

Abstract
Megasatellites are large DNA tandem repeats, originally described in Candida glabrata, in protein-coding genes. Most of the genes in which megasatellites are found are of unknown function. In this work, we extended the search for megasatellites to 20 additional completely sequenced fungal genomes and extracted 216 megasatellites in 203 out of 142,121 genes, corresponding to the most exhaustive description of such genetic elements available today. We show that half of the megasatellites detected encode threonine-rich peptides predicted to be intrinsically disordered, suggesting that they may interact with several partners or serve as flexible linkers. Megasatellite motifs were clustered into several families. Their distribution in fungal genes shows that different motifs are found in orthologous genes and similar motifs are found in unrelated genes, suggesting that megasatellite formation or spreading does not necessarily track the evolution of their host genes. Altogether, these results suggest that megasatellites are created and lost during evolution of fungal genomes, probably sharing similar functions, although their primary sequences are not necessarily conserved.
INTRODUCTION
Tandem repeats are a common component of all eukaryotic genomes sequenced so far (1). Besides the ubiquitous presence of microsatellites and the frequent occurrence of minisatellites, megasatellites represent a new class of larger tandem repeats that were initially identified in yeast genomes (2). Megasatellites were defined as tandem repeats whose base motif is longer than 100 nucleotides (whereas minisatellite motifs seldom reach this size [3]), tandemly repeated at least three times (to distinguish them from local duplications), and inserted within protein-coding genes. They are frequent in the pathogenic yeast Candida glabrata, in which two large families, called “SHITT” and “SFFIT,” respectively (due to the conservation of these five amino acids within the motif), have been described in about 30 genes (2, 4). Another yeast genome, that of the well-studied baker's yeast Saccharomyces cerevisiae, contains eight tandem repeats that qualify as megasatellites: in the FLO1 (YAR050w), FLO5 (YHR211w), and FLO9 (YAL063c) paralogous genes encoding cell wall proteins involved in yeast cell flocculation; in FIT1 (YDR534c) and HPF1 (YOL155c), two other cell wall genes; in NUM1 (YDR150w), a cytoskeleton organization gene; and in YIL169c, a gene of unknown function sharing high similarity with HPF1 (YOL155c). The FLO1 megasatellite was experimentally shown to play a role in cell flocculation and adhesion, with longer repeats being associated with better adhesion and flocculation (5). Kluyveromyces lactis subtelomeric regions were shown to contain several genes encoding large tandem repeats (6), with four of them qualifying as megasatellites (KLLA0A11935g, KLLA0B14916g, KLLA0C19316g, and KLLA0D00264g) (see Table S1 in the supplemental material). There is no experimental evidence of their putative function in this yeast, but based on sequence similarity, they might be good candidates to be cell wall genes. The genome of Candida albicans, an opportunistic pathogenic yeast, contains eight ALS genes, each of them with 108-bp tandem motifs, corresponding to the megasatellite definition (7–9). The ALS genes are involved in adhesion of C. albicans to epithelial host cells by a mechanism involving binding to a large variety of ligands, including carbohydrates and peptides (8–10). Twenty-one different allele sizes have been found for the tandem array of the ALS7 gene (CAL0005421) among different C. albicans strains (11), but it is not known whether some of them are associated with higher adhesion. However, tandem repeats of ALS5 (CAAL5736) and ALS3 (CAAL1816) were shown to be important for yeast cellular adhesion to epithelial cells or to fibronectin (12, 13). Aspergillus fumigatus was also shown to contain large tandem repeats, some of them included in genes proposed to encode cell wall components (14), but none of these megasatellites was shown to be directly involved in cellular adhesion. Among other tandemly repeated motifs detectable by our analysis, WD repeats are a family of tandem arrays frequently encountered in eukaryotic genes, having a structural function in proteins involved in functions as diverse as RNA processing, transcription, cytoskeleton assembly, vesicle trafficking, cell division, or sulfur metabolism in fungi (reviewed in: reference 15). WD motifs contain two highly variable regions, separating more conserved domains; therefore, all the motifs of a given tandem repeat do not necessarily share the same size, although their final structures are very similar.
Previous intraspecific comparisons between paralogous megasatellite-containing genes showed that megasatellite motifs are under purifying selection and that this selection is stronger in C. glabrata than in S. cerevisiae (16). It was proposed that megasatellites propagate by three different mechanisms: (i) duplication of a megasatellite-containing gene, (ii) gene conversion between homologous sequences, and (iii) “jumping” of one or several motifs from one megasatellite-containing gene to another gene (16). However, besides the intraspecific analyses carried out in S. cerevisiae and C. glabrata, very few studies in yeast or other fungal species are available to extensively characterize and compare megasatellite distribution.
In order to do so, we developed a methodology, based on the Tandem Repeat Finder (TRF) program (17, 18), to analyze 21 Dikarya genomes covering a large phylogenetic spectrum (mostly Ascomycota). Our analysis covered 15 ascomycetous yeasts (from S. cerevisiae to Schizosaccharomyces pombe), five filamentous ascomycetes (Podospora anserina and four Aspergillus species), and one basidiomycete (Ustilago maydis) (Fig. 1 and Table 1) (19–33). Megasatellites were classified according to their motif sequence similarity and characterized by their corresponding amino acid composition. About half of them encoded peptides particularly enriched in threonine residues. In silico structure prediction of the peptides encoded by these megasatellites indicates that most of them are not structured, suggesting that they do not form stable structures in vivo. Finally, we demonstrate that large tandem repeats are constantly created (and sometimes lost) during evolution, suggesting a rather fast molecular mechanism(s) that creates new functions in each fungal lineage.
Fig 1.

Distribution of megasatellites in the 21 genomes studied. Left, tree topology (68, 69). Branch lengths are arbitrary. Motif families are represented by a color code. Motifs drawn on the tree indicate their proposed time of appearance during evolution, under a parsimony hypothesis. Right, protein clusters containing two or more proteins are represented by vertical columns. Nonunique motifs are indicated by their number in a black box (see Table S1 in the supplemental material). Unique motifs are shown in gray. P2.n, all clusters containing only two proteins.
Table 1.
Fungal species analyzed
| Species | Genome size, Mb (reference) | No. of: |
|
|---|---|---|---|
| Coding sequences | Megasatellites | ||
| Saccharomyces cerevisiae | 12,068 (19) | 5,862 | 7 |
| Candida glabrata | 12,280 (20) | 5,202 | 28 |
| Zygosaccharomyces rouxii | 9,765 (21) | 4,991 | 11 |
| Lachancea thermotolerans | 10,393 (21) | 5,092 | 8 |
| Lachancea kluyveri | 11,346 (21) | 5,321 | 9 |
| Kluyveromyces lactis | 10,631 (20) | 5,076 | 7 |
| Eremothecium gossypii | 8,765 (22) | 4,768 | 0 |
| Debaryomyces hansenii | 12,221 (20) | 6,272 | 10 |
| Millerozyma sorbitophila | 21,460 (23) | 11,252 | 6 |
| Scheffersomyces stipitis | 15,400 (24) | 5,816 | 4 |
| Candida albicans | 14,855 (25) | 6,112 | 8 |
| Candida dubliniensis | Partiala (26) | 5,860 | 15 |
| Komagataella pastoris | 9,430 (27) | 5,040 | 5 |
| Yarrowia lipolytica | 20,503 (20) | 6,448 | 15 |
| Schizosaccharomyces pombe | 12,463 (28) | 4,993 | 12 |
| Podospora anserina | Partial (29) | 10,219 | 19 |
| Aspergillus nidulans | 30,069 (30) | 10,701 | 17 |
| Aspergillus oryzae | 37,000 (31) | 12,074 | 19 |
| Aspergillus fumigatus | 27,981 (30) | 9,926 | 9 |
| Aspergillus clavatus | 29,400 (32) | 4,574 | 1 |
| Ustilago maydis | 20,500 (33) | 6,522 | 6 |
| Total | >326,530 | 142,121 | 216 |
Partial genome sequence; the genome size is therefore not precisely known.
MATERIALS AND METHODS
Megasatellite detection.
A database containing 142,121 annotated genes from 21 fungal genomes was built (Table 1), and the Tandem Repeat Finder (TRF) program (17) was used to extract all motifs from this set of genes, using the following parameters: match weight = 2, mismatch penalty = 7, insertion/deletion penalty = 7, match probability = 80, insertion/deletion probability = 10, minimum alignment score to report = 50, and maximum period size to report = 2,000. For each genome, the following iterative approach was used to determine the minimal size of tandemly repeated motifs. Gene sequences were searched using TRF. Motifs with sizes equal to or greater than 90 bp were further analyzed for possible inclusion of repeated submotifs until no repeated motif was found. Submotifs of less than 90 bp were discarded. The presence of each megasatellite was confirmed by constructing self dot plots of the corresponding protein (34) and by comparing each corresponding gene sequence versus itself using bl2seq and blastn (35). At this step, only motifs repeated at least three times were retained as megasatellites.
Extraction of peptides encoded by megasatellites.
Using the starting position of the megasatellite and its motif size (in nucleotides), each polypeptide was extracted from the translated gene sequence. Motif starting position and motif size were obtained by dividing by 3 the megasatellite starting position and motif size, as determined by TRF on the DNA sequence. Repeated polypeptides were further validated using bl2seq and blastp to compare the translated megasatellite to its corresponding polypeptide sequence. Manual inspection was often needed to find the precise border of each motif (computer programs are inefficient at finding the precise border of a tandem repeat, since any amino acid within the motif may be chosen as the beginning of the motif). In most cases, megasatellites correspond to tandemly directly repeated motifs, but some megasatellites are separated by amino acid segments varying from one to a few amino acids. Some megasatellites needed the insertion of one or two gaps to keep the periodicity of the tandem repeat, whereas in some cases, a few amino acids needed to be removed to keep the periodicity of the megasatellite. Each megasatellite was given a unique identification number, defined by its gene name (see Table S1 in the supplemental material). When a gene carries more than one megasatellite, each megasatellite within this gene bears an additional rank number (for example, CADU0C86150-1 and CADU0C86150-2 define the two megasatellites found in CADU0C86150).
Amino acid compositions and correspondence analysis.
Amino acid compositions were computed for translation products of each megasatellite, for the set of proteins from which megasatellites were precisely removed, and for the 142,121 proteins of the 21 studied proteomes. Correspondence analysis is a multivariate method for exploration of large numerical data tables. It allows the projection of high-dimensional information onto low-dimensional spaces. Visual inspection of such projections onto a plane allows the detection of significant trends, which are often difficult to grasp in high-dimensional spaces. The method builds an orthogonal system called factorial axes (F1, F2, F3, etc.), with each axis representing a fraction (displayed in decreasing order) of all of the information contained in the analyzed data table. The statistical significance of this fraction determines the relative confidence attached to the displayed axes (megasatellites or amino acids). The orthogonality of the factorial axes allows the summation of their corresponding fractions. The first factorial plane corresponding to the first (F1) and second (F2) factorial axes includes the highest fraction of the total information, obtained by summing the fractions corresponding to the first (F1) and to the second (F2) factorial axes. Note that megasatellites and amino acids are displayed simultaneously on each factorial plane, in such a way that neighborhood between megasatellites and amino acids is indicative of significant relationships. Conversely, greater distance between megasatellites and amino acids is indicative of weaker relationships. The methodology and its applications in a similar case have been extensively described by Tekaia and Yeramian (36).
Comparison and clustering of all megasatellites against themselves.
All peptidic motifs were compared to each other, using blastp (35). A blastp similarity score was considered significant when the corresponding E value was equal or lower than 10−2. Nonunique peptides (i.e., those having significant similarity with at least one other peptide) were classified into clusters using mcl (37) with “−log(blastp(e-value))” and an inflation index I of 3.0. Each nonunique peptide was assigned to a cluster denoted Mp.q (for motif clusters), with p the number of peptides contained and q an arbitrary index number (38). Peptides included in each of the determined clusters were aligned using the ClustalW program (39), and conserved blocks were determined using the Gblock program (40).
Comparison and clustering of all proteins against themselves.
All proteins, from which the tandemly repeated peptides were removed, were compared to each other, using blastp (35). A blastp similarity score was considered significant when the corresponding E value was equal to or lower than 10−9, as previously described (41). Nonunique proteins were clustered using the mcl (37) program with the same options as indicated above for the megasatellites and assigned to a cluster denoted Pp.q (with p being the number of proteins contained and q an arbitrary index number).
Motif consensus and structure.
When three or more megasatellites were found in a given family (ALS, FLO, SHITT, SFFIT, WD, etc.), motif consensus were determined by alignment of all the motifs using the Jalview program (42). All motifs sequences are given in Table S1 in the supplemental material. Subsequently, each of the motifs (or motif consensus) was analyzed using the metaserver MeDor (43) in order to determine whether any part of the motif was predicted to be disordered. The eight motifs that were predicted probably not to be disordered (see Results) were compared to the Protein Data Bank (PDB) (44) in order to determine if their structures were already known. In addition, megasatellite-containing proteins and peptidic motifs were also compared to the Conserved Domain Database (CDD) (45) version 3.02 (December 2011), including 40,815 domain sequences. Motif families (ALS, FLO, SHITT, SFFIT, WD, etc.) were also compared to several databases of known motifs (PROSITE, BLOCKS, ProDom, PRINTS, and Pfam) using the motif analysis tool found at http://www.genome.jp/tools/motif/.
RESULTS
Fungal genomes contain a large diversity of megasatellites.
We have determined the complete set of tandem repeats detected in a total of 142,121 sequence-predicted protein-coding genes belonging to 21 fungal genomes (Dikarya) (Table 1). Out of more than 13,000 tandem repeats, we extracted 216 megasatellites (see Materials and Methods). The number of megasatellites detected ranges from 28 in Candida glabrata to none in Eremothecium gossypii and is correlated neither to genome size nor to gene content (Table 1). Motif sizes range from 90 bp (9 megasatellites in 5 species) to 735 bp (one megasatellite in Yarrowia lipolytica, YALI0B09867g). As expected for tandem repeats located within protein-coding genes, all motif sizes found are multiples of three nucleotides.
The most common motif found is the FLO motif, which was encountered in 35 megasatellites in 11 species, from S. cerevisiae to Komagataella pastoris, making it the most widespread of all megasatellite motifs. This motif encodes a Thr/Ser-rich sequence, often containing the Trp-Thr-Gly tripeptide (see Table S1 in the supplemental material). The FLO motif size is highly variable, ranging from 90 bp to 150 bp. By comparison, other frequent motifs, such as ALS, tetratricopeptide repeat (TPR), or WD, all share the same size (108 bp for ALS and 126 bp for TPR and WD). ALS motifs are only found in C. albicans and Candida dubliniensis, in eponymous genes and their homologues. TPR motifs occur in tandem arrays in more than 800 genes, from bacteria to humans. The motif corresponds to two antiparallel alpha helices separated by a turn (46). Megasatellites containing TPR motifs are particularly frequent in Aspergillus nidulans and P. anserina. WD repeats are generally encountered in proteins belonging to the whole eukaryotic world (15); however, in the present study, they were detected only in filamentous fungi (P. anserina and Aspergillus species).
Megasatellites found in C. glabrata and containing SHITT and SFFIT motifs were previously described and are widely spread in this genome (2, 4, 16). SHITT and SFFIT are sometimes encountered as motifs of slightly different sizes, suggesting that, like for FLO motifs, their containing proteins may accommodate some tandem repeat flexibility. Note that despite similar motif sizes, there is no detectable homology between SHITT and FLO motifs, suggesting that SHITT motifs either were de novo created in C. glabrata or rapidly evolved from an ancestral sequence.
Smaller families were also detected, such as the ankyrin (ANK) family, found within five megasatellites in Aspergillus orizae, A. fumigatus, and P. anserina, or the TTITL family, found in two megasatellites in C. glabrata. Ankyrin repeats consist of two alpha helices separated by loops and are involved in protein-protein interactions (47). Nothing is known about the structure or function of TTITL motifs. In addition to these, 21 other motifs belonging to small families (2 or 3 members) were found, and 79 other motifs did not share any detectable homology (Fig. 2).
Fig 2.

Distribution of megasatellites according to motif size. Upper panel, total number of megasatellites for each motif size. Lower panel, total number of megasatellites in each species, classified by families. The color code is the same in both panels.
Amino acid compositions of megasatellites.
The amino acid compositions of all translated peptidic motifs were computed. Compared to the average amino acid composition of more than 60 million amino acids making altogether the 21 proteomes, Thr and Ser are often overrepresented, whereas Leu, Arg, Lys, Met, and Gln are often underrepresented. Correspondence analysis was used to determine possible amino acid composition biases of megasatellites (36). Megasatellites are displayed mostly in two groups along the first factorial axis (Fig. 3, horizontal axis F1, covering 27% of the total information in the analyzed data). One group (left) is characterized by high composition biases in Thr and by underrepresentation of Arg, Leu, Lys, and Gln. The second group (right) is characterized by high composition biases in Leu, Lys, Arg, and Gln and underrepresentation of Thr. Therefore, megasatellite composition according to the first axis is directly correlated with threonine content. It is interesting to note that few megasatellites are characterized by average compositions (few are plotted close to the axis origin), meaning that most of them exhibit biased amino acid composition. Note that FLO, ALS, SHITT, SFFIT, and TTITL are Thr rich, whereas ANK, WD, and TPR repeats are Thr poor.
Fig 3.

Correspondence analysis showing the distribution of megasatellites (blue dots) according to the 20 amino acids on the first factorial plane. F1 and F2 are the first and second factorial axes and represent, respectively, 27% and 11% of the total information included in the analyzed data table (observed megasatellites versus their amino acid composition).
Structure prediction for megasatellite peptidic motifs.
Primary sequences of peptidic motifs encoded by megasatellites are generally not conserved, despite the existence of families as described above. This, however, does not exclude the possibility that common secondary structures exist. To address this question, several secondary structure and disorder predictors were used on each motif (see Materials and Methods). Out of 97 different peptidic motifs analyzed, 88 show an extensive level of disorder (50 to 100% of the motif) and no obvious secondary structure (data not shown). The eight remaining motifs, showing lower levels of disorder, were compared to the Protein Data Bank (PDB), and seven of them (all threonine-poor motifs) were found to correspond to known secondary structures. Motifs M8 (in PODANSg6698 and AN8019), M10 (in AN3543, AN8085 and AO090166000058), M21 (in PODANSg8665), and M56 (in AO090102000421) all correspond to ankyrin motifs, a common repeat in eukaryotic proteins, but also found in bacteria and archaea (PDB ID 2L6B). It is interesting to note that primary sequence similarity with the ANK motif described above in other megasatellites (Fig. 2) was not detected, but only the three-dimensional (3D) structure predicted that these four motifs should share the tertiary structure of ankyrin repeats. M69 (in ZYRO0G06028g) has a match in PDB with the structure of a carbohydrate epimerase from Pseudomonas aeruginosa (PDB ID 2IXI), hence linking this motif to carbohydrate metabolism (48). Finally, M49 (in AO090009000369) corresponds to a putative l-allo-threonine aldolase from Listeria monocytogenes (PDB ID 3PJ0).
In addition, each megasatellite peptidic motif was compared to the CDD (45). Only eight significant hits were detected, corresponding to ankyrin repeats, WD repeats (twice), TPR repeats, the cohesin-HEAT domain (associated with chromosome cohesion and condensation), DUF3659 (a 70-amino-acid domain of unknown function found in bacteria and eukaryotes), a putative 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase domain, and a deoxyhypusine synthase domain essential for translation initiation in eukaryotes. The overall conclusion is, therefore, that most peptidic motifs (88/97, ca. 91%) encoded by megasatellites are disordered and unstructured.
Formation and propagation of megasatellites during evolution.
One of the main questions of the present work was to determine whether megasatellite motifs were species specific or lineage specific or whether they were distributed randomly, suggesting a possible propagation among fungi. Our results very clearly show that both happened during fungal evolution. SFFIT, SHITT, and TTITL motifs are restricted to C. glabrata (Fig. 1). The FLO motif is widespread in all hemiascomycetous yeasts, from S. cerevisiae to Y. lipolytica, although it is found in seven different clusters (see Table S1 in the supplemental material). Some FLO motifs are shorter, on average, than others (Fig. 2), but despite these size discrepancies, there is little doubt that both “short” and “long” FLO motifs, recognizable by their Trp-Thr-Gly tripeptide, come from a common ancestor. The ALS motif seems to be restricted to C. albicans and C. dubliniensis, and the WD, TPR, and ANK motifs are themselves restricted to the branch leading to Pezizomycotina (P. anserina to Aspergillus species). Thr-rich and -poor motifs are widespread among all fungi. No evidence for a case of horizontal gene transfer between two distant fungal species could be detected (49).
Subsequently, megasatellites were extracted from their containing genes and translated, and peptidic motif families were compared to protein families after megasatellite extraction (see Materials and Methods). Some large protein families were found, such as P18.1, found exclusively in P. anserina and Aspergillus species. Most of its members contain a TPR motif, but three of them contain a unique motif (M21, M22, and M23) (see Table S1 in the supplemental material). P6.1 and P4.1 contain genes that carry only the FLO motif, whereas P5.1 contains orthologous genes carrying only the WD motif (Fig. 1). All the other clusters contain at least two different kinds of motifs. Hence, similar megasatellites may be found in nonhomologous genes (Fig. 4), whereas orthologous genes often carry different megasatellites (Fig. 5).
Fig 4.
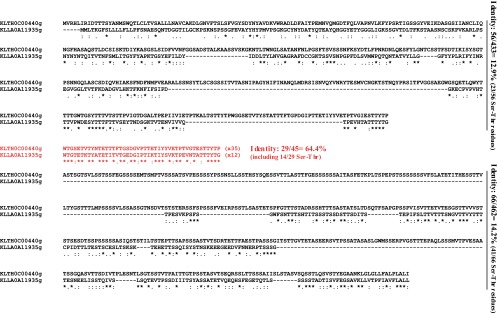
Example of similar megasatellites in two nonhomologous genes. Alignment of KLTH0C00440g and KLLA0A11935g translation products, two proteins belonging to two different clusters (P6.3 and P8.1, respectively) and containing the same peptidic motif (FLO, motif cluster M17.1 [see Table S1 in the supplemental material]), is shown. The peptidic motif is shown in red, along with the number of repeats in each protein. The N-terminal and C-terminal parts of both proteins exhibit little identity (12.9% and 14.2%, respectively), with most of the identical amino acids being serine and threonine residues, due to the compositional bias of both proteins. In comparison, both FLO motifs are very similar, despite a comparable compositional bias.
Fig 5.

Example of different megasatellites in two homologous genes. Alignment of PODANSg8665 and AN1071 translation products, two homologous proteins (P18.1) containing different megasatellite motifs, is shown. Both proteins show very similar N-terminal parts (42.6% identity) followed by less conserved regions (12.3% identity) containing the repeated peptides. There is no homology between the peptidic motifs.
DISCUSSION
In the present work, we present the first exhaustive comparative genomic analysis of megasatellite distribution in the genomes of 21 fungi. Peptidic motifs encoded by megasatellites were extracted from their containing proteins and compared to each other. Using the present approach, only megasatellites whose motifs have similar lengths can be detected. Therefore, none of the 55 WD repeats encoded by the S. cerevisiae genome was detected, since motif lengths are very different from each other.
Megasatellite-containing genes show hallmarks of plasma membrane or cell wall genes.
We have identified 216 megasatellites spread in 18 different families and 79 unique megasatellites, with half of those encoding proteins including motifs enriched in threonine residues. In S. cerevisiae cell wall proteins, such residues are sites of O mannosylations, which occur in the endoplasmic reticulum and are essential for localization of such proteins at the cell surface (50, 51). FLO1 (YAR050w), FLO5 (YHR211w), and FLO9 (YAL063c) encode flocculins involved in cell-to-cell adhesion and yeast flocculation (52), HPF1 (YOL155c) encodes a surface mannoprotein involved in protein aggregates in white wine fermentation, and NUM1 (YDR150w) and FIT1 (YDR534c) encode a cytoskeleton organization protein and a glycosylphosphatidylinositol (GPI) anchor-containing cell wall protein, respectively. Megasatellites are also found in adhesins in C. albicans (ALS1 to ALS7 and ALS9 genes) (9) and in C. glabrata (EPA1, EPA2, and EPA13 genes) (53, 54). In the well-studied model organism Schizosaccharomyces pombe, megasatellites are detected in the MAP2 P-factor pheromone gene (SPCC1795.06), the MAP4 gene (SPBC21D10.06c) (encoding an adhesin required for mating), and the MAM3 gene (SPAP11E10.02c) (involved in cell-to-cell adhesion). Comparison of peptide motifs to databases of known motifs shows the presence of possible phosphorylation and glycosylation sites found in flocculins, as well as putative sites of myristoylation. Addition of myristate (a 14-carbon fatty acid) to proteins is a common posttranslational modification of proteins generally associated with the plasma membrane and/or involved in signal cascades. The myristoyl part of the protein is directly involved in the interaction with membrane lipids, in a reversible manner, helping to localize the protein at the plasma membrane (55). It is therefore tempting to propose that megasatellites encoding such Thr-rich motifs belong to genes encoding proteins localized at the plasma membrane and/or cell wall and involved, directly or indirectly, in cell adhesion. However, there is no information about the function of megasatellite-containing genes except for the handful described above.
Possible function(s) of megasatellites in fungal genes.
The function of the megasatellite itself is puzzling. In S. cerevisiae, cell flocculation and adhesion to plasticware were correlated to the size of the FLO1 megasatellite (5), but there is no experimental evidence that this is also the case for its two paralogues, FLO5 and FLO9. In C. albicans, adhesion assays show reduced adhesiveness for strains with an ALS3 allele containing only nine motifs, compared to 12 (12), suggesting a role in adhesion for the megasatellite. Given that the number of allelic lengths of a megasatellite may be quite large (for example, 21 different lengths of the ALS7 megasatellite were found in patients infected with C. albicans [11]), megasatellite polymorphism offers the opportunity to modulate adhesion of such yeasts to their substrate. Finally, when EPA1 is expressed in C. glabrata or S. cerevisiae, adhesion to epithelial cells is partly dependent on the presence of its megasatellite (56). However, it is not known if the same holds true for other megasatellite-containing EPA genes.
The molecular mechanism by which peptidic motifs encoded by megasatellites modulate adhesion is unclear, but it was suggested that they may serve as variable spacers between the N-terminal part (bearing the binding domain) and the C-terminal part (anchored to the cell wall) of the protein. This spacer needs to reach a given length in order to properly expose the N-terminal ligand-binding domain to the cell surface (9, 10, 56). It was also proposed (10, 14) that the high variability of megasatellites would help pathogens to escape the host immune system by modifying their surface antigens. A similar strategy, based on the activation/inactivation of cell wall genes by small tandem repeat size changes and called “phase variation,” is extensively used by some human bacterial pathogens such as Haemophilus influenzae (57, 58) and Neisseria meningitidis (59–62). Similarly, the Mycobacterium tuberculosis genome contains two large families of proteins of unknown function called PE and PPE proteins; both have a disordered C-terminal domain made of tandemly repeated Pro-Glu or Pro-Pro-Glu motifs, which have been suggested to be a source of antigenic variation (63, 64).
It is commonly believed now that about 40% of human proteins contain long intrinsically disordered regions and that some 25% are probably disordered from beginning to end (65, 66). Predictions based on amino acid composition (43) suggest that 88 out of our 97 different megasatellite motifs (91%) are partially or fully disordered, a higher proportion than is commonly found for all eukaryotic proteins, suggesting that intrinsically disordered domains are a hallmark of megasatellites.
Formation and loss of megasatellites during evolution.
The relative distribution of megasatellites in fungi varies among species. For example, TPR repeats were found only in the branch leading to filamentous fungi (Pezizomycotina), whereas the FLO motif was only detected in Saccharomycotina (Fig. 1). These motifs show very different amino acid compositions (FLO motifs contain 35.5% Thr residues, while TPR motifs contain 7.1% Thr residues), and no sequence homology could be detected between them. Therefore, the most parsimonious hypothesis is that FLO and TPR repeats do not share a ancestor. The same holds true for other motif families, suggesting that megasatellites belonging to different families are created and lost during evolution of fungal genomes.
In a comparative analysis of intragenic tandem repeats among 10 Aspergillus genomes, it was concluded that such repeat sequences were highly variable (only 21% of intragenic tandem repeats found in a given species were also detected in another one) and that repeat-containing proteins were less conserved than other proteins (67). In another study, comparisons of SHITT and SFFIT motifs in C. glabrata led the authors to propose a new mechanism, tentatively called “motif jump,” to explain the presence of motifs belonging to a given family within a megasatellite-containing gene belonging to another family (16). Here, we can detect similar events occurring between nonorthologous gene families. For instance, FLO or WD repeats are found encoded by genes sharing no detectable homology, grouped in eight different protein clusters and two single proteins for FLO motifs and in three different protein clusters and four single proteins for WD motifs (see Table S1 in the supplemental material). To account for this observation, it may be proposed that the same megasatellite is recreated in different genes or that a megasatellite (or a discrete number of motifs) may “jump” from its original gene to another one, as proposed for SHITT and SFFIT motifs in C. glabrata. Alternatively, one may also propose that purifying selection operates more efficiently on megasatellites than on their containing genes, hence maintaining the same tandem repeat within genes that will eventually diverge to the point that any similarity between them will be erased. In support of the last hypothesis is the fact that megasatellite motifs in C. glabrata were found to be under a stronger purifying selection than their containing genes (16). Experiments aimed at determining how megasatellites appear and propagate within fungal genes are now needed to properly address this question.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by ANR blanc 2011 (DYGEVO) and by Institut Pasteur (PTR370 to F.T.).
We are grateful to M. Delepierre for advice on structure prediction for megasatellite motifs and to L. Chatre for pointing out to us the importance of myristoylation in membrane protein metabolism.
Footnotes
Published ahead of print 29 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00001-13.
REFERENCES
- 1. Richard GF, Kerrest A, Dujon B. 2008. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol. Mol. Biol. Rev. 72:686–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thierry A, Bouchier C, Dujon B, Richard G-F. 2008. Megasatellites: a peculiar class of giant minisatellites in genes involved in cell adhesion and pathogenicity in Candida glabrata. Nucleic Acids Res. 36:5970–5982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vergnaud G, Denoeud F. 2000. Minisatellites: mutability and genome architecture. Genome Res. 10:899–907 [DOI] [PubMed] [Google Scholar]
- 4. Thierry A, Dujon B, Richard GF. 20010. Megasatellites: a new class of large tandem repeats discovered in the pathogenic yeast Candida glabrata. Cell. Mol. Life Sci. 67:671–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Verstrepen KJ, Jansen A, Lewitter F, Fink GR. 2005. Intragenic tandem repeats generate functional variability. Nat. Genet. 37:986–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fairhead C, Dujon B. 2006. Structure of Kluyveromyces lactis subtelomeres: duplications and gene content. FEMS Yeast Res. 6:428–441 [DOI] [PubMed] [Google Scholar]
- 7. Nather K, Munro CA. 2008. Generating cell surface diversity in Candida albicans and other fungal pathogens. FEMS Microbiol. Lett. 285:137–145 [DOI] [PubMed] [Google Scholar]
- 8. Hoyer LL. 2001. The ALS gene family of Candida albicans. Trends Microbiol. 9:176–180 [DOI] [PubMed] [Google Scholar]
- 9. Lipke PN, Garcia MC, Alsteens D, Ramsook CB, Klotz SA, Dufrene YF. 2012. Strengthening relationships: amyloids create adhesion nanodomains in yeasts. Trends Microbiol. 20:59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Levdansky E, Sharon H, Osherov N. 2008. Coding fungal tandem repeats as generators of fungal diversity. Fungal Biol. Rev. doi: 10.106/j.fbr.2008.2008.2001 [DOI] [Google Scholar]
- 11. Zhang N, Harrex AL, Holland BR, Fenton LE, Cannon RD, Schmid J. 2003. Sixty alleles of the ALS7 open reading frame in Candida albicans: ALS7 is a hypermutable contingency locus. Genome Res. 13:2005–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oh SH, Cheng G, Nuessen JA, Jajko R, Yeater KM, Zhao X, Pujol C, Soll DR, Hoyer LL. 2005. Functional specificity of Candida albicans Als3p proteins and clade specificity of ALS3 alleles discriminated by the number of copies of the tandem repeat sequence in the central domain. Microbiology 151:673–681 [DOI] [PubMed] [Google Scholar]
- 13. Rauceo JM, De Armond R, Otoo H, Kahn PC, Klotz SA, Gaur NK, Lipke PN. 2006. Threonine-rich repeats increase fibronectin binding in the Candida albicans adhesin Als5p. Eukaryot. Cell 5:1664–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Levdansky E, Romano J, Shadkchan Y, Sharon H, Verstrepen KJ, Fink GR, Osherov N. 2007. Coding tandem repeats generate diversity in Aspergillus fumigatus genes. Eukaryot. Cell 6:1380–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smith TF, Gaitatzes C, Saxena K, Neer EJ. 1999. The WD repeat: a common architecture for diverse functions. Trends Biochem. Sci. 24:181–185 [DOI] [PubMed] [Google Scholar]
- 16. Rolland T, Dujon B, Richard GF. 2010. Dynamic evolution of megasatellites in yeasts. Nucleic Acids Res. 38:4731–4739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27:573–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Benson G, Waterman MS. 1994. A method for fast database search for all k-nucleotide repeats. Nucleic Acids Res. 22:4828–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG. 1996. Life with 6000 genes. Science 274:546–567 [DOI] [PubMed] [Google Scholar]
- 20. Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, De Montigny J, Marck C, Neuveglise C, Talla E, Goffard N, Frangeul L, Aigle M, Anthouard V, Babour A, Barbe V, Barnay S, Blanchin S, Beckerich JM, Beyne E, Bleykasten C, Boisrame A, Boyer J, Cattolico L, Confanioleri F, De Daruvar A, Despons L, Fabre E, Fairhead C, Ferry-Dumazet H, Groppi A, Hantraye F, Hennequin C, Jauniaux N, Joyet P, Kachouri R, Kerrest A, Koszul R, Lemaire M, Lesur I, Ma L, Muller H, Nicaud JM, Nikolski M, Oztas S, Ozier-Kalogeropoulos O, Pellenz S, Potier S, Richard GF, Straub ML, et al. 2004. Genome evolution in yeasts. Nature 430:35–44 [DOI] [PubMed] [Google Scholar]
- 21. Souciet JL, Dujon B, Gaillardin C, Johnston M, Baret PV, Cliften P, Sherman DJ, Weissenbach J, Westhof E, Wincker P, Jubin C, Poulain J, Barbe V, Segurens B, Artiguenave F, Anthouard V, Vacherie B, Val ME, Fulton RS, Minx P, Wilson R, Durrens P, Jean G, Marck C, Martin T, Nikolski M, Rolland T, Seret ML, Casaregola S, Despons L, Fairhead C, Fischer G, Lafontaine I, Leh V, Lemaire M, de Montigny J, Neuveglise C, Thierry A, Blanc-Lenfle I, Bleykasten C, Diffels J, Fritsch E, et al. 2009. Comparative genomics of protoploid Saccharomycetaceae. Genome Res. 19:1696–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dietrich FS, Voegeli S, Brachat S, Lerch A, Gates K, Steiner S, Mohr C, Pohlmann R, Luedi P, Choi S, Wing RA, Flavier A, Gaffney TD, Philippsen P. 2004. The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science 304:304–307 [DOI] [PubMed] [Google Scholar]
- 23. Leh Louis V, Despons L, Friedrich A, Martin T, Durrens P, Casarégola S, Neuvéglise C, Fairhead C. 2012. Pichia sorbitophila, an interspecies yeast hybrid, reveals early steps of genome resolution after polyploidization. Genes Genomes Genet. 2:299–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jeffries TW, Grigoriev IV, Grimwood J, Laplaza JM, Aerts A, Salamov A, Schmutz J, Lindquist E, Dehal P, Shapiro H, Jin YS, Passoth V, Richardson PM. 2007. Genome sequence of the lignocellulose-bioconverting and xylose-fermenting yeast Pichia stipitis. Nat. Biotechnol. 25:319–326 [DOI] [PubMed] [Google Scholar]
- 25. Jones T, Federspiel NA, Chibana H, Dungan J, Kalman S, Magee BB, Newport G, Thorstenson YR, Agabian N, Magee PT, Davis RW, Scherer S. 2004. The diploid genome sequence of Candida albicans. Proc. Natl. Acad. Sci. U. S. A. 101:7329–7334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jackson AP, Gamble JA, Yeomans T, Moran GP, Saunders D, Harris D, Aslett M, Barrell JF, Butler G, Citiulo F, Coleman DC, de Groot PW, Goodwin TJ, Quail MA, McQuillan J, Munro CA, Pain A, Poulter RT, Rajandream MA, Renauld H, Spiering MJ, Tivey A, Gow NA, Barrell B, Sullivan DJ, Berriman M. 2009. Comparative genomics of the fungal pathogens Candida dubliniensis and Candida albicans. Genome Res. 19:2231–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Schutter K, Lin YC, Tiels P, Van Hecke A, Glinka S, Weber-Lehmann J, Rouze P, Van de Peer Y, Callewaert N. 2009. Genome sequence of the recombinant protein production host Pichia pastoris. Nat. Biotechnol. 27:561–566 [DOI] [PubMed] [Google Scholar]
- 28. Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, Sgouros J, Peat N, Hayles J, Baker S, Basham D, Bowman S, Brooks K, Brown D, Brown S, Chillingworth T, Churcher C, Collins M, Connor R, Cronin A, Davis P, Feltwell T, Fraser A, Gentles S, Goble A, Hamlin N, Harris D, Hidalgo J, Hodgson G, Holroyd S, Hornsby T, Howarth S, Huckle EJ, Hunt S, Jagels K, James K, Jones L, Jones M, Leather S, McDonald S, McLean J, Mooney P, Moule S, Mungall K, Murphy L, Niblett D, Odell C, Oliver K, O'Neil S, Pearson D, Quail MA, Rabbinowitsch E, Rutherford K, et al. 2002. The genome sequence of Schizosaccharomyces pombe. Nature 415:871–880 [DOI] [PubMed] [Google Scholar]
- 29. Espagne E, Lespinet O, Malagnac F, Da Silva C, Jaillon O, Porcel BM, Couloux A, Aury JM, Segurens B, Poulain J, Anthouard V, Grossetete S, Khalili H, Coppin E, Dequard-Chablat M, Picard M, Contamine V, Arnaise S, Bourdais A, Berteaux-Lecellier V, Gautheret D, de Vries RP, Battaglia E, Coutinho PM, Danchin EG, Henrissat B, Khoury RE, Sainsard-Chanet A, Boivin A, Pinan-Lucarre B, Sellem CH, Debuchy R, Wincker P, Weissenbach J, Silar P. 2008. The genome sequence of the model ascomycete fungus Podospora anserina. Genome Biol. 9:R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Galagan JE, Calvo SE, Cuomo C, Ma LJ, Wortman JR, Batzoglou S, Lee SI, Basturkmen M, Spevak CC, Clutterbuck J, Kapitonov V, Jurka J, Scazzocchio C, Farman M, Butler J, Purcell S, Harris S, Braus GH, Draht O, Busch S, D'Enfert C, Bouchier C, Goldman GH, Bell-Pedersen D, Griffiths-Jones S, Doonan JH, Yu J, Vienken K, Pain A, Freitag M, Selker EU, Archer DB, Penalva MA, Oakley BR, Momany M, Tanaka T, Kumagai T, Asai K, Machida M, Nierman WC, Denning DW, Caddick M, Hynes M, Paoletti M, Fischer R, Miller B, Dyer P, Sachs MS, Osmani SA, Birren BW. 2005. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 438:1105–1115 [DOI] [PubMed] [Google Scholar]
- 31. Machida M, Asai K, Sano M, Tanaka T, Kumagai T, Terai G, Kusumoto K, Arima T, Akita O, Kashiwagi Y, Abe K, Gomi K, Horiuchi H, Kitamoto K, Kobayashi T, Takeuchi M, Denning DW, Galagan JE, Nierman WC, Yu J, Archer DB, Bennett JW, Bhatnagar D, Cleveland TE, Fedorova ND, Gotoh O, Horikawa H, Hosoyama A, Ichinomiya M, Igarashi R, Iwashita K, Juvvadi PR, Kato M, Kato Y, Kin T, Kokubun A, Maeda H, Maeyama N, Maruyama J, Nagasaki H, Nakajima T, Oda K, Okada K, Paulsen I, Sakamoto K, Sawano T, Takahashi M, Takase K, Terabayashi Y, Wortman JR, Yamada O, et al. 2005. Genome sequencing and analysis of Aspergillus oryzae. Nature 438:1157–1161 [DOI] [PubMed] [Google Scholar]
- 32. Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS, Arroyo J, Berriman M, Abe K, Archer DB, Bermejo C, Bennett J, Bowyer P, Chen D, Collins M, Coulsen R, Davies R, Dyer PS, Farman M, Fedorova N, Feldblyum TV, Fischer R, Fosker N, Fraser A, Garcia JL, Garcia MJ, Goble A, Goldman GH, Gomi K, Griffith-Jones S, Gwilliam R, Haas B, Haas H, Harris D, Horiuchi H, Huang J, Humphray S, Jimenez J, Keller N, Khouri H, Kitamoto K, Kobayashi T, Konzack S, Kulkarni R, Kumagai T, Lafon A, Latge JP, Li W, Lord A, Lu C, Majoros WH, May GS, Miller BL, et al. 2005. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438:1151–1156 [DOI] [PubMed] [Google Scholar]
- 33. Kamper J, Kahmann R, Bolker M, Ma LJ, Brefort T, Saville BJ, Banuett F, Kronstad JW, Gold SE, Muller O, Perlin MH, Wosten HA, de Vries R, Ruiz-Herrera J, Reynaga-Pena CG, Snetselaar K, McCann M, Perez-Martin J, Feldbrugge M, Basse CW, Steinberg G, Ibeas JI, Holloman W, Guzman P, Farman M, Stajich JE, Sentandreu R, Gonzalez-Prieto JM, Kennell JC, Molina L, Schirawski J, Mendoza-Mendoza A, Greilinger D, Munch K, Rossel N, Scherer M, Vranes M, Ladendorf O, Vincon V, Fuchs U, Sandrock B, Meng S, Ho EC, Cahill MJ, Boyce KJ, et al. 2006. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444:97–101 [DOI] [PubMed] [Google Scholar]
- 34. Sonnhammer EL, Durbin R. 1995. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 167:GC1–GC10 [DOI] [PubMed] [Google Scholar]
- 35. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tekaia F, Yeramian E. 2006. Evolution of proteomes: fundamental signatures and global trends in amino acid compositions. BMC Genomics 7:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Enright AJ, Van Dongen S, Ouzounis CA. 2002. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30:1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tekaia F, Yeramian E. 2012. SuperPartitions: detection and classification of orthologs. Gene 492:199–211 [DOI] [PubMed] [Google Scholar]
- 39. Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17:540–552 [DOI] [PubMed] [Google Scholar]
- 41. Tekaia F, Dujon B. 1999. Pervasiveness of gene conversion and persistence of duplicates in cellular genomes. J. Mol. Evol. 49:591–600 [DOI] [PubMed] [Google Scholar]
- 42. Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. 2009. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lieutaud P, Canard B, Longhi S. 2008. MeDor: a metaserver for predicting protein disorder. BMC Genomics 9(Suppl. 2):S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. 2011. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39:D225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I. 2000. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 101:199–210 [DOI] [PubMed] [Google Scholar]
- 47. Mosavi LK, Minor DL, Jr, Peng ZY. 2002. Consensus-derived structural determinants of the ankyrin repeat motif. Proc. Natl. Acad. Sci. U. S. A. 99:16029–16034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dong C, Major LL, Srikannathasan V, Errey JC, Giraud MF, Lam JS, Graninger M, Messner P, McNeil MR, Field RA, Whitfield C, Naismith JH. 2007. RmlC, a C3′ and C5′ carbohydrate epimerase, appears to operate via an intermediate with an unusual twist boat conformation. J. Mol. Biol. 365:146–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rolland T, Neuveglise C, Sacerdot C, Dujon B. 2009. Insertion of horizontally transferred genes within conserved syntenic regions of yeast genomes. PLoS One 4:e6515 doi: 10.1371/journal.pone.0006515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ecker M, Mrsa V, Hagen I, Deutzmann R, Strahl S, Tanner W. 2003. O-mannosylation precedes and potentially controls the N-glycosylation of a yeast cell wall glycoprotein. EMBO Reports. 4:628–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Latgé J-P, Calderone R. 2005. The fungal cell wall, p 73–104 In Esser K, Fischer R. (ed), The Mycota XIII. Springer, Berlin, Germany. [Google Scholar]
- 52. Klis FM, Mol Hellingwerf PK, Brul S. 2002. Dynamics of cell wall structure in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 26:239–256 [DOI] [PubMed] [Google Scholar]
- 53. Cormack BP, Ghori N, Falkow S. 1999. An adhesin of the yeast pathogen Candida glabrata mediating adherence to human epithelial cellls. Science 285:578–582 [DOI] [PubMed] [Google Scholar]
- 54. De Las Penas A, Pan SJ, Castano I, Alder J, Cregg R, Cormack BP. 2003. Virulence-related surface glycoproteins in the yeast pathogen Candida glabrata are encoded in subtelomeric clusters and subject to RAP1- and SIR-dependent transcriptional silencing. Genes Dev. 17:2245–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wright MH, Heal WP, Mann DJ, Tate EW. 2010. Protein myristoylation in health and disease. J. Chem. Biol. 3:19–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Frieman MB, McCaffery JM, Cormack BP. 2002. Modular domain structure in the Candida glabrata adhesin Epa1p, a β1,6 glucan-cross-linked cell wall protein. Mol. Microbiol. 46:479–492 [DOI] [PubMed] [Google Scholar]
- 57. Bayliss CD, Field D, Moxon ER. 2001. The simple sequence contingency loci of Haemophilus influenzae and Neisseria meningitidis. J. Clin. Invest. 107:657–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bayliss CD, van de Ven T, Moxon ER. 2002. Mutations in polI but not mutSLH destabilize Haemophilus influenzae tetranucleotide repeats. EMBO J. 21:1465–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Martin P, van de Ven T, Mouchel N, Jeffries AC, Hood DW, Moxon ER. 2003. Experimentally revised repertoire of putative contingency loci in Neisseria meningitidis strain MC58: evidence for a novel mechanism of phase variation. Mol. Microbiol. 50:245–257 [DOI] [PubMed] [Google Scholar]
- 60. Saunders NJ, Jeffries AC, Peden JF, Hood DW, Tettelin H, Rappuoli R, Moxon ER. 2000. Repeat-associated phase variable genes in the complete genome sequence of Neisseria meningitidis strain MC58. Mol. Microbiol. 37:207–215 [DOI] [PubMed] [Google Scholar]
- 61. Snyder LA, Butcher SA, Saunders NJ. 2001. Comparative whole-genome analyses reveal over 100 putative phase-variable genes in the pathogenic Neisseria spp. Microbiology 147:2321–2332 [DOI] [PubMed] [Google Scholar]
- 62. Tettelin H, Saunders NJ, Heidelberg J, Jeffries AC, Nelson KE, Eisen JA, Ketchum KA, Hood DW, Peden JF, Dodson RJ, Nelson WC, Gwinn ML, DeBoy R, Peterson JD, Hickey EK, Haft DH, Salzberg SL, White O, Fleischmann RD, Dougherty BA, Mason T, Ciecko A, Parksey DS, Blair E, Cittone H, Clark EB, Cotton MD, Utterback TR, Khouri H, Qin H, Vamathevan J, Gill J, Scarlato V, Masignani V, Pizza M, Grandi G, Sun L, Smith HO, Fraser CM, Moxon ER, Rappuoli R, Venter JC. 2000. Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science 287:1809–1815 [DOI] [PubMed] [Google Scholar]
- 63. Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544 [DOI] [PubMed] [Google Scholar]
- 64. Tekaia F, Gordon SV, Garnier T, Brosch R, Barrell BG, Cole ST. 1999. Analysis of the proteome of Mycobacterium tuberculosis in silico. Tuber. Lung Dis. 79:329–342 [DOI] [PubMed] [Google Scholar]
- 65. Uversky VN, Dunker AK. 2010. Understanding protein non-folding. Biochim. Biophys. Acta 1804:1231–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Uversky VN. 2011. Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 43:1090–1103 [DOI] [PubMed] [Google Scholar]
- 67. Gibbons JG, Rokas A. 2009. Comparative and functional characterization of intragenic tandem repeats in 10 Aspergillus genomes. Mol. Biol. Evol. 26:591–602 [DOI] [PubMed] [Google Scholar]
- 68. Dujon B. 2010. Yeast evolutionary genomics. Nat. Rev. Genet. 11:512–524 [DOI] [PubMed] [Google Scholar]
- 69. Wang H, Xu Z, Gao L, Hao B. 2009. A fungal phylogeny based on 82 complete genomes using the composition vector method. BMC Evol. Biol. 9:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.