

Abstract
Purpose
Vismodegib, an orally bioavailable small-molecule Smoothened inhibitor, is approved for treatment of advanced basal cell carcinoma (BCC). We conducted a pharmacokinetic study of vismodegib in advanced solid tumor patients to explore the effects of food on drug exposure.
Experimental design
In part I, patients were randomized to fasting overnight (FO), a high fat meal (HF), or a low fat meal (LF) before a single dose of vismodegib 150 mg. Plasma concentrations of vismodegib were determined by a validated liquid chromatography-tandem mass spectrometry assay. Primary endpoints were Cmax and AUC0–168. In part II, patients randomized to FO or HF in part I took vismodegib 150 mg daily after fasting; those randomized to LF took it after a meal. Primary endpoints after two weeks were Cmax and AUC0–24.
Results
60 (22 FO, 20 HF, 18 LF) and 52 (25 fasting, 27 fed) patients were evaluable for primary endpoints in parts I and II, respectively. Mean Cmax and AUC0–168 after a single dose were higher in HF than FO patients (ratios of geometric means (90% CI) = 1.75 (1.30, 2.34) and 1.74 (1.25, 2.42), respectively). There were no significant differences in Cmax or AUC0–24 between fasting and fed groups after daily dosing. The frequencies of drug-related toxicities were similar in both groups.
Conclusions
A high fat meal increases plasma exposure to a single dose of vismodegib, but there are no pharmacokinetic or safety differences between fasting and fed groups at steady-state. Vismodegib may be taken with or without food for daily dosing.
Keywords: food effect, vismodegib, pharmacokinetics
Introduction
The increasing use of oral anticancer drugs has made it important to discern whether or not concomitant food intake has a significant effect on the pharmacokinetics and safety profile of these drugs. Although this has been the case with oral drugs in other fields for many years, food effects have generally not been considered in the labeling of anticancer drugs.(1) Drugs with low water solubility and high cell membrane permeability, such as tyrosine kinase inhibitors, are particularly susceptible to food effects, especially when a high fat meal is consumed. In recent years, it has been documented that a number of kinase inhibitors approved by the U.S. Food and Drug Administration (FDA) are subject to significant food effects on plasma exposure. The most striking example is lapatinib, which has a 150% increase in exposure (area-under-the-curve; AUC) with food.(2) Other kinase inhibitors with significant food effects include erlotinib, pazopanib, and nilotinib.(3–5)
Food may affect pharmacokinetics by any or all of the following mechanisms: delaying gastric emptying, stimulating bile flow, changing the pH of the gastrointestinal tract, increasing splanchnic blood flow, changing luminal metabolism of a drug, and physically/chemically interacting with a dosage form or drug.(6) FDA recommends that studies report the ratio of geometric means between groups for exposure parameters such as Cmax and AUC, together with 90% confidence intervals, with a ratio of 80–125% establishing bioequivalence and ratios outside of that range establishing a significant food effect (FDA Guidance for Industry entitled “Food Effect Bioavailability and Fed Bioequivalence Studies”, 2002).
There are multiple reasons for wanting to better understand the effects of food on drug exposure. First, there may be safety risks from increased exposure due to lack of adherence with a requirement for fasting. For example, since nilotinib has a known risk of QT interval prolongation, the FDA has required the drug sponsor to institute a Risk Evaluation and Mitigation Strategy to assess and mitigate the potential risk of sudden death due to concomitant food intake.(7) Second, there may be a financial incentive for payors to conduct food effect studies. For example, abiraterone acetate is labeled to be taken fasting despite the presence of a large (five to 10-fold, depending on fat content of the meal) food effect. An ongoing, noninferiority study (NCT01543776) is randomizing patients to the labeled dose (1,000 mg fasting) versus a lower dose (250 mg with food). If the lower dose is noninferior with respect to the early change in PSA endpoint, a follow-up study could be done to establish whether the lower dose with food could be used routinely with significant potential cost savings for payors and patients.(8)
Vismodegib is an orally bioavailable small-molecule Smoothened inhibitor that is thought to target cancer stem cells through the Hedgehog signaling pathway. At an oral dose of 150 mg daily, the drug has an acceptable safety profile and considerable activity in locally advanced (response rate of 43%) and metastatic (response rate of 30%) basal cell carcinoma (BCC).(9) On January 30, 2012, the drug was approved by FDA “for the treatment of adults with metastatic basal cell carcinoma, or with locally advanced basal cell carcinoma that has recurred following surgery or who are not candidates for surgery, and who are not candidates for radiation” (vismodegib prescribing information).
The pharmacokinetics of vismodegib are characterized by less than dose-proportional increases in plasma concentration with increasing dose and lower than expected accumulation after continuous daily dosing, suggesting nonlinear pharmacokinetics. The nonlinear pharmacokinetics of vismodegib result from two separate, nonlinear processes: (1) saturable absorption and (2) high-affinity, saturable protein binding. Nonlinear absorption is consistent with the poor solubility of vismodegib at physiologic pH and likely resulted in a lack of dose-proportional increase in exposure after single doses of 270 mg and 540 mg. After multiple doses, vismodegib exhibits saturable binding to alpha-1-acid glycoprotein (AAG), which results in concentration-dependent changes in the pharmacokinetics of vismodegib. Under steady state conditions with daily dosing, the concentration of unbound drug is a constant proportion of total drug concentration.(10, 11) Since the drug is a Biopharmaceutics Classification System class 2 compound (high permeability, low solubility) and has solubility-limited absorption, it is at risk for having a significant food effect. We designed and conducted a pharmacokinetic study to test the hypothesis that there is a significant food effect with single and/or daily dosing of vismodegib. The results of this study were the basis for the instructions regarding food in the prescribing information for vismodegib.
Methods
Study design
The primary objectives of this study were to evaluate the effects of prandial state and fat content of meals on the pharmacokinetic parameters of vismodegib. The secondary objectives were to evaluate the effects of prandial state on the safety profile of vismodegib, as well as to describe the anticancer activity of vismodegib in patients with advanced solid tumors. Key inclusion criteria were as follows: histologically confirmed advanced cancer (except for leukemias) refractory to standard of care therapy, or for which no standard of care therapy is available; measurable or non-measurable disease; Karnofsky performance status >70%; and normal organ and marrow function (leukocytes ≥ 3,000/µL; absolute neutrophil count ≥ 1,500/ µL; platelets ≥ 100,000/µL; total bilirubin within normal institutional limits; AST/ALT ≤ 2.5 times institutional upper limit of normal; creatinine ≤ 1.5 times institutional upper limit of normal). Due to the potential for drug-drug interactions, patients with medical conditions requiring treatment with a strong inhibitor or inducer of CYP3A4 or CYP2C9 were excluded.
The original study design was as follows. In part I, which lasted for seven days, patients were randomized 1:1 to fasting overnight (FO) or a high fat meal (HF) before a single dose of vismodegib 150 mg. In both groups, vismodegib was taken with 240 mL (8 fluid ounces) of water and no food was allowed for at least 4 hours post dose. Patients in the FO group took vismodegib after fasting overnight for at least 10 hours. Patients in the HF group took vismodegib 30 minutes after a meal consisting of two eggs, two strips of bacon, toast with butter, 113 g hash brown potatoes and 228 g whole milk; this meal provides approximately 800 to 1000 calories with approximately 50% of the calories from fat. In part II, which lasted for 28 days, patients randomized to FO in part I took vismodegib 150 mg daily after fasting (as above); those randomized to HF took vismodegib 30 minutes after a recommended meal (a guideline with several examples of a “healthy breakfast” was given to patients). Patients were asked to record the time and date of all vismodegib doses in a medication diary and the time, date and content of all meals in a food diary and submit both diaries at each visit.
The study was amended after enrollment of 24 patients to additionally test the hypothesis that a low fat meal (LF) before a single dose would have a significant food effect compared to FO. Calorie and fat content of meals may have significant implications for the bioavailability of a drug, as different types of meals are likely to have varying impacts on gastrointestinal physiology in relation to a drug disposition.(6) If there is a food effect with vismodegib, it is important to know if the magnitude of effect (compared to fasting) is similar for low fat and high fat meals. After the amendment, the study design was as depicted in Figure 1. In part I, which lasted for seven days, patients were randomized 1:1:2 to FO, HF, or LF before a single dose of vismodegib 150 mg. Patients in the LF group took vismodegib 30 minutes after a meal consisting of one cup of cereal, 8 ounces of skim milk, one piece of toast with jam, apple juice, and a cup of coffee or tea; this meal provides approximately 520 calories. In part II, which lasted for 28 days, patients randomized to FO or HF in part I took vismodegib 150 mg daily after fasting; those randomized to LF took it 30 minutes after a recommended meal.
Figure 1.

Study schema (post-amendment).
The two subsets of patients (before and after amendment) were analyzed separately and, since there were no significant differences between endpoints for the two subsets, pooled for analysis. The primary endpoints for part I were Cmax and AUC0–168, while secondary endpoints were Tmax and Tlag. The coefficients of variation (CV) of Cmax, AUC and Tmax were approximately 30% for each parameter in a single dose study with healthy volunteers.(12) Assuming a CV of 30%, a sample size of 48 patients (12 in the LF group and 18 each in the HF and FO groups) would provide 85% and 80% power to detect a true ratio of geometric means of <67% or >150% (allowing for 15% inevaluable) with regard to the HF vs. FO and LF vs. FO comparisons, respectively, based on a two-sided test at the 0.05 significance level. The study was not powered to detect a difference between the HF and LF arms. The primary endpoints for part II were Cmax and AUC0–24, while secondary endpoints were Ctrough and Tmax. Assuming a CV of 30%, a sample size of 48 patients (24 fasting and 24 fed) would provide very high power (over 95%) to detect a true ratio of geometric means of <67% or >150% (allowing for 15% inevaluable), based on a two-sided test at the 0.05 significance level. At the time the study was designed, vismodegib seemed to be relatively well tolerated and did not appear to have a narrow therapeutic index, as there was no clear relationship between exposure and toxicity. Accordingly, the 67% and 150% thresholds were chosen rather than the more stringent thresholds recommended by FDA for bioequivalence (80% and 125%) because the investigators felt that the food effect would have to be more substantial to be clinically important.
Following the completion of part II, all patients took vismodegib after fasting for the remainder of the study. Toxicities were assessed according to the Common Terminology Criteria for Adverse Events (CTCAE, version 4.0), and response was assessed according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1).(13)
Biosampling
Part I: On day 1, each patient received vismodegib in capsule form at a dose of 150 mg, followed by a 7-day observation period. Blood samples were collected at 0, 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 24, 48, 120 and 168 hours after the dose. These samples were used to determine plasma concentrations of total vismodegib.
Part II: On day 1, patients began taking vismodegib 150 mg daily, either after fasting or after food. On day 14, each patient received vismodegib in capsule form at a dose of 150 mg, followed by a 24-hour observation period. Blood samples were collected at 0, 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, and 24 hours after the dose. These samples were used to determine plasma concentrations of total vismodegib. The samples at 0.25, 1, 4, and 24 hours after the dose were also used to determine plasma concentrations of unbound vismodegib. The samples at 0, 8, and 24 hours after the dose were also used to determine plasma concentrations of AAG.
Bioanalysis of vismodegib in plasma
Total vismodegib plasma concentrations were determined by Tandem Labs (Salt Lake City, UT) using a validated solid-phase extraction liquid chromatography/tandem mass spectrometry (LC/MS-MS) method.(12) Human plasma (K2EDTA) samples containing vismodegib were analyzed in 200 µL aliquots. Vismodegib concentrations were calculated using a 1/x2 quadratic regression over a concentration range of 5.00 to 5,000 ng/mL, with vismodegib-d5 as an internal standard. The API 3000 was operated in the selected reaction monitoring mode under optimized conditions for the detection of vismodegib and vismodegib-d5 positive ions formed by electrospray ionization.
Selected plasma samples were assayed for unbound vismodegib, which was measured in dialysate from plasma samples that underwent equilibrium dialysis (QPS, Newark, DE). Unbound plasma concentrations were determined using a LC/MS-MS method validated over a calibration curve range of 0.100 to 100 ng/mL (Tandem Labs, Salt Lake City, UT).(14)
Alpha-1-acid glycoprotein (AAG) analytic methods
Concentrations of AAG in human K2EDTA plasma were determined using a commercially available kit (Dade Behring Marburg, Germany) modified for assessment by 96-well ELISA.
Pharmacokinetic and statistical analyses
Cmax was defined as the highest observed concentration. Ctrough was defined as the concentration prior to the dose on day 14 during daily dosing. Individual patient AUC values were derived using noncompartmental analysis (WinNonlin 6.3, Pharsight) of total vismodegib concentration-time data. Exposure parameters (Cmax, Ctrough, and AUC) were log-transformed for statistical analysis. Standard statistical software (STATA, version 12.1) was used to compare groups with respect to the various endpoints. For exposure parameters, a two-sample t test with equal variances was used, and the ratios of geometric means were calculated. For Tmax and Tlag, a Wilcoxon rank-sum test was performed. Ninety-percent confidence intervals (CI) for the ratios of geometric means of the exposure parameters were obtained by back transformation. Multivariate regression analyses were performed to explore the influence of covariates (age, gender, weight, mean AAG concentration) on the pharmacokinetic parameters and group comparisons with daily dosing. The allocation ratio was 1:1:2 in the post-amendment phase in order to obtain an adequate number of patients in the LF arm. Similarly, patients in the HF arm took the drug fasted in part II after the amendment in order to maintain balance for the fasting vs. fed comparisons during the two phases. When pooling the pre- and post-amendment data, it must be assumed that there was no systematic shift in the study population, as this could admit bias into the comparison of the LF vs. FO arms. While this possibility can never be ruled out, comparisons of outcomes of patients pre- and post-amendment did not suggest that this had occurred, as mean levels of pharmacokinetic parameters were very similar (data not shown).
Given the flat pharmacokinetic profile at steady state, concentrations of unbound vismodegib at the four timepoints were averaged for each patient, and used to confirm the expectation that total concentration is an appropriate surrogate for unbound concentration under daily dosing conditions. Unbound concentrations were not compared statistically between the fasting and fed groups due to the sparse sampling and variability associated with the measurement assay. Mean AAG concentrations (mean of concentrations at 0, 8 and 24 hours) were calculated for each patient and the fasting and fed groups were compared by a two-sample t-test with equal variances.
Results
Patient characteristics are described in Table 1. A total of 63 patients were enrolled. The most common tumor type was colorectal cancer (41%). 60 patients were evaluable for the primary endpoints in the single dose part of the study: 22, 20, and 18 patients randomized to the FO, HF and LF, respectively. These numbers exceeded the accrual goals of 18, 18, and 12 patients in the respective groups because eight of these patients were not evaluable for the primary endpoints in the daily dosing part of the study. 52 patients were evaluable for the primary endpoints in the daily dosing part of the study: 25 and 27 patients were randomized to the fasting and fed groups, respectively. During daily dosing prior to pharmacokinetic sampling, 17% of patients missed at least one dose of vismodegib and 28% had missing or incomplete food diaries.
Table 1.
Characteristics of enrolled patients by cohort for a single dose of vismodegib 150 mg.
| Fasting | High fat | Low fat | ||
|---|---|---|---|---|
| No. of subjects | 23 | 20 | 20 | |
| Age (yrs), median (range) | 53 (34 – 74) | 57 (24 – 71) | 64 (46 – 84) | |
| Sex (%) | Male | 8 (35) | 9 (45) | 14 (70) |
| Female | 15 (65) | 11 (55) | 6 (30) | |
| Race (%) | White | 16 (70) | 17 (85) | 16 (80) |
| Black | 6 (26) | 3 (15) | 2 (10) | |
| Hispanic | 1 (4) | 0 | 2 (10) | |
| ECOG performance status (%) | 0 | 16 (70) | 12 (60) | 11 (55) |
| 1 | 7 (31) | 7 (35) | 9 (45) | |
| 2 | 0 | 1 (5) | 0 | |
| Tumor type (%) | Colorectal | 7 (30) | 11 (55) | 8 (40) |
| Pancreatic | 6 (26) | 1 (5) | 4 (20) | |
| Breast | 2 (9) | 1 (5) | 0 | |
| Biliary | 0 | 2 (10) | 1 (5) | |
| Renal cell | 1 (4) | 0 | 2 (10) | |
| Adenoid cystic | 0 | 2 (10) | 0 | |
| Basal cell | 1 (4) | 1 (5) | 0 | |
| Chondrosarcoma | 1 (4) | 1 (5) | 0 | |
| Urothelial | 1 (4) | 0 | 1 (5) | |
| Other* | 4 (17) | 1 (5) | 4 (20) |
a variety of tumor types with only a single patient enrolled
Mean concentration-time profiles for total vismodegib after a single dose and daily doses are presented in Figures 2a and 2b, respectively. Mean pharmacokinetic parameters based on noncompartmental analysis after a single dose and daily doses are presented in Tables 2a and 2b, respectively. Exposure parameters after a single dose and daily doses are also depicted in box plots in Figures 3a and 3b, respectively. Mean Cmax and AUC0–168 after a single dose were higher in HF than in FO patients (p = 0.003 and 0.008, respectively); ratios of geometric means (90% CI) were 1.75 (1.30, 2.34) and 1.74 (1.25, 2.42), respectively. Mean Cmax and AUC0–168 after a single dose were not significantly different in LF than in FO patients; ratios of geometric means (90% CI) were 1.08 (0.81, 1.44) and 1.12 (0.84, 1.49), respectively. There were no significant differences between groups for mean Tmax. Tlag was significantly higher in HF and LF than in FO patients, although the absolute difference was relatively small. Mean Ctrough, Cmax, and AUC0–24 after daily dosing were similar in fasting and fed patients; ratios of geometric means (90% CI) were 1.16 (0.97, 1.38), 1.13 (0.95, 1.35), and 1.22 (1.00, 1.48), respectively. Tmax was not significantly different in fasting than in fed patients. Mean ± SD of unbound vismodegib concentrations after daily dosing were 79 ± 53 ng/mL and 103 ± 49 ng/mL in the fasting and fed groups, respectively. Mean ± SD of AAG concentrations after daily dosing were similar in the fasting and fed groups (34.6 ± 13.0 µM vs. 33.6 ± 12.6 µM; p = 0.78). Furthermore, the strength of the linear correlation between mean AAG concentration and exposure (log(AUC), log(Cmax), log(Ctrough)) was similar in the fasting and fed groups. In multivariate regression analyses, no other covariates had a significant effect on exposure or fasting vs. fed comparisons after adjusting for mean AAG concentration (data not shown).
Figure 2.
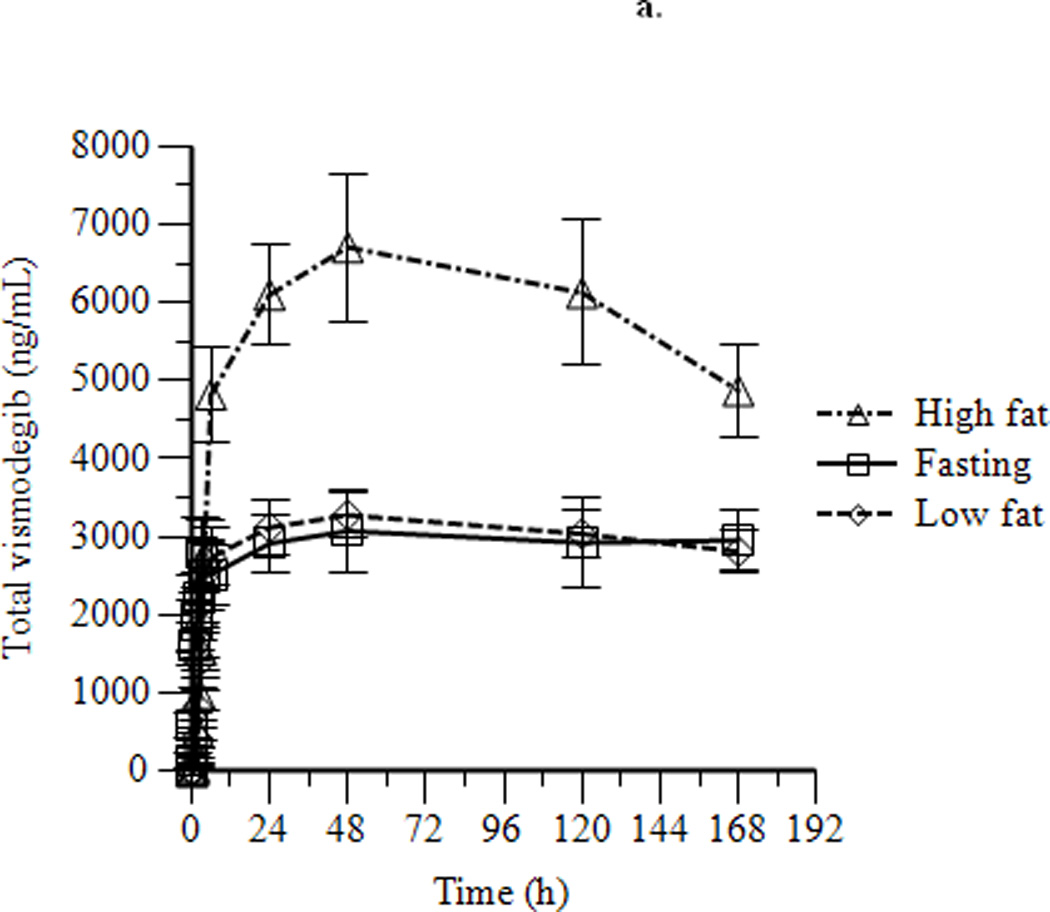
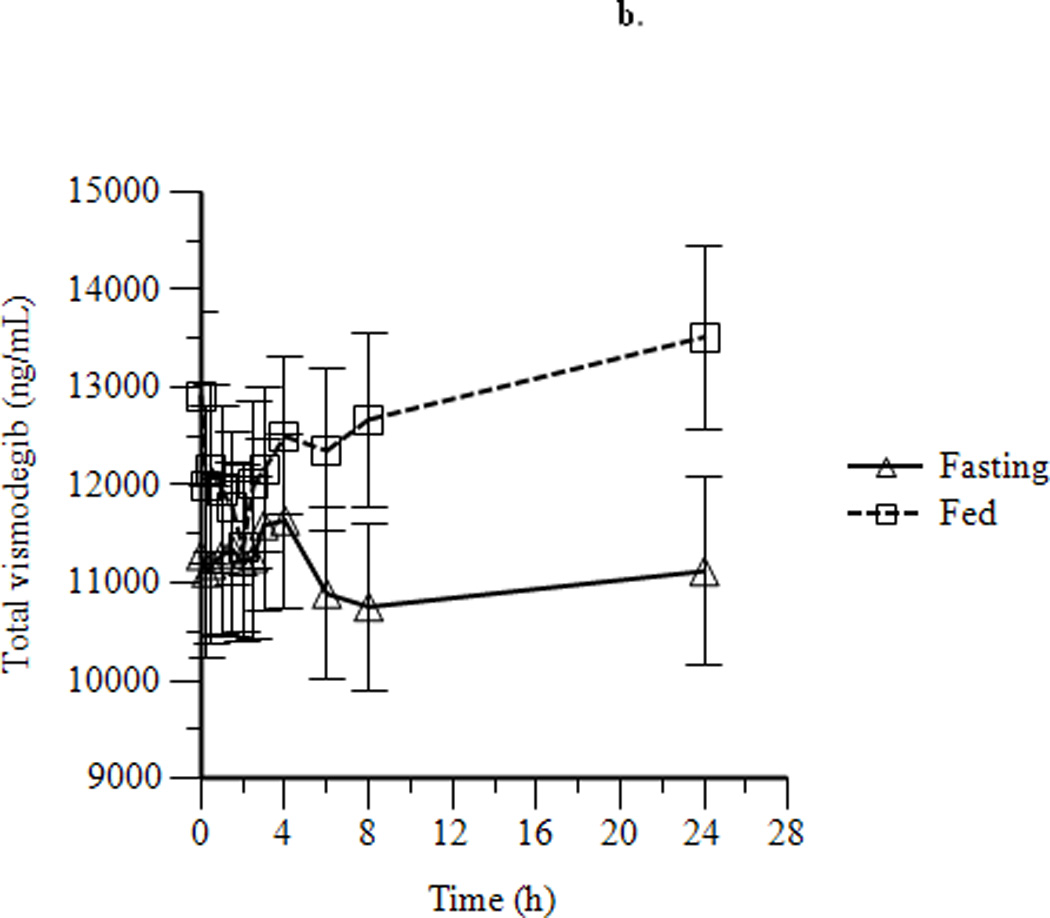
Mean ± SEM concentration-time profiles for vismodegib after a single dose of vismodegib 150 mg (a) and after 14 days of vismodegib 150 mg daily (b).
Table 2.
Summary of pharmacokinetic parameters after a single dose of vismodegib 150 mg (a) and after 14 days of vismodegib 150 mg daily (b). Exposure parameters (Ctrough, Cmax and AUC) are reported as geometric mean (CV%), while Tmax and Tlag are reported as mean ± SD.
| a. | ||||||
|---|---|---|---|---|---|---|
| Fasting (FO) (n=22) |
High fat (HF) (n=20) |
Low fat (LF) (n=18) |
p- value |
Ratio of geometric means (90% CI) |
||
| Cmax (ng/ml) | 3244 (55) | 5666 (45) | 3511 (40) | HF/FO | 0.003 | 1.75 (1.30, 2.34) |
| LF/FO | 0.65 | 1.08 (0.81, 1.44) | ||||
| AUC0–168 (ng*h/mL) | 416635 (56) | 723822 (54) | 466566 (38) | HF/FO | 0.008 | 1.74 (1.25, 2.42) |
| LF/FO | 0.51 | 1.12 (0.84, 1.49) | ||||
| Tmax (hours) | 57 ± 62 | 44 ± 48 | 62 ± 57 | HF/FO | 0.83 | -- |
| LF/FO | 0.57 | -- | ||||
| Tlag (hours) | 0.11 ± 0.17 | 0.50 ± 0.29 | 0.58 ± 0.45 | HF/FO | 0.0001 | -- |
| LF/FO | 0.0001 | -- | ||||
| b. | ||||
|---|---|---|---|---|
| Fasting (n=25) | Fed (n=27) | p-value | Ratio of geometric means (90% CI) |
|
| Ctrough (ng/ml) | 10493 (37) | 12171 (35) | 0.17 | 1.16 (0.97, 1.38) |
| Cmax (ng/ml) | 11449 (37) | 12950 (34) | 0.25 | 1.13 (0.95, 1.35) |
| AUC0–24 (ng*h/mL) | 238740 (40)a | 290896 (35)b | 0.096 | 1.22 (1.00, 1.48) |
| Tmax (hours) | 6.7 ± 9.1 | 10.2 ± 10.3 | 0.27 | -- |
n = 22;
n = 25
Figure 3.
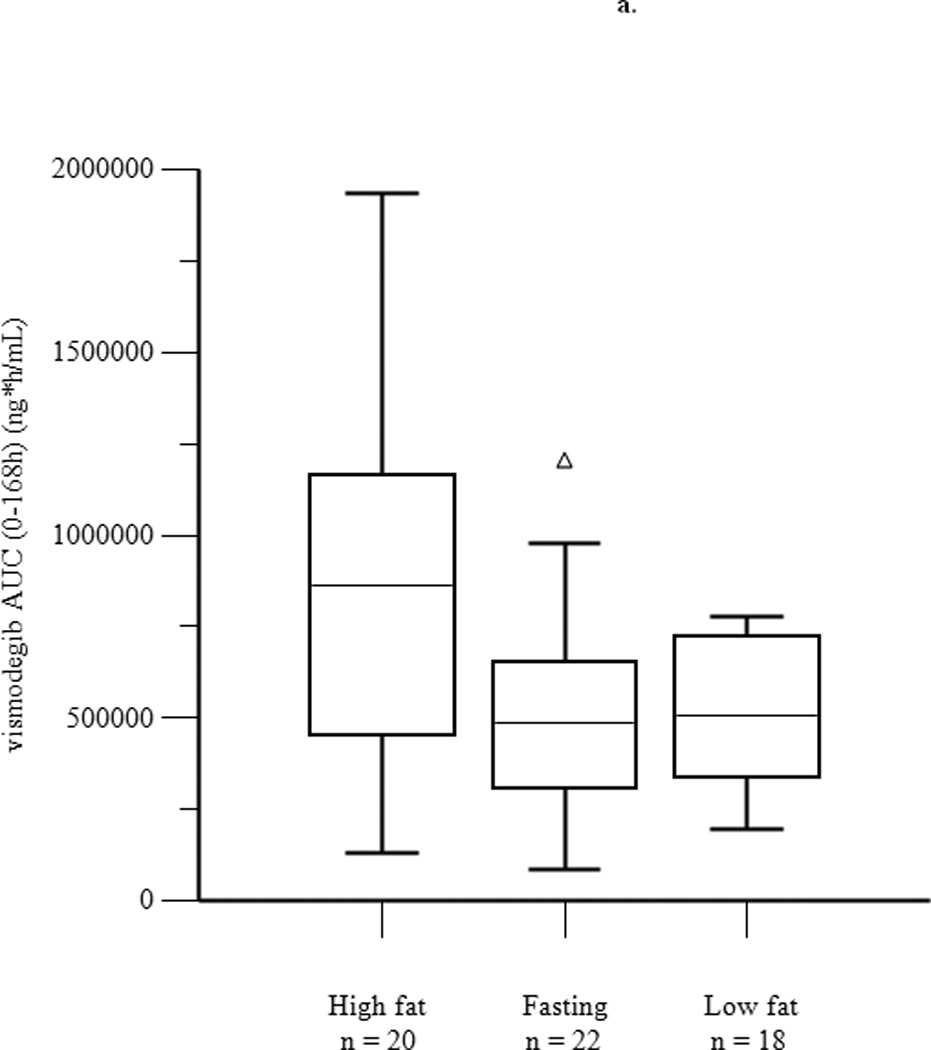
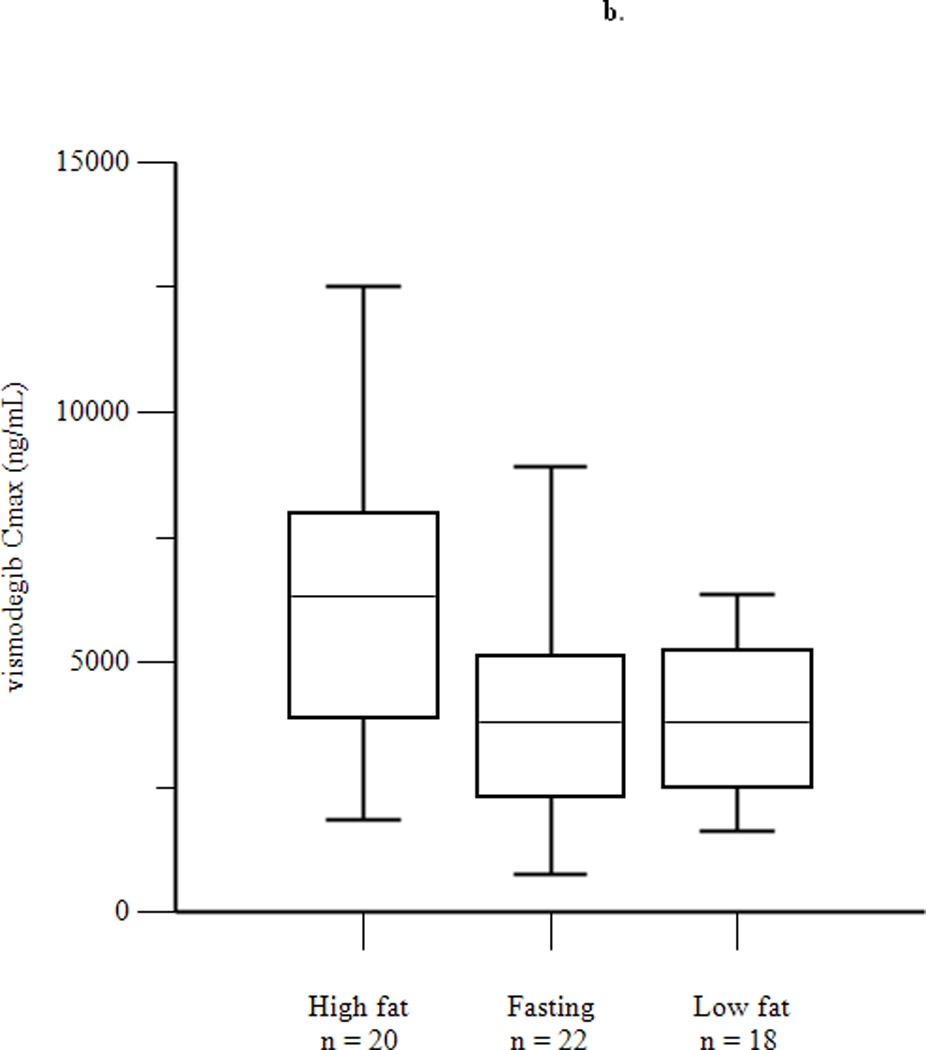
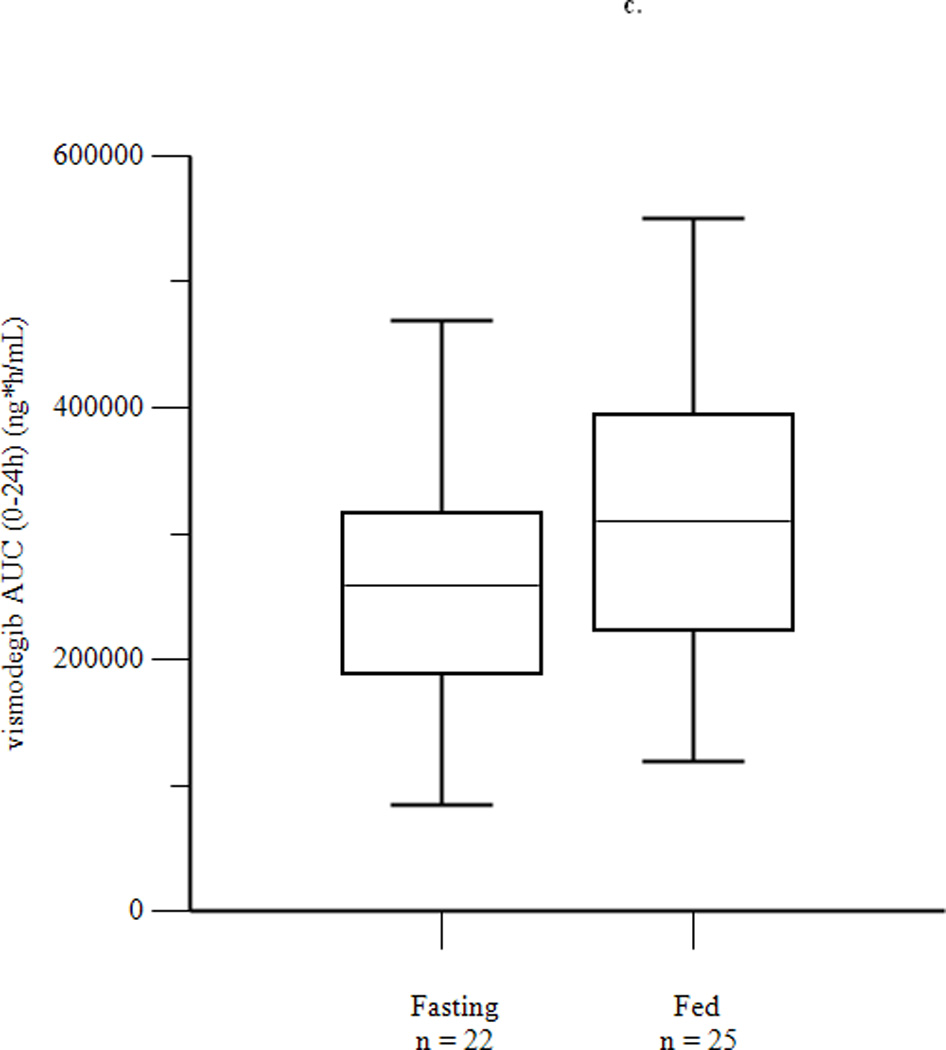
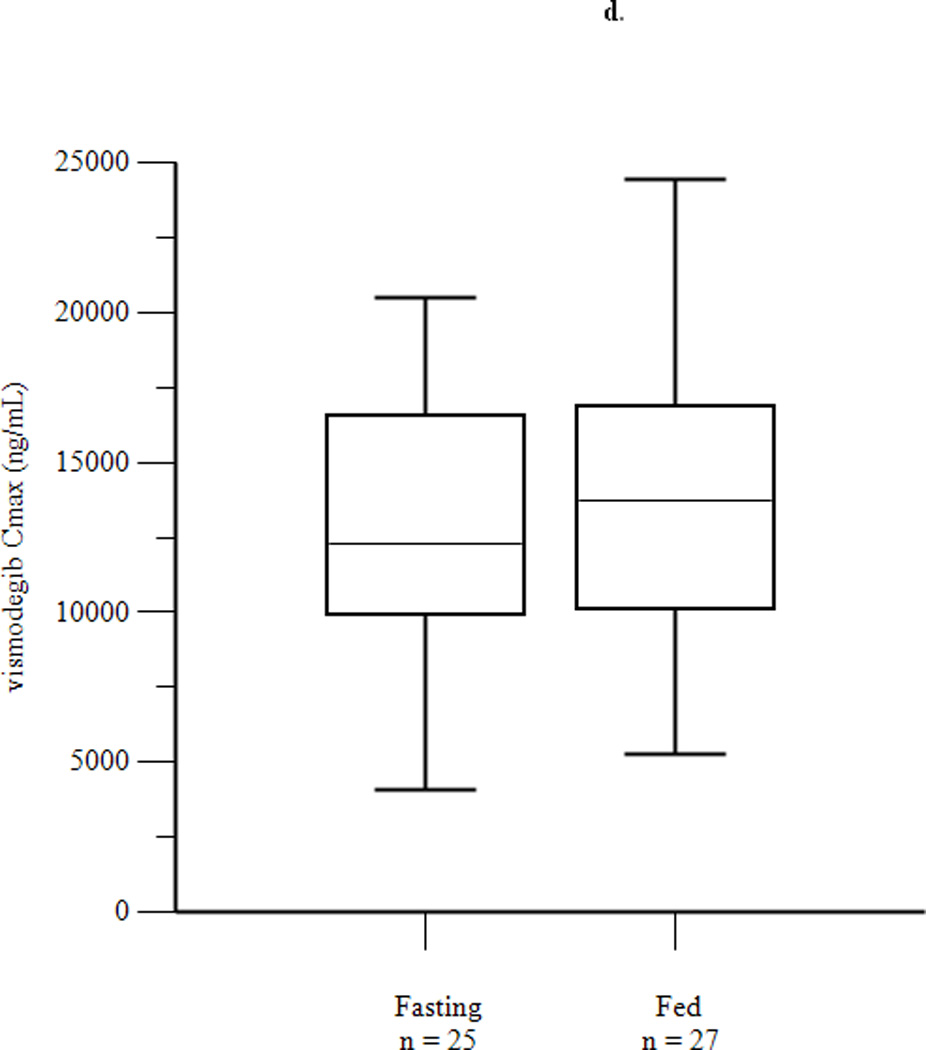
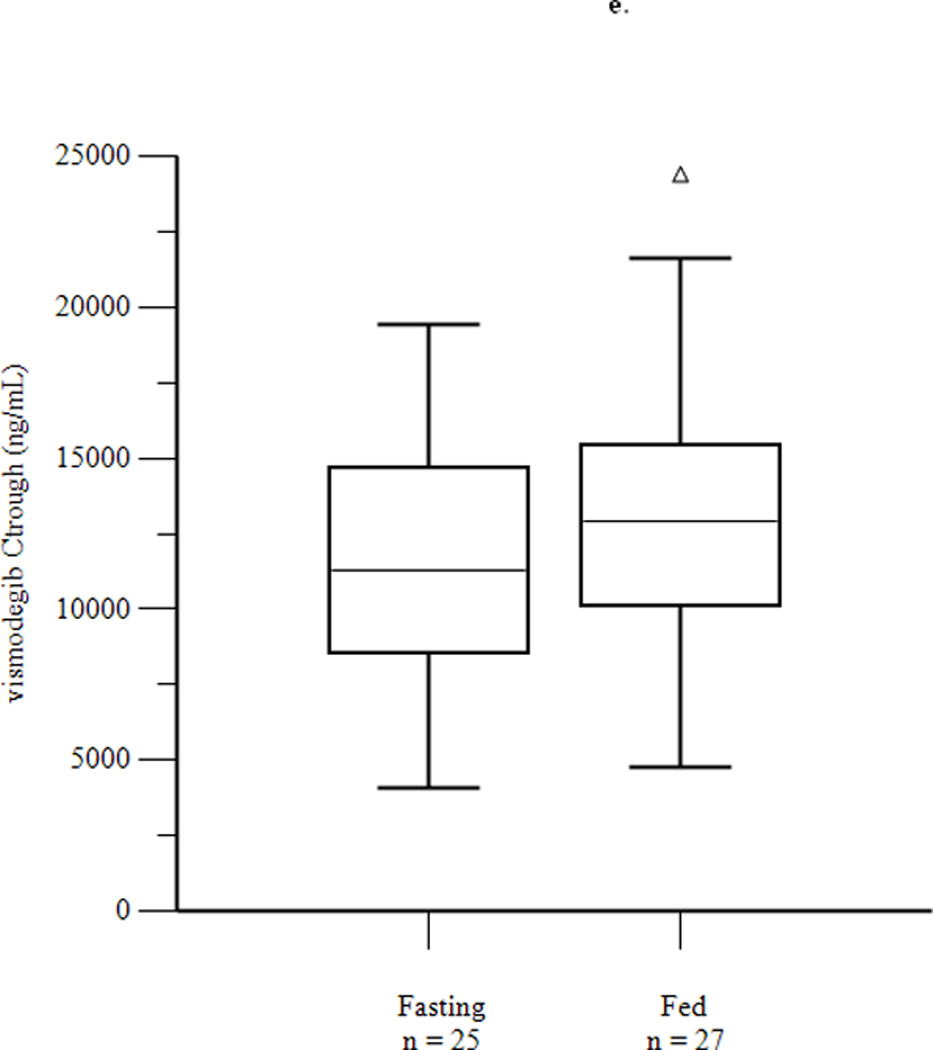
Box plots of AUC0–168 (a) and Cmax (b) after a single dose of vismodegib 150 mg, and of AUC0–24 (c), Cmax (d), and Ctrough (e) after 14 days of vismodegib 150 mg daily. Triangles denote outliers.
Toxicity data for adverse events at least possibly attributable to vismodegib during daily dosing are presented in Table 3. The most common toxicities of any grade were fatigue (36%), anorexia (36%), dysgeusia (34%) and nausea (31%). Grade 3 toxicities included fatigue (5%), hyperkalemia (2%), hypophosphatemia (2%) and neutropenia (2%). There were no significant differences between the frequencies of toxicities in the fasting and fed groups by Fisher’s exact test.
Table 3.
Adverse events by group. Patients were included if they received at least one dose in the daily dosing part of the study. Events were included if at least possibly related to vismodegib and either at least Grade 3 or present in at least three patients between the two groups. No drug-related adverse events above Grade 3 were reported. There were no statistically significant differences between groups by Fisher’s exact test.
| Adverse Event | Fasting (n=31) | Fed (n=30) | ||
|---|---|---|---|---|
| Grade 3 (%) | All grades (%) | Grade 3 (%) | All grades (%) | |
| Alopecia | 4 (13) | 1 (3) | ||
| Anorexia | 9 (29) | 13 (43) | ||
| Diarrhea | 4 (13) | 3 (10) | ||
| Dizziness | 3 (10) | 2 (7) | ||
| Dysgeusia | 8 (26) | 13 (43) | ||
| Dyspnea | 3 (10) | |||
| Fatigue | 2 (6) | 11 (35) | 1 (3) | 11 (37) |
| Hyperglycemia | 2 (6) | 1 (3) | ||
| Hyperkalemia | 1 (3) | 1 (3) | 1 (3) | |
| Hypophosphatemia | 1 (3) | 3 (10) | ||
| Myalgia | 6 (19) | 7 (23) | ||
| Nausea | 10 (32) | 9 (30) | ||
| Neutropenia | 1 (3) | 1 (3) | ||
| Vomiting | 4 (13) | 3 (10) | ||
| Weight loss | 5 (16) | 2 (7) | ||
Of 46 patients evaluable for response, there were no objective responses. Nine patients had stable disease for a median duration of 28 weeks (range: 8 to 152 weeks). The patient with the longest duration of stable disease (152 weeks) has BCC and remains on study.
Discussion
The results of this study demonstrate that there is no clinically relevant effect of prandial state on vismodegib pharmacokinetics. Following a single dose, a high fat meal increased exposure to the drug as measured by mean Cmax and AUC0–168, compared to fasting overnight. Food also appears to have a small but significant impact on delaying absorption of the drug, as evidenced by the higher Tlag. However, even if patients were to consistently consume high fat meals before taking the drug, it is unlikely that there would be a relevant food effect with daily dosing. Vismodegib exposure may be influenced by food when AAG binding is not saturated in plasma (under single dose conditions), but with continuous dosing, steady-state vismodegib exposure is simply related to levels of AAG in plasma. Even when single dose exposure is increased, the increase is not a safety concern because plasma concentrations are at or below steady-state concentrations. This is due to accumulation of vismodegib with continuous dosing. As expected, the magnitude of change in exposure parameters was smaller after a low fat meal compared to a high fat meal, and was not statistically significant when compared with fasting.
During daily dosing, there were no statistically significant differences between fasting and fed groups with respect to steady state Ctrough, Cmax, or AUC0–24 with lower limits of the 90% confidence bounds exceeding 80% and upper limits ranging from 135% to 148%. Although the upper bounds of the 90% confidence intervals exceed 125%, this small food effect is very unlikely to be of clinical significance for a drug that does not have a narrow therapeutic index. Toxicities observed in the current study were similar to those previously reported (9, 15), and durable stable disease (greater than 32 weeks) was only observed in a patient with BCC. Overall, the results of this study formed the basis for the instructions in the prescribing information that the drug may be taken either with or without food.
Although there were significant differences between groups with respect to certain demographic variables (age and sex), these did not have a significant effect on results of the study after adjusting for mean AAG concentration. This is consistent with a population pharmacokinetic analysis on 225 patients in five studies, which showed that AAG concentrations explain most (>70%) of the observed pharmacokinetic variability and that age, weight, sex, and creatinine clearance do not have a clinically meaningful effect on systemic exposure. (16)
One can speculate that the absence of a food effect may be explained by the pharmacokinetics of the drug, although the true reasons are impossible to determine from this study. The long terminal half-life of vismodegib was observed in earlier studies and confirmed in the current study, as plasma concentrations at 168 hours were often close to Cmax. Steady-state unbound drug concentrations were consistently less than 1% of total concentrations and were comparable between fasting and fed groups, consistent with previous evidence that the drug is highly bound to AAG.(10) In contrast to vismodegib, oral anticancer drugs with significant food effects have shorter terminal half-lives (prescribing information for lapatinib, erlotinib, pazopanib, and nilotinib). Although absorption of vismodegib may be limited by solubility and enhanced by intake of a meal, this factor is likely negligible compared to protein-binding and slow metabolic elimination that more substantially impact exposure to total and unbound vismodegib.
The general design strategy of randomizing patients to three groups (FO, HF, LF) for single dosing followed by two groups (fasting and fed) for daily dosing is one that could easily be applied to other oral anticancer drugs. For the fed group with daily dosing, the healthy breakfast was chosen because it was not practical to require patients to consume a high-fat, high-calorie breakfast daily for 14 days. The recommended healthy breakfast chosen for this study is expected to be representative of the real-life situation under which patients may take vismodegib on a daily basis. While a crossover design randomizing half the patients to fasting followed by fed and half the patients to fed followed by fasting for multiple dosing would be more efficient, this design is not feasible for drugs with a long terminal half-life and would not decrease the sample size needed for single dose pharmacokinetic endpoints. In the current study, 63 patients were enrolled but only 47 were evaluable for AUC0–24 with daily dosing, suggesting that an inevaluable rate as high as 25% should be factored into sample size calculations.
There are a few limitations to the current study. First, non-adherence to the assigned prandial state is a potential confounding factor. Although medication and food diaries were requested from patients during this study, they were either not returned or incomplete in a substantial minority of patients, making an accurate assessment of adherence difficult. Second, non-adherence to the rigorous pharmacokinetic sampling program led to missing samples at specific time points for some patients and limited the sample sizes available for statistical comparisons. For example, four patients did not return for pharmacokinetic sample collection on day 15 of daily dosing (24 hours after the day 14 dose), making it impossible to calculate a steady-state AUC0–24. Finally, the validity of our conclusions rests on the assumption that the subset of evaluable patients with advanced solid tumors is representative of patients for whom the drug is indicated.
In conclusion, although there are statistically significant effects of food on vismodegib pharmacokinetics after a single dose, these findings do not affect vismodegib exposure with daily dosing. The drug can be administered with or without food for standard use and in ongoing/future clinical trials, a fact that makes drug administration convenient for patients, physicians, and investigators alike. Furthermore, there does not appear to be any risk of food-induced serious toxicity due to an inadvertent increase in exposure.
Statement of Translational Relevance.
Vismodegib was recently approved by the U.S. Food and Drug Administration for the treatment of advanced basal cell carcinoma, and is being studied in clinical trials for other solid tumors. Prior to its approval, we initiated a pharmacokinetic study in advanced solid tumor patients to determine whether there is a significant effect of food on drug exposure, an issue of growing awareness and importance for all oral anticancer drugs. We found that a high fat meal increases exposure after a single dose of vismodegib compared with fasting, but there are no significant differences in drug exposure between fasting and fed groups after daily dosing. These data were the basis of the statement in the prescribing information that vismodegib may be taken with or without food.
Acknowledgments
Acknowledgments/Grant Support
The study was sponsored by the Cancer Therapy Evaluation Program and supported by NCI grant 5U01-CA69852-12 (PI: MJ Ratain). MRS was supported by award number K12CA139160 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Footnotes
Previous presentation:
Presented, in part, at the 48th Annual Meeting of the American Society of Clinical Oncology, June 1–5, 2012, Chicago, IL.
Conflicts of interest: MRS, TGK, BK, KW, MT, DG, NT, MLM, EEWC have no conflicts of interest. SPK is an employee of Merck & Co., Inc.. RAG is an employee of Genentech (a member of the Roche Group) and shareholder of Roche. RLS has an equity interest in Foundation Medicine and Universal Oncology, and is a scientific advisor for Foundation Medicine and GlaxoSmithKline. MJR has a consulting agreement with Genentech.
References
- 1.Kang SP, Ratain MJ. Inconsistent labeling of food effect for oral agents across therapeutic areas: differences between oncology and non-oncology products. Clin Cancer Res. 2010;16:4446–4451. doi: 10.1158/1078-0432.CCR-10-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ratain MJ, Cohen EE. The value meal: how to save $1,700 per month or more on lapatinib. J Clin Oncol. 2007;25:3397–3398. doi: 10.1200/JCO.2007.12.0758. [DOI] [PubMed] [Google Scholar]
- 3.Ling J, Fettner S, Lum BL, Riek M, Rakhit A. Effect of food on the pharmacokinetics of erlotinib, an orally active epidermal growth factor receptor tyrosine-kinase inhibitor, in healthy individuals. Anticancer Drugs. 2008;19:209–216. doi: 10.1097/CAD.0b013e3282f2d8e4. [DOI] [PubMed] [Google Scholar]
- 4.Heath EI, Chiorean EG, Sweeney CJ, Hodge JP, Lager JJ, Forman K, et al. A phase I study of the pharmacokinetic and safety profiles of oral pazopanib with a high-fat or low-fat meal in patients with advanced solid tumors. Clin Pharmacol Ther. 2010;88:818–823. doi: 10.1038/clpt.2010.199. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka C, Yin OQ, Sethuraman V, Smith T, Wang X, Grouss K, et al. Clinical pharmacokinetics of the BCR-ABL tyrosine kinase inhibitor nilotinib. Clin Pharmacol Ther. 2010;87:197–203. doi: 10.1038/clpt.2009.208. [DOI] [PubMed] [Google Scholar]
- 6.Singh BN, Malhotra BK. Effects of food on the clinical pharmacokinetics of anticancer agents: underlying mechanisms and implications for oral chemotherapy. Clin Pharmacokinet. 2004;43:1127–1156. doi: 10.2165/00003088-200443150-00005. [DOI] [PubMed] [Google Scholar]
- 7. [Accessed February 15, 2013]; http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022068s001rems.pdf.
- 8.Ratain MJ. Flushing oral oncology drugs down the toilet. J Clin Oncol. 2011;29:3958–3959. doi: 10.1200/JCO.2011.37.1617. [DOI] [PubMed] [Google Scholar]
- 9.Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171–2179. doi: 10.1056/NEJMoa1113713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graham RA, Lum BL, Cheeti S, Jin JY, Jorga K, Von Hoff DD, et al. Pharmacokinetics of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with locally advanced or metastatic solid tumors: the role of alpha-1-acid glycoprotein binding. Clin Cancer Res. 2011;17:2512–2520. doi: 10.1158/1078-0432.CCR-10-2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham RA, Hop CE, Borin MT, Lum BL, Colburn D, Chang I, et al. Single and multiple dose intravenous and oral pharmacokinetics of the hedgehog pathway inhibitor vismodegib in healthy female subjects. Br J Clin Pharmacol. 2012;74:788–796. doi: 10.1111/j.1365-2125.2012.04281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding X, Chou B, Graham RA, Cheeti S, Percey S, Matassa LC, et al. Determination of GDC-0449, a small-molecule inhibitor of the Hedgehog signaling pathway, in human plasma by solid phase extraction-liquid chromatographic-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:785–790. doi: 10.1016/j.jchromb.2010.01.039. [DOI] [PubMed] [Google Scholar]
- 13.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 14.Deng Y, Wong H, Graham RA, Liu W, Shen HS, Shi Y, et al. Determination of unbound vismodegib (GDC-0449) concentration in human plasma using rapid equilibrium dialysis followed by solid phase extraction and high-performance liquid chromatography coupled to mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879:2119–2126. doi: 10.1016/j.jchromb.2011.05.048. [DOI] [PubMed] [Google Scholar]
- 15.LoRusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res. 2011;17:2502–2511. doi: 10.1158/1078-0432.CCR-10-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. [Accessed March 12, 2013]; http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203388Orig1s000ClinPharmR.pdf.