
Fig. 8.
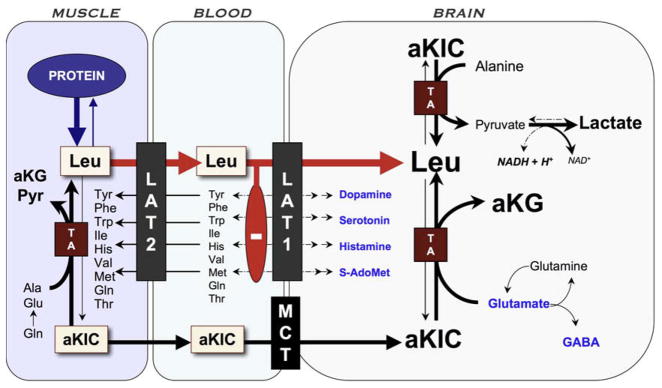
Pathophysiology of brain disease in MSUD. Episodic elevations of BCAAs occur when muscle degrades protein in response to infection or other physiologic stress and releases BCAAs and their ketoacid derivatives. High aKIC in muscle can reverse flow through cytosolic transaminases and deplete tissue of alanine and other amino acids. As leucine (Leu) exits tissue via the large amino acid transporter type 2 (LAT2), it drives import of other amino acids (heteroexchange). This process increases the relative concentration of leucine to other amino acids in plasma. At the blood–brain barrier, leucine, which has a low Km for LAT1, saturates the transporter and blocks uptake of its competitors, including precursors for neurotransmitters (dopamine, norepinephrine, serotonin, and histamine) and S-adenosylmethionine (S-AdoMet), the brain’s major methyl donor. Alpha-ketoisocaproic acid (aKIC) enters the brain via the monocarboxylate transporter (MCT) and reverses flux through cerebral transaminases (TA). This depletes brain glutamate, GABA, and glutamine while increasing production of leucine and alpha-ketoglutarate (aKG). Glutamate and GABA are the most abundant excitatory and inhibitory neurotransmitters, respectively, in the human brain. MSUD encephalopathy may also block oxidative phosphorylation through an as yet unknown mechanism; the increased NADH/NAD ratio could explain high cerebral lactate levels observed in both mice and humans during metabolic crisis.