al. 2010; Marín 2010) (supplementary table S1, Supplementary Material online). Phylogenomic analysis of the DGs in chordate, vertebrate, tetrapod, amniote, and mammalian genomes has provided direct evidence that the major diversification inside the DGs occurred in the ancestor of placental mammals.
Bayesian Phylogenies Clarified the Evolution of DGs
The sequence data available in genome databases can resolve the long-standing questions about the origin and evolution of the RDDGs. We used our large collection of RDDGs to perform numerous phylogenetic analyses. Maximum-likelihood (ML) and neighbor-joining (NJ) methods were found to produce similar resolutions of evolutionary relationships in RDDGs (supplementary fig. S1, Supplementary Material online). However, the Bayesian phylogeny of RDDGs provided better resolution than the previous small sample studies (Brandt, Schrauth, et al. 2005; Campillos et al. 2006) (fig. 2; supplementary figs. S2–S5, Supplementary Material online). Bayesian analysis also clarified evolutionary relationships and provided evidence about the RDDG originations. We found that integrase-derived DGs have remained as single genes or as small multigene families throughout the vertebrates (GIN1, NYNRIN, KRBA2, and SCAND3). In contrast, gag-derived DGs have undergone more complex and dynamic evolution by numerous domestications and subsequent gene duplications.
Fig. 2.

Bayesian phylogeny of the sushi family of RDDGs. The tree was inferred by MrBayes 3 program under a Poisson + G4 model from the N-terminal capsid of gag domain. Only posterior probabilities larger than 0.5 are shown. The scale bar corresponds to 0.1 substitutions per site. The N-terminal capsid of gag domain from Ciona intestinalis Cigr2 retroelement was used to root this tree.
Only a few studies of TE-derived DGs have been made (Brandt, Schrauth, et al. 2005; Campillos et al. 2006; Volff 2006; Feschotte and Pritham 2007; Sinzelle et al. 2009) and, due to the limited availability of genome data at the time of study, the origin of DGs in the vast majority of cases could not be identified. The evolutionary relationships of currently known RDDGs have also not been well resolved, due to the poor taxonomic sampling (Brandt, Schrauth, et al. 2005; Brandt, Veith, et al. 2005; Campillos et al. 2006). The new data, obtained in this study from the genomes of monotremes, marsupials, and placental superorders Afrotheria and Xenarthra, have resolved the RDDG evolutionary relationships. These are crucial for elucidating the chromosomal gene movements of RDDGs, as well as for the timing of their domestication. By using the extensive genome data, we obtained a global insight into the origin, distribution, diversity, and evolution of all the available RDDGs in vertebrate and mammalian genomes.
Numerous Origins of the RDDGs
Analysis of the RDDGs has provided strong evidence about where and when they originated and from which progenitor. Phylogenomic analysis of the RDDGs in Tetrapoda has shown few originations in the ancestor of tetrapods (ARC) (supplementary fig. S2, Supplementary Material online), the ancestor of mammals (ASPRV1) (supplementary fig. S3, Supplementary Material online), and the ancestor of Theria (PNMA progenitor, PEG10, SIRH12, NYNRIN, KRBA2, and SCAND3) (fig. 2; supplementary figs. S4 and S5, Supplementary Material online). Only a few cases of bursts in functional diversification of DGs have occurred, a major one being in the ancestor of placental mammals (sushi-derived DGs and PNMA-derived DGs) (fig. 2; supplementary fig. S4, Supplementary Material online). The ARC gene was domesticated in the ancestor of land vertebrates and is widespread in all land vertebrates (supplementary fig. S2, Supplementary Material online). In marsupials, only a single PNMA progenitor can be found, indicating that the placental PNMA gene family is a direct descendant of this gene (supplementary fig. S4, Supplementary Material online). The age of the PNMA family is therefore more than 160 My (Kordiš 2011) and that of the ARC gene is approximately 370 My. In contrast to the low level of functional diversification of the RDDGs in amphibians, sauropsids, monotremes, and marsupials, diversification was much greater in the ancestor of placental mammals (fig. 2; supplementary figs. S2–S5 and table S1, Supplementary Material online). Analysis of the RDDGs in the monotremes (origin ∼230 Ma) has demonstrated the presence of ARC, ASPRV1, and GIN1 genes only (supplementary figs. S2, S3, and S5 and table S1, Supplementary Material online). The first diversification of sushi and PNMA gene families occurred in the ancestor of placental mammals, when all 20 orthologous genes emerged (fig. 2; supplementary fig. S4, Supplementary Material online). They differ in their expression profiles and tissue specificities (Brandt, Schrauth, et al. 2005; Takaji et al. 2009). Phylogenetic and sequence analysis of Chromovirus-derived DGs provides strong evidence that they originated independently several times in the ancestor of placentals (fig. 2; supplementary table S1, Supplementary Material online). The greatest number of orthologous RDDGs is present in the genomes of placental mammals. They possess at least 27 orthologous RDDGs (fig. 2; supplementary figs. S2–S5 and table S1, Supplementary Material online). These orthologous genes have remained conserved throughout the placental mammals (fig. 2; supplementary figs. S2–S5 and table S1, Supplementary Material online). Within the mammals, a large difference between placentals and ancestral mammalian lineages (Prototheria and Metatheria) is clearly evident, because the latter possess only 3–10 orthologous RDDGs (fig. 2; supplementary figs. S2–S5 and table S1, Supplementary Material online).
The Burst of RDDG Originations Took Place in the Ancestor of Placental Mammals
The phylogenomic analysis of RDDGs in all extant mammalian lineages has provided a definitive answer to the timing of domestication of retroelements (Kordiš 2011). The long-standing question as to the origin of mammalian RDDGs (Llorens and Marin 2001; Brandt, Schrauth, et al. 2005; Brandt, Veith, et al. 2005; Campillos et al. 2006; Volff 2006) has been resolved. RDDGs originated in several steps by independent domestication events and were later diversified by gene duplications. Phylogenomic analysis of RDDGs has shown for the first time which RDDGs are present in the genomes of monotremes, marsupials, and all three placental superorders, and which of the RDDGs are also present in other vertebrate genomes (fig. 2; supplementary table S1 and figs. S2–S5, Supplementary Material online). The analysis of all RDDGs in chordates and mammals has shown that the greatest number of RDDGs were fixed in the ancestor of placentals, as demonstrated by their presence and sequence conservation in all placental superorders (fig. 2; supplementary table S1 and figs. S2–S5, Supplementary Material online). This kind of analysis has provided a temporal component, because we can determine precisely when a particular DG or RDDG family originated.
Conserved Synteny of RDDGs in Placental Mammals
Synteny analyses can be a powerful tool for establishing gene homology relationships and providing clues about the mechanisms of origins of new genes. We therefore conducted an analysis of the genetic neighborhoods of all chordate RDDGs. Specifically, the identity and relative genomic position of the genes that map to genomic regions immediately adjacent to either side (5′ vs. 3′) of all chordate RDDGs were recorded using Ensembl and Entrez genome databases. In instances where the gene nomenclature did not readily reveal the identity of a gene, BLAST searches were conducted to establish possible relationships to other genes recorded in this manner. Synteny analyses provided a framework for comparing the evolutionary links between RDDGs. Examinations of genetic neighborhoods revealed robust synteny within ortholog comparisons of different RDDGs (fig. 3; supplementary figs. S6–S9, Supplementary Material online). We compared the loci of RDDGs and looked for genes that are conserved in synteny. For a more in-depth analysis, we used numerous mammalian and vertebrate species to take into account syntenic conservation within these slowly evolving genes. We searched for genes that are in synteny between placentals (and other chordates) and found that, for the majority of RDDGs, the conservation of the genome loci is very high (Kordiš 2011), for noncoding sequences as well as for syntenic genes. The distribution of the syntenic genes surrounding the diverse RDDG loci in all species considered in this analysis is shown in figure 3 and supplementary figures S6–S9, Supplementary Material online. From the comparison of syntenic positions between multiple mammalian lineages, we reconstructed the ancestral states of the RDDG chromosomal positions.
Fig. 3.

Conserved synteny in the sushi family of RDDGs. Chromosomal regions carrying all the RDDGs in the species considered in this analysis were compared, and neighbouring genes with conserved synteny were identified. Horizontal lines denote orthologous relationships. Each RDDG gene is represented in bold as a horizontal orange line on the chromosome. Neighbouring genes that are in synteny are shown with a schematic indication of their distance (not to scale). Ancestral states of the RDDG chromosomal positions were reconstructed from comparison of syntenic positions between multiple mammalian lineages.
Analysis of syntenic loci has enabled clear orthology distinction within DGs and provides the evidence for lineage-specific gene origins (fig. 3; supplementary figs. S6–S9, Supplementary Material online). For example, mouse 2410018M08Rik/Scand3 has been mapped to chr. 5G1.3 and human Scand3 to chr. 6p22.1. Human and mouse Scand3 share sequence similarity and have a partially shared domain structure (2410018M08Rik lack SCAN and integrase domains), but they are clearly not orthologs, because the mouse 2410018M08Rik/Scand3 genetic locus is not syntenic to the Scand3 genetic locus of human and other placental mammals. Therefore, it is most likely that the 2410018M08Rik is the product of gene duplication and subsequent loss of Scand3 in the rodent lineage. In the case of the ZCCHC12/ZCCHC18 gene pair, it was not possible to infer orthology by phylogenetic analysis alone. However, synteny analysis has enabled the identification of true orthologs and clearly shows the presence of both genes in the ancestor of placentals. The analysis of syntenic loci inside the PNMA family in marsupials and placentals, combined with phylogenetic and gene structure analysis, provided evidence for a single exaptation event of the PNMA progenitor sequence in the therian ancestor. In marsupial genomes, we identified only a single PNMA homolog (M-PNMA gene) that is located in antisense orientation inside the intron sequence of the LAMA3 gene (Monodelphis domestica, chr. 3). PNMA homologs at the LAMA3 syntenic loci in placentals (Homo sapiens, chr 18q11.2) could not be identified, indicating that the M-PNMA gene progenitor underwent a translocation in the ancestor of placentals, followed later by gene duplications and diversifications.
Analysis of conserved syntenies has shown clearly that the initial emergence and subsequent diversification of numerous RDDGs occurred in the ancestor of placental mammals (fig. 3; supplementary figs. S6–S9, Supplementary Material online). Analysis of conserved synteny has demonstrated that diverse RDDGs and their chromosomal positions were fully established in the ancestor of placental mammals. The combined use of Bayesian phylogenetics and conserved synteny has enabled analysis of the chromosomal gene movements of RDDGs in the ancestor of placentals.
DGs Originated from Retroelement Remains
The evolutionary scenarios leading to retroelement domestication in mammals have not been fully reconstructed. The number of events in the domestication of gag multigene families (multiple domestications or one unique event followed by serial duplications of the recruited gene) remains unresolved. The evolution of function from the ancestral retroelement to the gag- or integrase-derived DG is also not well understood (Llorens and Marin 2001; Brandt, Schrauth, et al. 2005; Brandt, Veith, et al. 2005; Campillos et al. 2006; Volff 2006). TE domestication has frequently not been fully reconstructed, because the ancestral TE was difficult to identify unambiguously. Establishing a molecular phylogeny and comparing the distribution of host and TE sequences can solve the problem and orient the evolution of the sequences. The timing of TE inactivation versus domestication is also another unexplored field of research. Already “dead” TEs can be recruited by the host before complete decay and “resurrected” to fulfil a new function. Frequently, the ancestral TE does not cohabit with the derived domesticated sequence and has been eliminated during evolution (Volff 2006). We have used all the above recommendations in solving the problem of the origin of RDDGs and finding the transition point from Metaviridae to RDDGs.
Active Metaviridae Progenitors of DGs Are Still Present in Diverse Reptilian Genomes
Analysis of diverse Metaviridae lineages in Deuterostomia has shown that numerous active (represented in the genome by the full-length elements) lineages are still present in diverse reptilian (e.g., in Anolis and turtles) but not in any of bird or mammalian genomes (fig. 4). As the active Metaviridae lineages are present in reptiles (sauropsids), the sister group of synapsids, they were present also in the ancestor of Amniota (Kordiš et al. 2006; Kordiš 2009). Synapsids—with the mammals as the only surviving lineage of this large taxonomic group—have evidently lost all the active Metaviridae elements, although they were present for approximately 300 Ma in their genomes. However, in the ancestors of modern mammals only rare Metaviridae remains (in the form of highly fragmented molecular fossils) have persisted.
Fig. 4.

Tracing the RDDGs progenitors in Amniota. The rooted NJ tree was inferred by MEGA 5.0 program under a p-distance + pairwise deletion model from the combined Metaviridae RT and RNAse H domains. Reliability for the internal branches was assessed using the 1,000 bootstrap replications; nodes with confidence values greater than 50% are indicated. The DIRS1 element from Lytechinus (AC131494) has been used to root this tree. Most sequences were obtained from the GenBank; genus names and accession numbers are included.
In contrast to previous studies (Brandt, Schrauth, et al. 2005; Brandt, Veith, et al. 2005; Campillos et al. 2006), we used a different approach to find progenitors of RDDGs and the transition point from Metaviridae to DGs. We asked which Metaviridae clades are still present and active in reptiles, the sister group of mammals. Such an approach has enabled us to trace the progenitors of RDDGs in amniotes (supplementary table S2, Supplementary Material online). In diverse reptilian genomes (tuatara, squamates, turtles and crocodiles), we found the following Metaviridae clades: Gmr1, Barthez, Cigr2, Chromovir, and three Mag lineages (fig. 4). As they are still represented in reptilian genomes by the full-length elements, we may infer that numerous active Metaviridae lineages (progenitors of DGs) were present in the ancestor of Amniota (supplementary table S2 and file S2, Supplementary Material online).
DGs Were Not Responsible for the Silencing of Active Metaviridae Lineages in Mammals
We have examined critically the speculations that some RDDGs, such as GIN1 and RTL1 (Llorens and Marin 2001; Lynch and Tristem 2003; Brandt, Schrauth, et al. 2005; Brandt, Veith, et al. 2005; Campillos et al. 2006), can restrict colonization of mammalian genomes by Ty3/gypsy retrotransposons (Metaviridae), might have caused the “death” of their parental retrotransposons, and might also protect the genome against infection by related viruses and retrotransposons (Lynch and Tristem 2003). Our analysis of DGs has important implications regarding the earlier mentioned speculations about the silencing of active Metaviridae lineages in mammals. We have shown unequivocally that the gag- and integrase-derived DGs originated from Metaviridae remains (molecular fossils), because no active Chromovirus or Barthez lineages of Metaviridae are present in any mammalian genome. Therefore, all previous speculations about the silencing of active Metaviridae lineages by DGs in mammals (Llorens and Marin 2001; Lynch and Tristem 2003; Brandt, Schrauth, et al. 2005; Brandt, Veith, et al. 2005; Campillos et al. 2006; Volff 2006) are incorrect. As the synapsid ancestor also possessed a very rich TE repertoire, very similar to the sauropsids, this repertoire was extensively modified after the end Permian extinction in cynodont ancestors of modern mammals (Kordiš et al. 2006; Kordiš 2009).
The Genesis of RDDGs Is Connected to the Origin of Diverse Phenotypic Novelties in Placental Mammals
Our comprehensive analysis has demonstrated that the burst of RDDG origination took place in the ancestor of placentals (Kordiš 2011) (figs. 2 and 3; supplementary figs. S2–S9, Supplementary Material online). This has important implications for explaining the origin of numerous novelties in placentals. It is increasingly evident that the RDDGs—originating from the junk DNA or from the molecular fossils of Metaviridae—have been crucially involved in, or even promoted development of, phenotypic novelties such as placenta (Kaneko-Ishino and Ishino 2012) and neocortex (Oldham 2006). It is somehow surprising that RDDGs participated in so many brain/central nervous system (CNS)-connected functions and in reproduction (supplementary table S3, Supplementary Material online). Some of the very important functions emerged even earlier in the ancestor of Tetrapoda with the ARC gene, that plays a crucial role in synaptic plasticity and long-term memory (Korb and Finkbeiner 2011).
The emergence of the orthologous RDDG families in placental mammals (figs. 2 and 3; supplementary table S1 and figs. S2–S9, Supplementary Material online) is most probably connected to the origin of their innovations and adaptations, such as placenta and newly evolved brain functions (Campillos et al. 2006; Oldham 2006; Volff 2006; Kaneko-Ishino and Ishino 2012). In the majority of orthologous RDDGs, the prevalent trend was loss of the ancestral activity and acquisition of a novel function (neofunctionalization). Although some of these orthologous RDDGs still possess the conserved gag, integrase, and protease domains, they have lost ancestral activity due to mutations in structurally important regions. The number of newly gained functions in the RDDGs indicates that the gag, integrase, and protease domains are highly versatile protein–protein interaction modules that can readily interact with novel targets (Campillos et al. 2006; Volff 2006; Feschotte 2008).
Newly emerged DGs may evolve new functional roles through adaptive evolution of encoded proteins and/or by developing new spatial or temporal expression patterns. A recent study has provided evidence for the association-area-selective gene expression pattern of the PNMA5 gene in the primate neocortex (Takaji 2009). PNMA5 gene belongs to the group of several genes previously shown to have a neocortex-specific expression pattern (either in the primary-sensory/visual cortex, association area, or motor area) in primates, and are thus proposed to be involved critically in the evolution and expansion of the primary visual, parietotemporal, and prefrontal association areas of the neocortex in the primate lineage (Yamamori 2011). There is growing evidence that some DGs (e.g., LDOC1) are involved in the gradual growth of CNS interaction networks in the particularly active regions of brain (neocortex)—not only during the evolution of placentals, but in very recent times, that is, after the split of Homo and chimpanzee lineages (Oldham et al. 2006).
Chromosomal Gene Movements of RDDGs Were Highly Dynamic only in the Ancestor of Placentals
The chromosome locations of diverse RDDGs have been relatively well studied. These genes show different patterns of chromosome locations, some being located preferentially on autosomes and the remainder on the X chromosome (Llorens and Marin 2001; Brandt, Schrauth, et al. 2005; Brandt, Veith, et al. 2005; Campillos et al. 2006). By a phylogenomic analysis and highly improved resolution of evolutionary relationships, we have elucidated the dynamics of chromosomal gene movements in diverse RDDGs.
MART and PNMA families are very interesting because, among the RDDGs, only they show diverse and mixed patterns of chromosomal location on X chromosomes and on autosomes (Brandt, Schrauth, et al. 2005; Brandt, Veith, et al. 2005; Campillos et al. 2006). However, in previous studies, it was not elucidated whether the ancestral MART and PNMA DG families were located on X chromosomes or on autosomes, so the dynamics of their chromosomal gene movements has remained unexplored. We have examined the chromosome locations of MART and PNMA DGs in the light of their improved evolutionary relationships. By mapping their chromosomal positions on the phylogenetic trees of RDDGs, we inferred the putative ancestral chromosome locations and the potential cases of RDDG movements from the autosomes to the X-chromosome, or from the X-chromosome to the autosomes (fig. 5). Our analysis has demonstrated that the chromosomal gene movements of RDDGs were highly dynamic only in the ancestor of placental mammals. However, after rapid fixation of RDDGs in the ancestor of placentals, their chromosomal positions have remained highly conserved in placentals, which is also evident from their conserved syntenies. As a number of RDDGs show testis-specific expression (Kaessmann 2010), it is apparent that they behave similarly to the mammalian retrogenes, which tend to leave the X chromosome and integrate into the autosomes, evolving male-biased expression patterns (Emerson et al. 2004; Vinckenbosch et al. 2006; Shiao et al. 2007).
Fig. 5.

Chromosomal gene movements of RDDGs occurred only in the ancestor of placental mammals. The putative ancestral chromosomal locations and potential cases of movement of RDDGs from autosomes to the X-chromosome, or from the X-chromosome to autosomes, were inferred from our phylogenetic and conserved synteny analyses. (A, B) Schematic representations of alternative hypotheses regarding the movement of PNMA family genes from the autosomes to the X-chromosome or from the X-chromosome to the autosomes. The therian PNMA gene progenitor is located on the autosome. A few independent originations from this progenitor may generate autosomal and X-chromosome (genes located on the X-chromosome are marked in red) gene movements. In the case of PNMA progenitor, the ancestral chromosomal position is unknown and can be either autosomal (scenario under hypothesis A) or on the X chromosome (scenario under hypothesis B). (C, D) Schematic representation of alternative hypotheses regarding the movement of the sushi family genes from the autosomes to the X-chromosome or from the X-chromosome to autosomes. The chromoviral full-length progenitor was most probably located on the autosome, because the RTL1 and PEG10 genes are also autosomal. Numerous independent originations from the chromoviral gag progenitor may generate autosomal and X-chromosome (genes located on the X-chromosome are marked in red) movements. In the case of the chromoviral gag progenitor, the ancestral chromosomal position is unknown and can be either autosomal (scenario under hypothesis C) or on the X chromosome (scenario under hypothesis D).
Numerous Complex Mechanisms Were Involved in the Process of Neofunctionalization
The time frame from 250 Ma (end Permian mass extinction) (Benton and Twitchett 2003) to 160 Ma (origin of placental mammals—when progenitors of RDDGs started to diversify) (Meredith et al. 2011) is very important for explaining the origin of RDDGs. It demonstrates that a very long time (90–100 My) was necessary for establishing the first RDDGs. Why was this process so slow and complex? In the transition phase from the retroelement remains to the first RDDGs, many nucleotide changes were necessary for the neofunctionalization (Lynch and Conery 2000; Long et al. 2003; Krull et al. 2007); such a process could be quite rapid, due to the initial functional diversification by adaptive evolution. By analyzing monotreme, marsupial, and placental genomes, we have demonstrated that these genes were fixed in the ancestor of placental mammals (figs. 2 and 3; supplementary table S1 and figs. S2–S9, Supplementary Material online) and evolved by a strong purifying selection (Brandt, Schrauth, et al. 2005) to preserve the important newly gained functions. One of the crucial steps in the process of neofunctionalization was the exonization (Sorek 2007; Schmitz and Brosius 2011) of retroelement domains (gag, protease, and integrase), which produced ready to use modules—Such a process was probably quite slow (fig. 6). As retroelement remains were without regulatory regions, the acquisition of regulatory regions (Castillo-Davis 2004; Kaessmann et al. 2009; Kaessmann 2010) such as 5′-untranslated regions (UTRs) and 3'-UTRs has been very important for survival of exapted sequences. Even more important was the simultaneous intron gain into 5'-UTRs and promoter acquisition—This process has enabled the regulatory wiring of RDDGs (Kordiš 2011; Kordiš and Kokošar 2012) (fig. 6). It is well documented that RDDGs exhibit highly restricted and specialized tissue-specific expression in brain, testis, placenta, and so forth (Brandt, Schrauth, et al. 2005; Schüller et al. 2005; Takaji et al. 2009; Kaneko-Ishino and Ishino 2012), which was only possible through the cis-regulatory evolution. Subsequent gene duplications and chromosomal gene movements have further diversified DGs and enabled the acquisition of novel (more specialized or more diversified) biological functions.
Fig. 6.

Mechanisms involved in the process of RDDG neofunctionalization. In the transition phase from retroelement remains to the first RDDGs, many nucleotide changes were necessary for the neofunctionalization. One of the crucial steps in the process of neofunctionalization was the exonization of retroelement domains (gag, protease, and integrase), which produced ready-to-use modules. Retroelement remains in mammalian genomes will normally turn into pseudogenes, due to lack of a promoter, and they can survive as a functional gene only if they recruit a new promoter sequence. To become expressed at a significant level and in the tissues where it can exert a selectively beneficial function, a new gene needs to acquire a core promoter and other structural elements that regulate its expression. Exons and introns are shown as orange (5'- and 3'-UTR regions) or gray (coding part of the exons) boxes and connecting lines. A de novo acquired promoter is shown in blue.
Some of the RDDGs that are specific to placental mammals, such as the Chromovirus-derived DGs, exhibit surprisingly large differences in the size of their proteins (Brandt, Schrauth, et al. 2005; Brandt, Veith, et al. 2005; Campillos et al. 2006; Volff 2006; Kordiš 2011) (supplementary table S4, Supplementary Material online). Although Metaviridae gag and integrase domains are relatively small, some RDDGs have increased very little, or even decreased substantially, in their size, whereas others have become very large, encoding proteins from 1,000 to nearly 2,000 amino acids long. As there are no signs of simple domain fusions in RDDGs (Kordiš 2011), other mechanisms responsible for such large differences in the sizes of the RDDGs, must be invoked, such as internal gene duplications (Brandt, Schrauth, et al. 2005) and exon extension by multiple internal duplications. Generation of premature stop codons in those RDDGs that are much shorter than the original Metaviridae gag domain could be responsible for the decrease in their sizes. An alternative and more plausible explanation, however, is that such a large variation in the sizes of their proteins is the result of several independent domestications of Chromovirus-derived DGs in the ancestor of placental mammals.
DGs Have Acquired Regulatory Regions De Novo
The regulatory evolution in mammalian RDDGs is one of the most noteworthy questions of this study and has previously been only partially explored (Kalitsis and Safferty 2009). New coding sequences can be generated by the recruitment of new regions (a new 5'-UTR, 3'-UTR, promoter, new introns) and gene fusions (Long et al. 2003; Fablet et al. 2009; Kaessmann et al. 2009; Kaessmann 2010). The resulting gene can remain in a genome and gain a new function. Retroelement remains in mammalian genomes will normally turn into pseudogenes, due to lack of a promoter, and they can survive as a functional gene only if they recruit a new promoter sequence. Because of the very small likelihood that retroelement remains (consisting either from the protease, gag, or integrase domains) will acquire a promoter sequence, either de novo or from a pre-existing gene (e.g., bidirectional promoters), the possibility that they become a new functional gene is limited. A mechanism by which a promoter sequence can be obtained is therefore critical for the generation of functional RDDGs (Long et al. 2003; Fablet et al. 2009; Kaessmann et al. 2009; Kaessmann 2010). As, in retroelements, gag, protease and integrase domains lack promoters and UTRs, they must have been acquired de novo in RDDGs. We have studied the origin and evolution of such de novo acquired promoters, 5'- and 3'-UTRs in diverse mammalian RDDGs by comparative analysis of orthologous gene loci (fig. 7 and tables 1 and 2). The mechanisms responsible appear to be very similar to those observed in retrogenes (Long et al. 2003; Vinckenbosch et al. 2006; Shiao et al. 2007; Fablet et al. 2009; Kaessmann et al. 2009; Kaessmann 2010) and are outlined in the following.
Fig. 7.
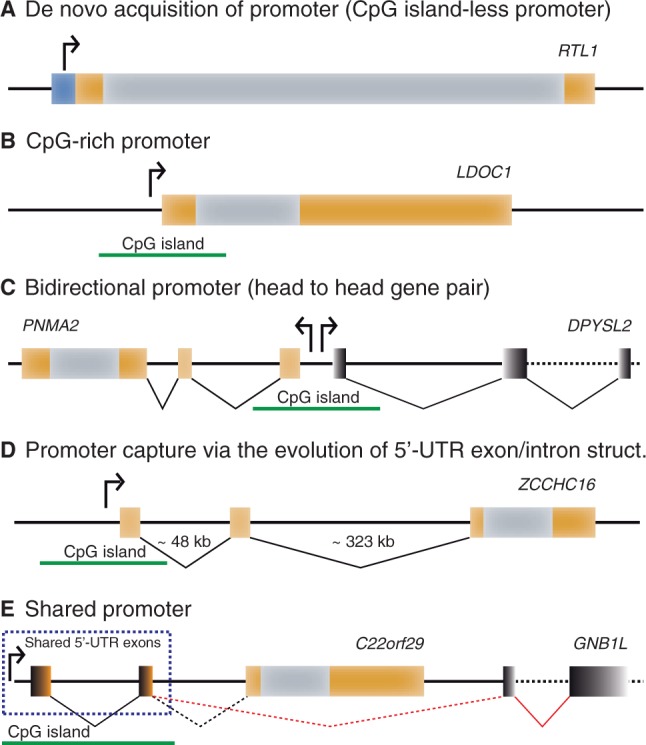
Diverse sources of RDDGs promoters. Various scenarios that lead to the transcription of RDDG gene copies are illustrated. (A) Recruitment of proto-promoters from the CpG island-less region. (B) Recruitment of proto-promoters from the CpG-rich island. (C) Recruitment of a bidirectional (CpG-enriched) promoter from neighboring gene in the vicinity of the RDDG gene. (D) Recruitment of distant promoters in the genomic neighbourhood via the acquisition of a new 5'-untranslated exon–intron structure. (E) Sharing of the unidirectional (CpG-enriched) promoter from a neighboring gene in the vicinity of the RDDG gene. Exons and introns are represented by orange and gray (RDDG genes) or black (neighbouring genes in the case of bidirectional promoters) boxes and connecting lines. Distances between exons are not to scale.
Table 1.
Newly Recruited Promoters of RDDGs.
| TE Progenitor | Gene Name | Presence of 5'-UTR Introns | CpG Island/Proto Promoter | Bidirectional Promoter | Not Associated with Promoter CpG Island |
|---|---|---|---|---|---|
| Chromovirus | RGAG1 | Yes | • | ||
| RGAG4 | No | • | |||
| PEG10 | Yes | • | |||
| RTL1 | No | • | |||
| LDOC1 | No | • | |||
| LDOC1L | Yes | • | |||
| FAM127A/B/C | No | • | |||
| C22orf29 | Yes | • | |||
| ZCCHC5 | Yes | • | |||
| ZCCHC16 | Yes | • | |||
| Barthez | PNMA1 | No | • | ||
| PNMA2 | Yes | • | |||
| MOAP1 | Yes | • | |||
| PNMA3 | Yes | • | |||
| PNMA5 | Yes | • | |||
| PNMA6A/B | Yes | • | |||
| ZCCHC12 | Yes | • | |||
| ZCCHC18 | Yes | • | |||
| PNMAL1 | Yes | • | |||
| PNMAL2 | No | • | |||
| CCDC8 | No | • | |||
| Gmr1 | GIN1 | Yes | • | ||
| GIN2 | No | • | |||
| KRBA2 | Yes | • | |||
| SCAND3 | No | • | |||
| Osvaldo | ARC | No | • | ||
| Cigr2 | ASPRV1 | No | • | ||
| ERV | NYNRIN | Yes | • | ||
Note.—The type of promoter is marked with the black dot.
Table 2.
Newly Recruited 3'-UTRs of RDDGs: 3'-UTR Lengths (in bp) in Human and Mouse RDDGs in Connection with Their Expression Profiles.
| Gene Name | Homo sapiens | Expression Profile | Mus musculus | Expression Profile |
|---|---|---|---|---|
| RTL1 | >2,000 | TS | N/A | TS |
| PEG10 | 5,161 | HK | 5,166 | TS |
| LDOC1L | 4,267 | HK | 3,064 | HK |
| C22ORF29 | 5,029 | HK | N/A | N/A |
| ZCCHC5 | 924 | TS | 911 | TS |
| ZCCHC16 | 1,584 | TS | 1,912 | TS |
| RGAG4 | 2,268 | HK/TS | 2,521 | HK/TS |
| LDOC1 | 836 | HK | 785 | TS |
| Fam 127a | 821 | HK | N/A | |
| Fam127b | 835 | HK | N/A | |
| Fam127c | 1,614 | HK/TS | N/A | |
| RGAG1 | 1,013 | TS | N/A | TS |
| PNMA1 | 795 | HK | 735 | TS |
| PNMA2 | 2,981 | TS | 2,835 | TS |
| PNMA3 | 2,023 | TS | 1,969 | TS |
| MOAP1 | 991 | HK | 2,400 | TS |
| PNMA5 | 1,428 | TS | 394 | TS |
| PNMA6a | 754 | TS | N/A | |
| PNMAL1 | 2,070 | HK | 284 | TS |
| PNMAL2 | 2,367 | HK/TS | 1,750 | TS |
| ZCCHC12 | 515 | TS | 518 | TS |
| ZCCHC18 | 519 | TS | 1,104 | TS |
| CCDC8 | 865 | HK/TS | N/A | TS |
| ARC | 1,551 | TS | 1,668 | TS |
| GIN1 | 1,898 | HK | 400 | HK/TS |
| KRBA2 | 479 | TS | N/A | |
| SCAND3 | 281 | TS | N/A | |
| NYNRIN | 1,842 | HK | 1,776 | HK/TS |
| ASPRV1 | 568 | TS | 517 | TS |
Note.—HK, housekeeping gene; TS, tissue-specific gene; N/A, not available. Underlined expression profiles reflect the change of the expression profile between human and mouse orthologous genes.
De Novo Acquisition of Promoters and 5'-UTRs in RDDGs
The presence of numerous functional DGs in mammals (Campillos et al. 2006; Volff 2006) immediately raises the question as to how they obtained the regulatory sequences that enable them to be transcribed—a precondition for gene functionality. To become expressed at a significant level and in the tissues where it can exert a selectively beneficial function, a new gene needs to acquire a core promoter and other structural elements that regulate its expression. Various sources of promoters and regulatory sequences exist and provide general insights into how new genes can acquire promoters and evolve new expression patterns (Fablet et al. 2009; Kaessmann et al. 2009; Kaessmann 2010). The expression of DGs may benefit from pre-existing regulatory machinery and expression capacities of genes in their vicinity. Transcribed DGs are often located close to other genes, suggesting that their transcription could be made possible by open chromatin and/or regulatory elements of nearby genes. This possibility is supported by the observations that DGs may be transcribed from the bidirectional CpG-rich promoters of genes in their proximity (Kalitsis and Safferty 2009).
Analysis of the promoters has shown that only a small proportion of RDDGs (5 genes, 18%) have captured bidirectional promoters. Our analysis has demonstrated that a large majority of RDDGs (19 genes, 68%) have recruited, from their genomic vicinity, CpG-rich proto-promoter sequences not previously associated with other genes for their transcription (table 1). Some of the RDDGs promoters may have evolved de novo by small substitutional changes under the influence of natural selection. In seven RDDGs (25%), the process of promoter acquisition has involved the evolution of new 5′ untranslated exon–intron structures, which often span substantial distances between the recruited promoters and RDDGs (table 1). By the acquisition of new 5′-UTR structures, DGs might also become transcribed from distant CpG-enriched sequences, that often have the inherent capacity to promote transcription, and were not previously associated with other genes. The primary role, and selective benefit, of newly gained 5′ UTR introns has been to span the substantial distances to potent CpG promoters, driving transcription of DGs and reducing the size of the UTR exons (Kordiš 2011; Kordiš and Kokošar 2012).
Analysis of transcription factor binding sites in promoters of RDDGs has shown a large diversity between genes or between human and mouse orthologous genes (supplementary file S3, Supplementary Material online), indicating that cis-regulatory evolution was responsible for the large differences in expression patterns of RDDGs. The frequent inheritance of CpG promoters could also help to explain why a significant number of DGs evolved paternally or maternally imprinted expression (Campillos et al. 2006; Volff 2006; Kaneko-Ishino and Ishino 2012).
De Novo Acquisition of 3'-UTRs in RDDGs
The analysis of all known DGs in chordates and mammals (fig. 1 and table 2; supplementary table S1, Supplementary Material online) shows that they contain newly acquired 3'-UTRs. The availability of human (fig. 1) and mouse RefSeq genes (Pruitt et al. 2009; Maglott et al. 2011) and numerous mammalian genomes (at NCBI WGS and at the Ensembl sites) has enabled the length of 3'-UTRs to be analyzed in RDDGs. De novo-acquired 3'-UTRs in placental mammals show large variation in length (table 2), the shortest being present in the human SCAND3 gene (281 bp) and the longest in the mouse PEG10 gene (5,166 bp). The mean 3'-UTR length in humans is approximately 520 bp (Grillo et al. 2010), but such lengths are present only in three human RDDGs (ZCCHC12, ZCCHC18, and ASPRV1) and only two RDDGs are shorter (SCAND3 and KRBA2). The great majority of human or mouse RDDGs have much longer 3'-UTRs, eight are shorter than 1,000 bp, seven are in the range of 1,000 to 2,000 bp, six in the range of 2,000 to 3,000 bp, one is longer than 4 kb, and two longer than 5 kb. Searching for TEs in the unusually long 3'-UTRs with RepeatMasker has shown the absence of species-specific repeats in the analyzed species.
What is the reason for such increased lengths of the 3'-UTRs of RDDGs? Although housekeeping genes possess significantly shorter coding and untranslated sequences than the tissue-specific genes (She et al. 2009), the lengths of the 3'-UTRs of tissue-specific genes have drastically increased (Stark et al. 2005). RDDGs may be an exception, because the longest 3'-UTRs are found in the housekeeping RDDGs that are expressed in the majority of tissues tested (data obtained from the Unigene EST profiles). It is likely, therefore, that all RDDGs have recruited nearby genomic regions as 5′ or 3′ UTRs. The consequence of the very long 3'-UTRs in some RDDGs is that the lengths of the 3' exons are greatly increased.
The 3'-UTRs are important post-transcriptional regulatory regions of mRNAs that are enriched for regulatory elements and are vital for correct spatial and temporal gene expression. They have been found to be involved in numerous regulatory processes, including transcript cleavage, stability and polyadenylation, translation and mRNA localization. RNA-binding proteins and miRNAs bind to cis-acting sequences within 3'-UTRs to influence mRNA stability, translation, and localization. 3'-UTRs are thus critical in determining the fate of an mRNA (Andreassi and Riccio 2009; Barrett et al. 2012). De novo recruitment of 3'-UTRs may therefore lead to the novel expression patterns of DGs. Because all the RDDGs have recruited adjacent sequences as their 3'-UTRs, these de novo-acquired 3'-UTRs may play an important role in establishing new regulatory functions.
Conclusions
The genesis and regulatory wiring of the RDDGs have been traced through the phylogenomic analyses of more than 90 chordate genomes. We have provided direct evidence for the main diversification of DGs having occurred in the ancestor of placental mammals. These RDDGs have been shown to have originated in several steps by independent domestication events and later diversified by gene duplications. We have demonstrated that placental mammal-specific DGs originated from retroelement remains. Analysis of syntenic loci has shown that diverse RDDGs and their chromosomal positions were fully established in the ancestor of placental mammals. The chromosomal gene movements of RDDGs were highly dynamic only in the ancestor of placental mammals. During the domestication process, de novo acquisition of regulatory regions is a prerequisite for survival of the DGs. The findings of this study thus provide a new view on the origin and evolution of the de novo acquired promoters, 5'- and 3'-UTRs, in diverse mammalian RDDGs. The regulatory wiring of DGs and their rapid fixation in the ancestor of placental mammals have played an important role in the origin of their innovations and adaptations, such as placenta and newly evolved brain functions. DGs could thus constitute an excellent system on which to analyze the mechanisms of regulatory evolution in placental mammals.
Materials and Methods
Data Mining
The databases analyzed were Ensembl (http://www.ensembl.org), the nonredundant, EST, GSS, HTGS, and WGS, as well as the diverse taxon-specific (mammalian, chordate, and metazoan) genome databases at the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov). Comparisons were performed using the diverse BLAST tools (Gertz et al. 2006), with the E-value cutoff set to 10−5 and other parameters to default settings. DGs, as well as diverse LTR retrotransposon domains (gag, protease, and integrase), have been used as queries. DNA sequences were translated using the Translate program (http://web.expasy.org/translate). Orthologs of DGs have been identified in Ensembl and Entrez/NCBI (WGS) genome databases. The reference set of all representatives of the DGs is available in supplementary table S1, Supplementary Material online.
Phylogenomic Analysis of DGs
The availability of RefSeq genes (Pruitt et al. 2009; Maglott et al. 2011) and numerous mammalian and chordate genomes (at the Ensembl and the NCBI WGS sites) has enabled the genome-wide analysis of RDDG sequences. The use of annotated human and mouse RDDGs has enabled their origin in mammals to be traced by genome-wide comparisons of orthologous genes in placentals, marsupials, and monotremes. Using phylogenomic analysis, we obtained and characterized RDDGs from all currently available mammalian genomes and from the genomes of the key tetrapods (amphibians and sauropsids) and the remaining chordate genomes (supplementary file S1, Supplementary Material online). The genome organization of RDDG loci, chromosomal localization, and chromosomal gene movements of RDDGs has been analyzed. Gene structures were systematically extracted for each gene of interest from Ensembl (release 67) and NCBI Entrez (GenBank release 190.0) genome databases.
Phylogenetic Analysis of Metaviridae in Deuterostomia
The amino acid sequences of the combined reverse transcriptase (RT) and ribonuclease H (RNAse H) domains of Metaviridae were aligned using Muscle program (Edgar 2004). Gap positions in aligned sequences were removed for the purpose of analysis. All the available correction models were tested, but the complex ones were outperformed by the simple correction models. Phylogenetic trees were inferred using the NJ method (Saitou and Nei 1987) implemented in MEGA 5.05 (Tamura et al. 2011) program. As an outgroup, we used DIRS1 element from Lytechinus (AC131494). The reliability of the resulting topologies was tested by the bootstrap method. To confirm that the novel elements belong to the Metaviridae, we included representatives of all known Metaviridae clades in Deuterostomia.
Phylogenetic Analysis of DGs
All the nonredundant representatives of the gag-, integrase-, and protease-derived DGs have been included in the analyses. Protein or nucleotide sequences were aligned using Muscle program (Edgar 2004). All the available correction models were tested, but the complex ones were outperformed by the simple correction models. Phylogenetic trees were reconstructed using the NJ (Saitou and Nei 1987), ML (Guindon et al. 2005), and Bayesian methods (Huelsenbeck and Ronquist 2001). The reliability of the resulting NJ tree topologies was evaluated by 10,000 bootstrap replications. Phylogenetic analyses were performed using MEGA 5.05 (Tamura et al. 2011), PhyML 3.0 (Guindon et al. 2005), and MrBayes 3.1.2 programs (Ronquist and Huelsenbeck 2003). MrBayes jobs were run on XSEDE using CIPRES Science Gateway (Miller et al. 2010) for 2 × 106 generations (sample freqency = 100) using either a Poisson or a JTT substitution model with among site rate heterogeneity, following a gamma invariant sites distribution. Bayesian posterior probabilities were estimated on the consensus of the last 10,000 trees. Diverse representatives of the Metaviridae were used as outgroups.
Synteny Analysis of DGs
For synteny analyses, the chromosomal locations, lengths, and the directionality of the neighboring genes upstream and downstream of all RDDGs were extracted from Ensembl and Entrez genome databases. In instances of uncertain identity (e.g., genes annotated with numerical identifiers), BLAST searches were conducted to establish possible homology relationships between the genes. Additional synteny comparisons were conducted using Genomicus (Muffato et al. 2010).
Analysis of De Novo Acquired Regulatory Regions in DGs
The origin and evolution of de novo acquired regulatory regions (promoters, 5'- and 3'-UTR regions) in diverse mammalian genomes was studied by comparative analysis of orthologous RDDGs. Sequence data used in the analysis were extracted from the Ensembl database. The lengths of the 3'-UTRs were obtained from the UTRdb (Grillo et al. 2010). The expression profiles of human and mouse RDDGs were obtained from the Unigene EST Profile Viewer (http://www.ncbi.nlm.nih.gov/unigene/). The distribution of CpG islands within RDDG promoter regions was analyzed using EpiGraph pre-computed data available through the UCSC genome browser interface (Bock et al. 2007). TE content was analyzed using the RepeatMasker website (http://www.repeatmasker.org). The most relevant transcription factor binding sites in the promoters of human and mouse DGs were predicted by the SABiosciences Champion ChiP Transcription Factor Search Portal (http://www.sabiosciences.com/chipqpcrsearch.php).
Supplementary Material
Supplementary figures S1–S9, tables S1–S4, and files S1–S3 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank Prof. Roger H. Pain for critical reading of the manuscript. This work was supported by the Slovenian Research Agency grant P1-0207.
References
- Almeida LM, Silva IT, Silva WA, Jr, Castro JP, Riggs PK, Carareto CM, Amaral ME. The contribution of transposable elements to Bos taurus gene structure. Gene. 2007;390:180–189. doi: 10.1016/j.gene.2006.10.012. [DOI] [PubMed] [Google Scholar]
- Andreassi C, Riccio A. To localize or not to localize: mRNA fate is in 3'UTR ends. Trends Cell Biol. 2009;19:465–474. doi: 10.1016/j.tcb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Bao W, Kapitonov VV, Jurka J. Ginger DNA transposons in eukaryotes and their evolutionary relationships with long terminal repeat retrotransposons. Mob DNA. 2010;1:3. doi: 10.1186/1759-8753-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett LW, Fletcher S, Wilton SD. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell Mol Life Sci. 2012;69: 3613–3634. doi: 10.1007/s00018-012-0990-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton MJ, Twitchett RJ. How to kill (almost) all life: the end-Permian extinction event. Trends Ecol Evol. 2003;18:358–365. [Google Scholar]
- Biémont C, Vieira C. Junk DNA as an evolutionary force. Nature. 2006;443:521–524. doi: 10.1038/443521a. [DOI] [PubMed] [Google Scholar]
- Bock C, Walter J, Paulsen M, Lengauer T. CpG island mapping by epigenome prediction. PLoS Comput Biol. 2007;3:e110. doi: 10.1371/journal.pcbi.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt J, Schrauth S, Veith AM, Froschauer A, Haneke T, Schultheis C, Gessler M, Leimeister C, Volff JN. Transposable elements as a source of genetic innovation: expression and evolution of a family of retrotransposon-derived neogenes in mammals. Gene. 2005;345:101–111. doi: 10.1016/j.gene.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Brandt J, Veith AM, Volff JN. A family of neofunctionalized Ty3/gypsy retrotransposon genes in mammalian genomes. Cytogenet Genome Res. 2005;110:307–317. doi: 10.1159/000084963. [DOI] [PubMed] [Google Scholar]
- Britten R. Transposable elements have contributed to thousands of human proteins. Proc Natl Acad Sci U S A. 2006;103:1798–1803. doi: 10.1073/pnas.0510007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosius J. RNAs from all categories generate retrosequences that may be exapted as novel genes or regulatory elements. Gene. 1999;238:115–134. doi: 10.1016/s0378-1119(99)00227-9. [DOI] [PubMed] [Google Scholar]
- Campillos M, Doerks T, Shah PK, Bork P. Computational characterization of multiple Gag-like human proteins. Trends Genet. 2006;22:585–589. doi: 10.1016/j.tig.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Castillo-Davis CI, Hartl DL, Achaz G. cis-Regulatory and protein evolution in orthologous and duplicate genes. Genome Res. 2004;14:1530–1536. doi: 10.1101/gr.2662504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churakov G, Kriegs JO, Baertsch R, Zemann A, Brosius J, Schmitz J. Mosaic retroposon insertion patterns in placental mammals. Genome Res. 2009;19:868–875. doi: 10.1101/gr.090647.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Parseval N, Heidmann T. Human endogenous retroviruses: from infectious elements to human genes. Cytogenet Genome Res. 2005;110:318–332. doi: 10.1159/000084964. [DOI] [PubMed] [Google Scholar]
- Delsuc F, Brinkmann H, Philippe H. Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet. 2005;6:361–375. doi: 10.1038/nrg1603. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JA. Phylogenomics: improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res. 1998;8:163–167. doi: 10.1101/gr.8.3.163. [DOI] [PubMed] [Google Scholar]
- Emerson JJ, Kaessmann H, Betrán E, Long M. Extensive gene traffic on the mammalian X chromosome. Science. 2004;303:537–540. doi: 10.1126/science.1090042. [DOI] [PubMed] [Google Scholar]
- Emerson RO, Thomas JH. Gypsy and the birth of the SCAN domain. J Virol. 2011;85:12043–12052. doi: 10.1128/JVI.00867-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fablet M, Bueno M, Potrzebowski L, Kaessmann H. Evolutionary origin and functions of retrogene introns. Mol Biol Evol. 2009;26:2147–2156. doi: 10.1093/molbev/msp125. [DOI] [PubMed] [Google Scholar]
- Feschotte C. Transposable elements and the evolution of regulatory networks. Nat Rev Genet. 2008;9:397–405. doi: 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–368. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz EM, Yu YK, Agarwala R, Schäffer AA, Altschul SF. Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol. 2006;4:41. doi: 10.1186/1741-7007-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorinšek B, Gubenšek F, Kordiš D. Evolutionary genomics of chromoviruses in eukaryotes. Mol Biol Evol. 2004;21:781–798. doi: 10.1093/molbev/msh057. [DOI] [PubMed] [Google Scholar]
- Gorinšek B, Gubenšek F, Kordiš D. Phylogenomic analysis of chromoviruses. Cytogenet Genome Res. 2005;110:543–552. doi: 10.1159/000084987. [DOI] [PubMed] [Google Scholar]
- Gotea V, Makałowski W. Do transposable elements really contribute to proteomes? Trends Genet. 2006;22:260–267. doi: 10.1016/j.tig.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Grillo G, Turi A, Licciulli F, et al. (11 co-authors) UTRdb and UTRsite (RELEASE 2010): a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 2010;38(Database issue):D75–D80. doi: 10.1093/nar/gkp902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Lethiec F, Duroux P, Gascuel O. PHYML online—a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 2005;33(Web Server issue):W557–W559. doi: 10.1093/nar/gki352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogeny. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Kohany O, Jurka MV. Repetitive sequences in complex genomes: structure and evolution. Annu Rev Genomics Hum Genet. 2007;8:241–259. doi: 10.1146/annurev.genom.8.080706.092416. [DOI] [PubMed] [Google Scholar]
- Kaessmann H. Origins, evolution, and phenotypic impact of new genes. Genome Res. 2010;20:1313–1326. doi: 10.1101/gr.101386.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaessmann H, Vinckenbosch N, Long M. RNA-based gene duplication: mechanistic and evolutionary insights. Nat Rev Genet. 2009;10:19–31. doi: 10.1038/nrg2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalitsis P, Saffery R. Inherent promoter bidirectionality facilitates maintenance of sequence integrity and transcription of parasitic DNA in mammalian genomes. BMC Genomics. 2009;10:498. doi: 10.1186/1471-2164-10-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko-Ishino T, Ishino F. The role of genes domesticated from LTR retrotransposons and retroviruses in mammals. Front Microbiol. 2012;3:262. doi: 10.3389/fmicb.2012.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH., Jr Mobile elements: drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- Kidwell MG, Lisch DR. Perspective: transposable elements, parasitic DNA, and genome evolution. Evolution. 2001;55:1–24. doi: 10.1111/j.0014-3820.2001.tb01268.x. [DOI] [PubMed] [Google Scholar]
- Korb E, Finkbeiner S. Arc in synaptic plasticity: from gene to behavior. Trends Neurosci. 2011;34:591–598. doi: 10.1016/j.tins.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordiš D. A genomic perspective on the chromodomain-containing retrotransposons: chromoviruses. Gene. 2005;347:161–173. doi: 10.1016/j.gene.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Kordiš D. Transposable elements in reptilian and avian (sauropsida) genomes. Cytogenet Genome Res. 2009;127:94–111. doi: 10.1159/000294999. [DOI] [PubMed] [Google Scholar]
- Kordiš D. Extensive intron gain in the ancestor of placental mammals. Biol Direct. 2011;6:59. doi: 10.1186/1745-6150-6-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordiš D, Kokošar J. What can domesticated genes tell us about the intron gain in mammals? Int J Evol Biol. 2012;2012:27898. doi: 10.1155/2012/278981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordiš D, Lovšin N, Gubenšek F. Phylogenomic analysis of the L1 retrotransposons in Deuterostomia. Syst Biol. 2006;55:886–901. doi: 10.1080/10635150601052637. [DOI] [PubMed] [Google Scholar]
- Krull M, Petrusma M, Makalowski W, Brosius J, Schmitz J. Functional persistence of exonized mammalian-wide interspersed repeat elements (MIRs) Genome Res. 2007;17:1139–1145. doi: 10.1101/gr.6320607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens C, Marin I. A mammalian gene evolved from the integrase domain of an LTR retrotransposon. Mol Biol Evol. 2001;18:1597–1600. doi: 10.1093/oxfordjournals.molbev.a003947. [DOI] [PubMed] [Google Scholar]
- Long M, Betrán E, Thornton K, Wang W. The origin of new genes: glimpses from the young and old. Nat Rev Genet. 2003;4:865–875. doi: 10.1038/nrg1204. [DOI] [PubMed] [Google Scholar]
- Lynch C, Tristem M. A co-opted gypsy-type LTR-retrotransposon is conserved in the genomes of humans, sheep, mice, and rats. Curr Biol. 2003;13:1518–1523. doi: 10.1016/s0960-9822(03)00618-3. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez gene: gene-centered information at NCBI. Nucleic Acids Res. 2011;39(Database issue):D52–D57. doi: 10.1093/nar/gkq1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A, Marín I. CGIN1: a retroviral contribution to mammalian genomes. Mol Biol Evol. 2009;26:2167–2170. doi: 10.1093/molbev/msp127. [DOI] [PubMed] [Google Scholar]
- Marín I. GIN transposons: genetic elements linking retrotransposons and genes. Mol Biol Evol. 2010;27:1903–1911. doi: 10.1093/molbev/msq072. [DOI] [PubMed] [Google Scholar]
- Mariño-Ramírez L, Lewis KC, Landsman D, Jordan IK. Transposable elements donate lineage-specific regulatory sequences to host genomes. Cytogenet Genome Res. 2005;110:333–341. doi: 10.1159/000084965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Miyamoto K, Kubo A, et al. (12 co-authors) SASPase regulates stratum corneum hydration through profilaggrin-to-filaggrin processing. EMBO Mol Med. 2011;3:320–333. doi: 10.1002/emmm.201100140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medstrand P, van de Lagemaat LN, Dunn CA, Landry JR, Svenback D, Mager DL. Impact of transposable elements on the evolution of mammalian gene regulation. Cytogenet Genome Res. 2005;110:342–352. doi: 10.1159/000084966. [DOI] [PubMed] [Google Scholar]
- Meredith RW, Janečka JE, Gatesy J, et al. (22 co-authors) Impacts of the cretaceous terrestrial revolution and KPg extinction on mammal diversification. Science. 2011;334:521–524. doi: 10.1126/science.1211028. [DOI] [PubMed] [Google Scholar]
- Mi S, Lee X, Li X, et al. (12 co-authors) Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature. 2000;403:785–789. doi: 10.1038/35001608. [DOI] [PubMed] [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES science gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE); 2010 Nov 14, New Orleans, LA.2010. [Google Scholar]
- Miller WJ, McDonald JF, Nouaud D, Anxolabéhère D. Molecular domestication—more than a sporadic episode in evolution. Genetica. 1999;107:197–207. [PubMed] [Google Scholar]
- Muffato M, Louis A, Poisnel CE, Roest Crollius H. Genomicus: a database and a browser to study gene synteny in modern and ancestral genomes. Bioinformatics. 2010;26:1119–1121. doi: 10.1093/bioinformatics/btq079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrutenko A, Li WH. Transposable elements are found in a large number of human protein-coding genes. Trends Genet. 2001;17:619–621. doi: 10.1016/s0168-9525(01)02445-3. [DOI] [PubMed] [Google Scholar]
- Nishihara H, Maruyama S, Okada N. Retroposon analysis and recent geological data suggest near-simultaneous divergence of the three superorders of mammals. Proc Natl Acad Sci U S A. 2009;106:5235–5240. doi: 10.1073/pnas.0809297106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC, Horvath S, Geschwind DH. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc Natl Acad Sci U S A. 2006;103:17973–17978. doi: 10.1073/pnas.0605938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Klimke W, Maglott DR. NCBI reference sequences: current status, policy and new initiatives. Nucleic Acids Res. 2009;37(Database issue):D32–D36. doi: 10.1093/nar/gkn721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Schmitz J, Brosius J. Exonization of transposed elements: a challenge and opportunity for evolution. Biochimie. 2011;93:1928–1934. doi: 10.1016/j.biochi.2011.07.014. [DOI] [PubMed] [Google Scholar]
- Schüller M, Jenne D, Voltz R. The human PNMA family: novel neuronal proteins implicated in paraneoplastic neurological disease. J Neuroimmunol. 2005;169:172–176. doi: 10.1016/j.jneuroim.2005.08.019. [DOI] [PubMed] [Google Scholar]
- She X, Rohl CA, Castle JC, Kulkarni AV, Johnson JM, Chen R. Definition, conservation and epigenetics of housekeeping and tissue-enriched genes. BMC Genomics. 2009;10:269. doi: 10.1186/1471-2164-10-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiao MS, Khil P, Camerini-Otero RD, Shiroishi T, Moriwaki K, Yu HT, Long M. Origins of new male germ-line functions from X-derived autosomal retrogenes in the mouse. Mol Biol Evol. 2007;24:2242–2253. doi: 10.1093/molbev/msm153. [DOI] [PubMed] [Google Scholar]
- Sinzelle L, Izsvák Z, Ivics Z. Molecular domestication of transposable elements: from detrimental parasites to useful host genes. Cell Mol Life Sci. 2009;66:1073–1093. doi: 10.1007/s00018-009-8376-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek R. The birth of new exons: mechanisms and evolutionary consequences. RNA. 2007;13:1603–1608. doi: 10.1261/rna.682507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM. Animal microRNAs confer robustness to gene expression and have a significant impact on 3'UTR evolution. Cell. 2005;123:1133–1146. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Takaji M, Komatsu Y, Watakabe A, Hashikawa T, Yamamori T. Paraneoplastic antigen-like 5 gene (PNMA5) is preferentially expressed in the association areas in a primate specific manner. Cereb Cortex. 2009;19:2865–2879. doi: 10.1093/cercor/bhp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg BG, Gotea V, Makałowski W. Transposable elements as a significant source of transcription regulating signals. Gene. 2006;365:104–110. doi: 10.1016/j.gene.2005.09.036. [DOI] [PubMed] [Google Scholar]
- Vinckenbosch N, Dupanloup I, Kaessmann H. Evolutionary fate of retroposed gene copies in the human genome. Proc Natl Acad Sci U S A. 2006;103:3220–3225. doi: 10.1073/pnas.0511307103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volff JN. Turning junk into gold: domestication of transposable elements and the creation of new genes in eukaryotes. Bioessays. 2006;28:913–922. doi: 10.1002/bies.20452. [DOI] [PubMed] [Google Scholar]
- Yamamori T. Selective gene expression in regions of primate neocortex: implications for cortical specialization. Prog Neurobiol. 2011;94:201–222. doi: 10.1016/j.pneurobio.2011.04.008. [DOI] [PubMed] [Google Scholar]
- Zdobnov EM, Campillos M, Harrington ED, Torrents D, Bork P. Protein coding potential of retroviruses and other transposable elements in vertebrate genomes. Nucleic Acids Res. 2005;33:946–954. doi: 10.1093/nar/gki236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.