

Abstract
Kenny-Caffey syndrome (KCS) and the similar but more severe osteocraniostenosis (OCS) are genetic conditions characterized by impaired skeletal development with small and dense bones, short stature, and primary hypoparathyroidism with hypocalcemia. We studied five individuals with KCS and five with OCS and found that all of them had heterozygous mutations in FAM111A. One mutation was identified in four unrelated individuals with KCS, and another one was identified in two unrelated individuals with OCS; all occurred de novo. Thus, OCS and KCS are allelic disorders of different severity. FAM111A codes for a 611 amino acid protein with homology to trypsin-like peptidases. Although FAM111A has been found to bind to the large T-antigen of SV40 and restrict viral replication, its native function is unknown. Molecular modeling of FAM111A shows that residues affected by KCS and OCS mutations do not map close to the active site but are clustered on a segment of the protein and are at, or close to, its outer surface, suggesting that the pathogenesis involves the interaction with as yet unidentified partner proteins rather than impaired catalysis. FAM111A appears to be crucial to a pathway that governs parathyroid hormone production, calcium homeostasis, and skeletal development and growth.
Main Text
In 1966, Kenny and Linarelli described a mother and son who had severe short stature (“dwarfism”), thin long bones with narrow diaphysis, and bouts of hypocalcemia.1 In 1967, Caffey described the radiographic features of the same individuals in more detail.2 The condition has since been known as Kenny-Caffey syndrome (KCS [MIM 127000]) (Figures 1A–1D). Among its clinical features are delayed closure of fontanels, defective dentition, small eyes with hypermetropia, and frontal bossing with a triangular face. Individuals with KCS often have recurrent hypocalcemia with inappropriately low levels of parathyroid hormone (PTH), and the majority of affected individuals require continuous treatment with activated vitamin D and calcium3–5 (see also Table 1). In 1994, Verloes and coworkers coined the name osteocraniostenosis (OCS [MIM 602361]) for a perinatally lethal condition characterized by gracile bones with thin diaphyses, premature closure of basal cranial sutures, and microphthalmia.6–8 The name refers to the narrowing (“stenosis”) of the medullary cavity of the long bones and of the skull (Figures 1E–1G and 2). In both KCS and OCS, skeletal radiology shows that bone density is increased, whereas the individual skeletal elements are small and thin. The two disorders also share other features, such as microphthalmia and a triangular face with frontal bossing. In addition, although hypocalcemia was not observed in the original description,6 most OCS-affected individuals who survive beyond the perinatal period develop hypocalcemia with low PTH (Table 1). On the basis of these observations, we speculated that OCS and KCS might be allelic disorders.
Figure 1.
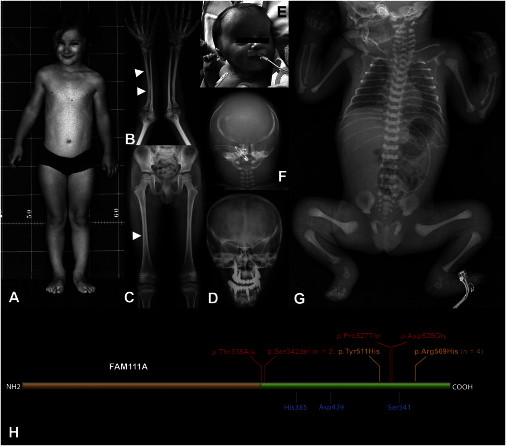
Phenotypic Features of KCS and OCS
(A) Individual 1 (c.1706G>A [p.Arg569His]) at age 8 years. Note short stature (95 cm) and frontal bossing with a triangular face and small eyes.
(B and C) Radiographs of individual 2 (c.1706G>A [p.Arg569His]) at age 14 years. The arrowheads indicate the increased thickness of the cortical bone (B) and the narrowing of the medullary cavity of tubular bones (C) (diaphyseal stenosis).
(D) Skull radiograph of individual 1 at age 25 years. Note V-shaped orbital roofs (secondary to basal craniosynostosis; compare to F) and extensive dental prosthetization.
(E) Facial appearance of individual 7 (c.1026_1028delTTC [p.Ser342del]) at a few days of age. Note frontal bossing, bitemporal narrowing with supra-auricular bulging, low-set ears, and small, deep-set eyes; compare to (A).
(F) Skull radiograph of individual 6 (c.1026_1028delTTC [p.Ser342del]). There is hypomineralization of the cranial vault, and the configuration of the orbital region indicates basal craniosynostosis (compare to D).
(G) Babygram of individual 6. The skeletal elements have a dense appearance. The long bones are thin, and the medullary cavity is not recognizable (“osteostenosis”).
(H) Simplified scheme of FAM111A. The green domain has homology to trypsin-like peptidases, and the residues indicated in blue constitute the conserved putative catalytic triad. The changes predicted by the mutations observed in individuals with KCS and OCS are indicated in orange (KCS) and red (OCS).
Table 1.
Clinical Features, Origin, and Molecular Findings in Subjects with KCS or OCS
| Subject | Gender | Origin | Clinical Diagnosis | Age at Follow-up | Presence of Hypocalcemia | Length or Staturea | Other Features |
FAM111A Mutationsb |
Predicted FAM111A Substitution | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Proband | Mother | Father | |||||||||
| 1 | F | Switzerland | KCS | 40 years | yes | −6 SDs | defective dentition, premature shedding, hypermetropia, cataracts, hypoacusis, high-pitched voice | c.1706G>A | WT | WT | p.Arg569His |
| 2 | M | India | KCS | 17 years | yes | −6 SDs | defective dentition, hypermetropia | c.1706G>A | WT | NA | p.Arg569His |
| 3 | M | Germany | KCS | 10 years | NA | −7 SDs | hypermetropia | c.1706G>A | NA | NA | p.Arg569His |
| 4 | F | Italy | KCS | 6 months | yes | −2 SDs at birth, −3 SDs at 6 months |
- | c.1706G>A | WT | WT | p.Arg569His |
| 5 | M | India | KCS | 7 years | NA | −5 SDs | open anterior fontanel, defective dentition, high-pitched voice | c.1531T>C | NA | NA | p.Tyr511His |
| 6 | M | Sweden | OCS | deceased at age 3 days | yes | −5 SDs | micropenis, asplenia at autopsy | c.1026_1028delTTC | WT | WT | p.Ser342del |
| 7 | M | Italy | OCS | deceased at age 25 days | yes | −3 SDs at birth | micropenis, spleen present, extramedullary haemopoiesis | c.1026_1028delTTC | WT | WT | p.Ser342del |
| 8 | M | Chile | OCS | deceased at age 2 months | yes | −5 SDs at birth | femoral fracture at birth, microphthalmia | c.1012A>G | WT | WT | p.Thr338Ala |
| 9 | M | Sweden | OCS | age 20 months | yes | −6 SDs at birth | hydrocephalus (requiring shunt), seizures, micropenis, bone fragility, severe failure to thrive, hepatopathy, developmental delay | c.1583A>G | WT | WT | p.Asp528Gly |
| 10 | M | Japan | OCS | deceased at age 8 months | yes | −3.5 SDs at birth | hydrocephalus, micropenis, | c.1579C>A | WT | WT | p.Pro527Thr |
Abbreviations are as follows: M, male; F, female; NA, not applicable; WT, wild-type; and NA, not available.
Length for babies and stature for children and adults.
In affected subjects, all FAM111A mutations were found to be heterozygous.
Figure 2.
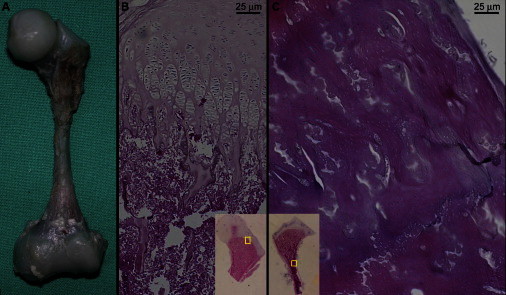
Skeletal Pathology of an OCS-Affected Newborn, Case 7, Who Died at Age 25 Days
(A) Macroscopic view of the left femur. Note the thin diaphysis with splayed metaphyses. The cartilagineous epiphyses appear macroscopically normal. Compare with the radiographic appearance in Figure 1G.
(B) Representative view of the growth-plate architecture at the proximal femur (magnification is approximately 200×). The overall architecture is preserved. The chondrocyte columns are slightly shorter and plumper than normal, and most of them have a polyclonal rather than monoclonal appearance. The inset (magnification is approximately 4×) shows the localization of the larger panel on the whole mount.
(C) Representative view of the diaphyseal cortex of the femur (magnification is approximately 200×); the inset (magnification is approximately 4×) shows the localization of the larger panel on the whole mount. The cortical bone is thicker than normal (as also appreciable on the radiographs). The bone trabeculae are thicker, and the lacunae are reduced. The osteoid lamellae have an unusual wavy appearance. These findings correlate well with the findings in parathyroid hormone (PTH)-ablated mice.9
All individuals in our series were ascertained by physician-initiated referral and diagnosed with KCS or OCS by the combination of radiographic changes, facial appearance, and, for most individuals (see Table 1), hypoparathyroidism and hypocalcemia. The diagnosis of OCS was given to those individuals who had apparent cranial deformity at birth and dense but thin bones with obliteration of the medullary cavity on radiographs (Figure 1); four of them did not survive beyond the newborn period, whereas individual 9 (Table 1) is alive at age 21 months. The five individuals with KCS all have severe short stature, a typical facial appearance with a prominent forehead and small eyes, and medullary stenosis on skeletal radiographs but no life-threatening complications except for hypocalcemia; notably, their mental development is strictly normal. The study was approved by the cantonal ethic committee of Lausanne, Switzerland, and informed consent for molecular studies was obtained from the affected individuals or their legal guardians.
Mother-to-son transmission in the original description of KCS and absence of affected siblings in our population of individuals diagnosed with KCS or OCS (Table 1) led us to hypothesize dominant inheritance with a majority of de novo mutations. Whole-exome data obtained on four affected individuals (three with OCS and one with KCS), revealed a single gene, FAM111A (RefSeq accession number NM_001142519.1), harboring heterozygous mutations in all of them (see Tables S1 and S2, available online). Direct mutation analysis in an additional group of six individuals (two with OCS and four with KCS) revealed the presence of FAM111A mutations in all (Table 1, Figure 1H, and Figure S1). The c.1706G>A (p.Arg569His) transition was detected in four of the five unrelated KCS-affected individuals, aged 6 months to 40 years; occurrence of this mutation at a CpG dinucleotide might partially explain the recurrence. The c.1026_1028delTTC (p.Ser342del) mutation (which occurs at a short triplet repeat) was detected in two of the five unrelated OCS-affected individuals, both of whom died in the newborn period.
All affected individuals in our series were simplex cases, and all parents were clinically unaffected and of normal stature. DNA samples were available from 15 of 20 parents (family relationships were confirmed through microsatellite testing): all tested parents were wild-type for the respective FAM111A mutation in their offspring, confirming the de novo status of mutations in seven probands, including three with the recurrent mutation c.1706G>A (p.Arg569His) and two with c.1026_1028delTTC (p.Ser342del) (Table 1). No individuals meeting our diagnostic criteria were found to be wild-type for FAM111A, confirming the specificity of the criteria and the genetic homogeneity of KCS and OCS. Importantly, none of the four individuals who were subjected to whole-exome sequencing had mutations in tubulin-folding cofactor E (TBCE [MIM 604934]), the gene associated with the recessive disorder Sanjad-Sakati syndrome (MIM 241410; hypoparathyroidism, short stature, intellectual disability, and seizures; also known as recessive KCS), which has partial clinical overlap with KCS.10 Thus, the genetic data indicate that heterozygous mutations in FAM111A are the genetic cause of both KCS and OCS, confirming the hypothesis of dominance and allelism of the two disorders and establishing a new dysplasia family. The observation that p.Arg569His is consistently associated with a moderate phenotype (closely corresponding to the original description of KCS1,2) but that p.Ser342del is associated with the lethal phenotype of OCS6 indicates a close genotype-phenotype correlation, comparable to that of FGFR3-related disorders.11
The role of FAM111A in the cell is unknown. All major transcripts of FAM111A encode a protein of 611 amino acids; the carboxy-terminal half of the protein has homology to trypsin-like peptidases, and the catalytic triad specific to such peptidases is conserved. Fine and coworkers recently reported that the LT antigen of the SV40 virus has a strong and specific interaction with FAM111A and that abolition of FAM111A expression results in reestablishment of the viral-host-range phenotype, indicating that FAM111A has antiviral properties.12 They found no evidence, however, that the LT antigen itself is cleaved by FAM111A.12 In all cell types used in their study, FAM111A was present in both the nucleus and the cytoplasm, and its expression was strongly cell-cycle dependent. As observed by Fine and coworkers,12 other LT-interacting proteins, such as RB, p53, FBXW7, and CDC73, are involved in gene transcription and are bona fide tumor suppressors. Nuclear localization, cell-cycle-dependent expression, and LT binding suggest that FAM111A might be involved in the regulation of gene transcription.
The mutations identified in the KCS and OCS individuals result in the substitution or deletion of single amino acids that are phylogenetically conserved (Figure S2), and they do not include frameshifts or premature terminations, suggesting that simple haploinsufficiency is not sufficient to produce the KCS and OCS phenotypes. This notion is supported by the observation of an early FAM111A frameshift mutation with a relatively high frequency of approximately 0.5% in the data from the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variation Server (EVS). None of the KCS and OCS mutations are listed in current databases (including the 1000 Genomes Project13 and the NHLBI EVS14) or in data from the CoLaus study.15 The Catalogue of Somatic Mutations in Cancer (COSMIC) lists a FAM111A c.1705C>G (p.Arg569Gly) transversion seen in one sample of ovarian cancer but not present in the >12,000 exomes on the NHLBI EVS; this variation could point to a critical function of residue Arg569.
Given the lack of data on the physiologic function of FAM111A, we used molecular modeling to obtain insight on the structural consequences of the KCS and OCS mutations, particularly on their possible impact on the putative proteolytic active site of FAM111A. A comparative model of the tertiary structure of the C-terminal domain of FAM111A was obtained and used for mapping the residues affected by the mutations. In this model, the KCS and OCS altered residues are found in the linker region preceding the protease domain and in the loops connecting the core secondary-structure elements of the protease domain; they do not map at or near the putative active site but rather cluster on one segment of the protein, at or close to its outer surface (Figure S3). Although a reduction of proteolytic activity through an effect on protein folding or the conformation of the active site cannot be formally excluded, it seems unlikely, also from the genetic considerations (see above). If the enzymatic activity is unlikely to be affected, how would the heterozygous mutations produce such remarkable phenotypes? As seen in the surface representations (Figure S3), Thr338, Ser341–Ser343, and Arg569 are adjacent to each other, and Tyr511, Pro527, and Asp528 are equally close to each other in an adjacent area; thus, all altered residues are at, or close to, the surface of a restricted area of the protein and would be well positioned to participate in intermolecular interactions, such as the binding between FAM111A and its physiologic partner(s). A plausible model to explain the findings would be to assume that FAM111A is active when unbound but inactive when bound by an (as yet unidentified) partner protein. If the KCS and OCS mutations weaken or prevent binding of FAM111A by its inactivating partner, the consequence would be increased and/or deregulated FAM111A activity, a gain-of-function effect that would be observed even in heterozygosity. This model would also fit the observations by Fine and coworkers: binding of FAM111A by the LT-antigen of SV40 occurs at the C-terminal domain of FAM111A and has similar biological consequences to FAM111A knockdown, i.e., reduced activity of FAM111A.12
The salient phenotypic features of KCS and OCS are skeletal (tiny bones with constricted medullary cavities that fail to grow properly, dysfunction of the cranial sutures, and defective dentition) and endocrine (primary hypoparathyroidism with hypocalcemia). This combination of skeletal and endocrine findings can be explained by prenatal-onset deficiency or dysfunction of PTH. In addition to its fundamental role in plasma calcium homeostasis after birth, PTH is necessary for the differentiation and proliferation of skeletal elements in the fetus. Pth−/− mice are smaller than wild-type mice at birth, and their long bones are shortened and display increased cortical bone density and a reduced number of osteoclasts,9 recapitulating the skeletal features of KCS and OCS. On the other hand, autosomal-recessive hypoparathyroidism (MIM 146200), due to mutations in either GCM2 (MIM 603716), a differentiation factor for parathyroid glands,16,17 or PTH (MIM 168450) itself,18 is not associated with skeletal dysplasia and related features of KCS and OCS, indicating that FAM111A mutations must induce additional perturbations in embryonic morphogenesis. Some phenotypic overlap between KCS and the autosomal-recessive disorder Sanjad-Sakati syndrome might indicate a functional relationship between FAM111A and TBCE. The pathogenic mechanism associated with TBCE mutations remains undetermined; it is tempting to speculate that the two proteins might be interlinked in a common regulatory pathway.
In summary, the genetic data indicate that recurrent heterozygous mutations in FAM111A are the cause of KCS and OCS and point to a hitherto unknown role of FAM111A in the activation of the PTH axis. The nature and localization of these mutations suggest that they disrupt the binding of FAM111A to partner protein(s). These proteins and their regulatory network remain to be discovered.
Acknowledgments
Our thanks go to the affected individuals and their families for donating biologic samples for this study. We are also indebted to Adi Fanconi, Sergio Fanconi, Thomas Fehr, Cecilia Giunta, and Beat Steinmann for additional clinical information on individual 1. The project was supported by the Swiss National Foundation (www.snf.ch), by the Faculty of Biology and Medicine of the University of Lausanne (Fonds de Developpement de la Recherche en Pédiatrie), and by the Leenaards Foundation in Lausanne (www.leenaards.ch). The authors would like to thank the National Heart, Lung, and Blood Institute Grand Opportunity (GO) Exome Sequencing Project and its ongoing studies, which produced and provided exome variant calls for comparison—the Lung GO Sequencing Project (HL-102923), the Women’s Health Initiative Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010)—as well as the exome-sequencing data from the CoLaus cohort, which was sequenced as part of a partnership among the Wellcome Trust Sanger Institute, the CoLaus principal investigators, and the Quantitative Sciences Department of GlaxoSmithKline. This work is dedicated to Tinetta and Gustav and their parents.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://browser.1000genomes.org
Catalogue of Somatic Mutations in Cancer (COSMIC), http://cancer.sanger.ac.uk/cosmic-search/s/
Ensembl, http://www.ensembl.org/index.html
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
RCSB Protein Data Bank, http://www.rcsb.org/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Kenny F.M., Linarelli L. Dwarfism and cortical thickening of tubular bones. Transient hypocalcemia in a mother and son. Am. J. Dis. Child. 1966;111:201–207. doi: 10.1001/archpedi.1966.02090050133013. [DOI] [PubMed] [Google Scholar]
- 2.Caffey J. Congenital stenosis of medullary spaces in tubular bones and calvaria in two proportionate dwarfs—mother and son; coupled with transitory hypocalcemic tetany. Am. J. Roentgenol. Radium Ther. Nucl. Med. 1967;100:1–11. doi: 10.2214/ajr.100.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Fanconi S., Fischer J.A., Wieland P., Atares M., Fanconi A., Giedion A., Prader A. Kenny syndrome: evidence for idiopathic hypoparathyroidism in two patients and for abnormal parathyroid hormone in one. J. Pediatr. 1986;109:469–475. doi: 10.1016/s0022-3476(86)80120-2. [DOI] [PubMed] [Google Scholar]
- 4.Majewski F., Rosendahl W., Ranke M., Nolte K. The Kenny syndrome, a rare type of growth deficiency with tubular stenosis, transient hypoparathyroidism and anomalies of refraction. Eur. J. Pediatr. 1981;136:21–30. doi: 10.1007/BF00441706. [DOI] [PubMed] [Google Scholar]
- 5.Wilson M.G., Maronde R.F., Mikity V.G., Shinno N.W. Dwarfism and congenital medullary stenosis (Kenny syndrome) Birth Defects Orig. Artic. Ser. 1974;10:128–132. [PubMed] [Google Scholar]
- 6.Verloes A., Narcy F., Grattagliano B., Delezoide A.L., Guibaud P., Schaaps J.P., Le Merrer M., Maroteaux P. Osteocraniostenosis. J. Med. Genet. 1994;31:772–778. doi: 10.1136/jmg.31.10.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elliott A.M., Wilcox W.R., Spear G.S., Field F.M., Steffensen T.S., Friedman B.D., Rimoin D.L., Lachman R.S. Osteocraniostenosis-hypomineralized skull with gracile long bones and splenic hypoplasia. Four new cases with distinctive chondro-osseous morphology. Am. J. Med. Genet. A. 2006;140:1553–1563. doi: 10.1002/ajmg.a.31326. [DOI] [PubMed] [Google Scholar]
- 8.Spear G.S. Parietal bone agenesis with gracile bones and splenic hypoplasia/aplasia: clinico-pathologic report and differential diagnosis with review of cranio-gracile bone syndromes, “osteocraniostenosis” and Kleeblattschädel. Am. J. Med. Genet. A. 2006;140:2341–2348. doi: 10.1002/ajmg.a.31473. [DOI] [PubMed] [Google Scholar]
- 9.Miao D., He B., Karaplis A.C., Goltzman D. Parathyroid hormone is essential for normal fetal bone formation. J. Clin. Invest. 2002;109:1173–1182. doi: 10.1172/JCI14817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parvari R., Hershkovitz E., Grossman N., Gorodischer R., Loeys B., Zecic A., Mortier G., Gregory S., Sharony R., Kambouris M., HRD/Autosomal Recessive Kenny-Caffey Syndrome Consortium Mutation of TBCE causes hypoparathyroidism-retardation-dysmorphism and autosomal recessive Kenny-Caffey syndrome. Nat. Genet. 2002;32:448–452. doi: 10.1038/ng1012. [DOI] [PubMed] [Google Scholar]
- 11.Warman M.L., Cormier-Daire V., Hall C., Krakow D., Lachman R., LeMerrer M., Mortier G., Mundlos S., Nishimura G., Rimoin D.L. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fine D.A., Rozenblatt-Rosen O., Padi M., Korkhin A., James R.L., Adelmant G., Yoon R., Guo L., Berrios C., Zhang Y. Identification of FAM111A as an SV40 host range restriction and adenovirus helper factor. PLoS Pathog. 2012;8:e1002949. doi: 10.1371/journal.ppat.1002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Firmann M., Mayor V., Vidal P.M., Bochud M., Pécoud A., Hayoz D., Paccaud F., Preisig M., Song K.S., Yuan X. The CoLaus study: a population-based study to investigate the epidemiology and genetic determinants of cardiovascular risk factors and metabolic syndrome. BMC Cardiovasc. Disord. 2008;8:6. doi: 10.1186/1471-2261-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding C., Buckingham B., Levine M.A. Familial isolated hypoparathyroidism caused by a mutation in the gene for the transcription factor GCMB. J. Clin. Invest. 2001;108:1215–1220. doi: 10.1172/JCI13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maret A., Ding C., Kornfield S.L., Levine M.A. Analysis of the GCM2 gene in isolated hypoparathyroidism: a molecular and biochemical study. J. Clin. Endocrinol. Metab. 2008;93:1426–1432. doi: 10.1210/jc.2007-1783. [DOI] [PubMed] [Google Scholar]
- 18.Parkinson D.B., Thakker R.V. A donor splice site mutation in the parathyroid hormone gene is associated with autosomal recessive hypoparathyroidism. Nat. Genet. 1992;1:149–152. doi: 10.1038/ng0592-149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.