

Abstract
Perfluorocarbon (PFC) double emulsions loaded with a water-soluble, therapeutic agent can be triggered by ultrasound in a process known as acoustic droplet vaporization (ADV). Elucidating the stability and biodistribution of these sonosensitive vehicles and encapsulated agents are critical in developing targeted drug delivery strategies using ultrasound. [18F]fluorodeoxyglucose (FDG) was encapsulated in a PFC double emulsion and the in vitro diffusion of FDG was assessed using a Franz diffusion cell. Using dynamic micro positron emission tomography (micro-PET) and direct tissue sampling, the biodistribution of FDG administered as a solution (i.e. non-emulsified) or as an emulsion was studied in Fisher 344 rats (n = 6) bearing subcutaneous 9L gliosarcoma. Standardized uptake values (SUVs) and area under the curve of the SUV (AUCSUV) of FDG were calculated for various tissues. The FDG flux from the emulsion decreased by up to a factor of 6.9 compared to the FDG solution. FDG uptake, calculated from the AUCSUV, decreased by 36% and 44% for brain and tumor, respectively, when comparing FDG solution versus FDG emulsion (p < 0.01). Decreases in AUCSUV in highly metabolic tissues such as brain and tumor demonstrated retention of FDG within the double emulsion. No statistically significant differences in lung AUCSUV were observed, suggesting minimal accumulation of the emulsion in the pulmonary capillary bed. The liver AUCSUV increased by 356% for the FDG emulsion, thus indicating significant hepatic retention of the emulsion.
Keywords: micro-PET, biodistribution, perfluorocarbon, emulsion, acoustic droplet vaporization, ultrasound, responsive agents
INTRODUCTION
Nano- or micron-sized perfluorocarbon (PFC) particles are commonly used in diagnostic and therapeutic applications of ultrasound. For example, surfactant-stabilized, PFC microbubbles (i.e. contrast agents) are clinically utilized for perfusion imaging (1,2) and can be specifically targeted to vascular receptors, such as vascular endothelial growth factor receptor (3,4). Microbubbles can also facilitate drug or gene delivery, in response to ultrasound, when a therapeutic agent is either co-administered with the contrast agent (5,6) or the agent is contained/bound within the contrast agent (7,8). Gas-filled contrast agents have also been used to enhance the effects of thermal ablation achieved by high intensity focused ultrasound (HIFU) (9). As highlighted in a recent review (10), various types of liquid PFC droplets have also been investigated as diagnostic imaging agents. Similarly, PFC emulsions have been used therapeutically for ultrasound-triggered drug delivery (11–14) and HIFU (15,16).
Understanding the pharmacokinetics of sonosensitive PFC particles and the biodistribution of therapeutic agents contained within the particles are critical in developing ultrasound-based, drug delivery therapies that are safe and efficacious. For example, lung retention of microbubbles within the pulmonary microvasculature is a size dependent phenomenon (17). The formulation of transpulmonary, micron-sized emulsions can minimize certain types of PFC-related bioeffects (18,19). Additionally, the pharmacokinetics of sonosensitive PFC particles are directly related to the time window between vascular administration of the particles and the application of ultrasound to activate the particles. For nanoparticles that are passively targeted to tumor tissue, sufficient extravasation must occur to achieve an optimal therapeutic outcome (16,20).
Various imaging modalities – such as positron emission tomography (PET) (21–24), magnetic resonance (MR) (20,25), and fluorescence (20,26–28) imaging - have been used to study the in vitro cellular localization and in vivo biodistribution of both PFC microbubbles (21–24,28) and droplets (20,25–27). Depending on the type of PFC particle (i.e. diagnostic versus therapeutic) and incorporated imaging marker (e.g. 19F for MRI or 18F for PET), these modalities enable the visualization of either the PFC phase, surfactant, or therapeutic payload. Small animal PET (i.e. micro-PET) enables the study of pre-clinical biodistribution in a serial or paired manner and does not require sacrificing animals for direct tissue sampling. PET is an attractive imaging technique given its high sensitivity and clinical translatability, when compared to optical imaging techniques. Additionally, PET enables treatment monitoring and planning (29), thereby potentially leading to more individualized medical care (30).
In the presented studies, micro-PET is used to determine the biodistribution of a water-soluble radiotracer contained within a micron-sized, sonosensitive double emulsion of the following structure: water-in-PFC-in-water (W1/PFC/W2); similar double emulsion formulations have been used both in vitro (31,32) and ex vivo (33). Water-soluble agents, contained within the W1 phase, can be released using ultrasound in a process known as acoustic droplet vaporization (ADV) (34,35). Upon exposure to ultrasound above a certain acoustic pressure amplitude, the PFC liquid within the sonosensitive emulsion is converted into a gas. Thus, this double emulsion belongs to a class of PFC emulsions that have been termed phase-shift (16,36) or phase-change (37) emulsions. Low boiling point PFCs - such as perfluorobutane (C4F10, - 2°C), perfluoropentane (C5F12, 29°C), or perfluorohexane (C6F14, 56°C) – are typically used in emulsions that undergo ADV in order to minimize the acoustic pressures that are required for the vaporization at normal body temperature (37°C) (38). Besides drug delivery (11–14,20,26,31,32,39,40), ADV has been utilized in medical applications such as embolotherapy (41–43), enhancement of HIFU (15,16), and phase aberration correction (41,44). The primary excretion route of vascularly-administered PFC emulsions is via the mononuclear phagocyte system (MPS), where the particles accumulate primarily in the liver and spleen (45,46); the PFC is then transported to the lung for exhalation (47).
Elucidating the biodistribution of sonosensitive emulsions, in the absence of ADV, is critical in the development of safe and effective ADV-based therapies. To our knowledge, this is the first study that uses PET to track the biodistribution of a radiolabeled compound encapsulated within a sonosensitive, PFC double emulsion. [18F]fluorodeoxyglucose (FDG) – a water-soluble, radiotracer used in glucose utilization studies for tumor, cardiac, and cerebral tissue – was incorporated into the W1 phase of the double emulsion. First, the stability of the FDG-loaded emulsion was studied in vitro. Second, the biodistribution of emulsified FDG was evaluated in rats using both dynamic micro-PET and traditional tissue sampling techniques. The biodistribution of the FDG emulsion and non-emulsified (i.e. free) FDG was studied sequentially in rats to assess the in vivo stability of the emulsion, with a focus on the retention of FDG within the emulsion.
RESULTS
Characterization and in vitro stability of emulsion
The size distributions of the FDG emulsion, obtained using a Coulter counter, are displayed in Figure 1. The mean droplet diameter is 1.5 ± 0.1 μm and the droplet number density is 2.6 ± 0.5 × 1010 droplets/mL. The vast majority of droplet volume (93%) is contained within droplets whose diameters are 6 μm or less. There were no statistically significant changes in the size distributions of the emulsion after 24 hours, thereby indicating in vitro stability of the emulsion in the presence of high energy β+ (positron) and gamma radiation (FDG).
Figure 1.

Number and volume-weighted distributions of the PFC double emulsion containing FDG as obtained by the Coulter counter.
In vitro release of FDG
The retention of FDG within the emulsion, as evaluated using a Franz diffusion cell, is shown in Figure 2. All values in Figure 2 were corrected for the aliquots of solution and hence FDG activity removed during sampling. A solution of FDG, equal in activity to the emulsion, was used as a control. It was confirmed, by mixing blank (i.e. without FDG) emulsion and FDG solution, that the presence of the droplets within the donor compartment did not statistically change the FDG diffusion across the membrane for the FDG solution.
Figure 2.

Normalized activity of FDG in the acceptor compartment of the Franz diffusion cell for the FDG solution and FDG emulsion. In both cases, an equal concentration of FDG was loaded into the donor compartment. The average activity (n = 3) and standard deviation is plotted for each data point.
The flux of FDG was calculated based on a linear regression of the data between 10 and 60 minutes, which yielded squared correlation coefficients greater than 0.99 in all cases. In saline, the fluxes for the FDG solution and emulsion were 0.14 ± 0.003 MBq/cm2/min and 0.02 ± 0.007 MBq/cm2/min, respectively. In plasma, the fluxes for the FDG solution and emulsion were 0.13 ± 0.004 MBq/cm2/min and 0.03 ± 0.01 MBq/cm2/min, respectively. Therefore, in saline and plasma, the flux of FDG is reduced, respectively, by a factor of 6.9 and 4.2, compared to the solution, when FDG is encapsulated in the double emulsion.
In vivo dynamic micro-PET of emulsion
Representative micro-PET images, scaled based on the standardized uptake value (SUV), are shown in Figure 3. Qualitatively, it can be observed that the uptake of FDG is higher for the FDG solution (Figure 3, left) than the FDG emulsion (Figure 3, right) for the denoted organs and tumor, except the liver. Figure 4 displays the time profiles of the SUV for brain, lung, liver, tumor, muscle, and blood for both FDG solution and FDG emulsion. Statistically significant decreases of 36% and 44% were observed for the area under the curve for the normalized time activity data (AUCSUV) when comparing FDG emulsion versus FDG solution for brain and tumor, respectively. The liver displayed a 356% increase in the AUCSUV when comparing FDG emulsion versus FDG solution. No statistically significant changes in AUCSUV were noted for lung, muscle, or blood. Additionally, activity levels in blood obtained from the left ventricular cavity were likely overestimated for both FDG emulsion and solution, due to the “spill-in” of activity from the myocardium. Statistically significant differences in the maximum AUC (AUCMAX) were observed with brain and liver; a 36% decrease and 120% increase in AUCMAX occurred in brain and liver, respectively. No statistically significant differences in the time to reach AUCMAX (tMAX) or the time required for the SUV to reach half of AUCMAX (t1/2) were observed except in liver, which displayed increases of 321% and 498%, respectively. The control rats that received FDG solution on day one followed by blank (i.e. without FDG) emulsion and FDG solution on day two did not display any statistically significant differences in AUCSUV, AUCMAX, tMAX, or t1/2 for the time profiles measured on day one and two.
Figure 3.

Coronal micro-PET images of a rat that received an injection of FDG solution (left) and FDG emulsion (right). The summed images (0–60 minutes) are SUV-scaled (scale bar on far right).
Figure 4.

Time-activity curves of intravenously injected FDG solution and FDG emulsion, measured by dynamic micro-PET, in Fisher 344 rats with 9L tumors (n = 6). The data represent mean and standard deviation of the SUV for the a) brain, b) lung, c) liver, d) tumor, e) muscle, and f) blood.
Direct tissue sampling studies
The biodistribution of FDG measured as percent injected dose per gram tissue (%ID/g) in various organs and tissues at 3 minutes post injection is shown in Figure 5. Uptake of FDG solution and emulsion are also shown in Table 1. For all organs and tissues, except heart and adrenal glands, a statistically significant difference in uptake was observed when comparing FDG solution versus FDG emulsion. Except for lung, liver, and spleen, the uptake was lower for the FDG emulsion than for the FDG solution. When comparing the activity for the FDG solution and FDG emulsion, decreases of 62%, 59%, and 57% were observed for brain, tumor, and muscle, respectively. For lung, liver, and spleen, the uptake (in %ID/g) increased by 384%, 122%, and 609% when comparing FDG emulsion versus FDG solution.
Figure 5.
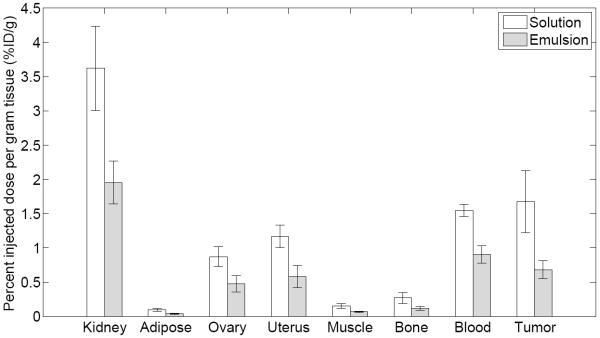

Biodistribution of FDG solution (n = 6) and FDG emulsion (n=6) in Fisher 344 rats with 9L tumors at 3 minutes. The data represent mean and standard deviation of each organ/tissue. In all cases except for the heart and adrenal glands, a statistically significant difference (p < 0.01) was observed when comparing the solution versus the emulsion.
Table 1.
FDG uptake in various organs and tissues obtained 3 minutes post intravenous injection. The average uptake (n = 6) and standard deviation are listed for each data point.
| Uptake (%ID/g) |
|||||
|---|---|---|---|---|---|
| FDG | Brain | Lung | Liver | Tumor | Muscle |
| Solution | 1.42 ± 0.19 | 1.39 ± 0.11 | 2.03 ± 0.18 | 1.67 ± 0.45 | 0.16 ± 0.04 |
| Emulsion | 0.54 ± 0.09 | 6.72 ± 1.66 | 4.52 ± 0.76 | 0.68 ± 0.13 | 0.07 ± 0.01 |
DISCUSSION
Focused ultrasound has been used to localize drug release from vascularly-administered sonosensitive emulsions (13,20,40,48). The timing between the administration of the emulsion and the subsequent application of the ultrasound to generate ADV and localized release is critical in achieving efficacious results. A sufficient delay following the injection of the emulsion can facilitate therapy via intended mechanisms of particle accumulation, such as the binding of actively targeted droplets to vascular receptors (25,49) or the extravasation of nano-sized emulsions in a tumor (20). However, the efficacy of localized drug delivery from sonosensitive emulsions can be negatively affected by unintended release (i.e. release in the absence of ultrasound sufficient to trigger ADV) or clearance of the emulsion via the MPS. Additionally, as observed with other emulsions (50), the formulation of a therapeutic agent as an emulsion can dramatically change the biodistribution and pharmacokinetics of the agent.
One main aim of this study was to demonstrate the stable retention of the payload (i.e. FDG) within the W1 phase such that the release of the payload could be triggered by ultrasound via ADV. By minimizing the amount of burst release from the emulsion upon injection, the spatial and temporal profiles of drug release can be localized by the application of focused ultrasound. This precise control of drug release is especially useful when delivering agents that are systemically toxic (ex. chemotherapeutic agents), such that drug release and uptake are localized to the intended target site (ex. tumor).
As demonstrated in Figure 2, the flux of FDG is reduced when encapsulated in the double emulsion. This decrease in diffusion rate, which is consistent with previous studies using fluorescein within the W1 phase (31), is attributed to the hydrophobic perfluoro-n-pentane (PFP) layer that surrounds the W1 phase (47). Given that the flux of FDG from the emulsion is non-zero, there are four potential mechanisms by which FDG could be diffusing from the emulsion: 1) spontaneous vaporization of the PFP within the emulsion, thereby releasing the W1 phase; 2) conversion of the double emulsion into a single emulsion (i.e. PFC/W2); 3) diffusion of FDG from the W1 phase into the bulk phase in the absence of PFP vaporization or emulsion destabilization; or 4) diffusion of residual FDG, likely present in the W2 phase, that remains post washing. Though fluorocarbon emulsions have been used as radiation dosimetry detectors (51,52), whereby the emulsion transitions from liquid droplets into gas bubbles when exposed to radiation, the double emulsion used in this work is stable in the presence of high-energy radiation resulting from the decay of fluorine-18. This is likely due to the boiling point of PFP (29°C), which is significantly higher than the boiling point of fluorocarbons typically used in dosimetry applications (i.e. < 0°C). Therefore, spontaneous vaporization is likely not causing release of FDG from the emulsion. Additionally, since the volume fraction of W1 in the double emulsion is large (31), the conversion of the double emulsion into a single emulsion – an instability issue with double emulsions (53) - should be detectable by measuring the droplet size distribution. Again, no differences in size distribution were observed when comparing the initial droplet size distribution to that one day later; therefore, this mechanism seems unlikely, at least in vitro. It is hypothesized that FDG release from the emulsion is due to FDG diffusing from the W1 phase, without emulsion destabilization, and/or the presence of residual FDG in the W2 phase that was not removed during washing.
The comparative in vivo uptake of FDG, whether in free form (i.e. solution) or emulsified, is an indicator of the degree to which FDG is stably retained within the emulsion. This retention can be clearly seen by comparing the time-activity curves for brain and tumor (Figure 4a and 4d) – two tissues that display high levels of glucose utilization and retention of FDG. In both cases, statistically significant decreases in the AUCSUV, an indicator of the exposure and uptake of the radiotracer (54), were observed (Figure 4), thus confirming that emulsification hinders uptake of FDG. For both FDG solution and FDG emulsion, the AUCSUV was higher in tumor than brain, which is likely due to the anesthetic isoflurane that decreases FDG uptake in rodent brain (55), but not in tumor tissue (56), versus the awake state. The decrease in FDG uptake due to emulsification is further supported by Figure 6, which displays relative SUVs between scans of data shown in Figure 4. These relative SUVs are calculated as the SUV of the `FDG emulsion' scan divided by the SUV of the `FDG solution' scan at each time point. Similarly, for the control rats, relative SUVs are given by the SUV of the `FDG solution + blank emulsion' scan divided by the `FDG solution' scan. For the control animals, the relative SUVs are close to one, which indicates that the presence of blank emulsion did not affect FDG metabolism. It is important to note that any affinity that FDG has for the emulsion (ex. binding to the the Poloxamer shell), which may be evident with the FDG emulsion, would not necessarily be present in this control given that the rats were injected with FDG solution followed by blank emulsion.
Figure 6.

Relative SUVs are calculated as the SUV of the `FDG emulsion' scan divided by the SUV of the `FDG solution' scan (Figure 4). For control rats, relative SUVs are given by the SUV of the `FDG solution + blank emulsion' scan divided by the `FDG solution' scan. The data represent mean and standard deviation for the a) brain, b) lung, c) liver, d) tumor, e) muscle, and f) blood.
For rats that received the FDG emulsion, the relative SUVs are less than one. The fraction of FDG retained within the emulsion versus the total FDG can be estimated using the data in Figure 6. When the blood-brain barrier (BBB) extraction is high and the tissue blood fraction is low, the fraction of FDG in the emulsion is simply 1 minus the values shown in Figure 6. However, as extraction decreases or blood volume increases, a simple correction factor is needed. Assuming that the brain blood volume fraction is 3%, the BBB extraction fraction is 0.2, and a relative SUV of 0.5, the fraction of FDG retained in the emulsion at 5 minutes is ~58%. The fraction of FDG retained in the emulsion is fairly consistent for brain and tumor. It can also be observed in Figure 6 that the relative SUVs in brain, tumor, and muscle increase slowly from 5 minutes through the end of the scan. This likely indicates that a smaller fraction of FDG becomes available for transport out of the blood due to FDG release from the droplets. Thus, by the end of the study, the fraction of FDG retained in the emulsion has decreased to around 40–45%. Comparatively, if all of the FDG were to be retained in the emulsion, the relative SUV would be 0.13.
Minimizing PFC accumulation in the lung is critical in preventing the disruption of pulmonary gas exchange (18,19), which can cause respiratory distress or death (43,57). One approach to minimize lung effects generated by PFC emulsions, as well as to ensure uniform ADV thresholds of the emulsions, is to generate monodisperse droplets (37). As supported by the AUCSUV (Figure 4), no lung retention of the polydisperse FDG emulsion was detected by micro-PET. Since 98% (by number) of the droplets are 3 μm in diameter or smaller (Figure 1), minimal lung accumulation was expected given that a previous study demonstrated clearance of 2 μm and 3 μm rigid microparticles in rat lungs (58). Based on the direct tissue sampling (Figure 5), there was a statistically significant increase in lung activity at 3 minutes for the FDG emulsion compared to the FDG solution. However, the AUCSUV for lung between 0–3 minutes indicates no statistically significant differences. The relative SUV (Figure 6b) between 0.75–6.5 minutes displays mean values greater than one, though the differences are not statistically significant relative to the control rats. Thus, it is hypothesized that the differences in observed lung uptake between the micro-PET and tissue sampling studies may be due to differences in the duration of isoflurane exposure, a vasodilator (59). Isoflurane was administered for 15–20 minutes and 5 minutes prior to the administration of the emulsion in the micro-PET and biodistribution tissue sampling studies, respectively.
Previous studies have demonstrated that PFC emulsions accumulate in organs of the MPS such as liver and spleen (45,46). Significant accumulation of the FDG emulsion was confirmed by both micro-PET imaging (Figures 4c and 6c) and direct tissue sampling (Figure 5) in both the liver and spleen. The largest increases in AUCSUV, AUCMAX, tMAX, t1/2, and uptake were observed in liver and spleen when comparing FDG emulsion versus FDG solution – another indicator that FDG is being retained within the double emulsion. Due to the faster rate of clearance (i.e. ~ 150% higher) of FDG from the liver for the FDG emulsion compared to the FDG solution, as determined by the slope of the time-activity curves in Figure 4c between 12.5 to 55 minutes, it may be likely that FDG is released from the emulsion while accumulated in the liver. Incorporating polyethylene glycol (PEG) chains or PEG-containing polymers into the shell stabilizing the double emulsion could reduce the rate at which the droplets are opsonized (i.e. the rate at which droplets are marked for phagocytic clearance) and removed by the MPS (60,61). Though Poloxamer 188, the water-soluble surfactant used to stabilize the double emulsion, is a copolymer containing PEG, Polaxamer 188 has not been found to reduce hepatic or splenic uptake of particles (62,63).
CONCLUSIONS
As demonstrated both in vitro and in vivo, small, water-soluble molecules such as FDG can be retained in sonosensitive double emulsions. Given that drug release from PFC emulsions can be triggered using ultrasound, the retention of the encapsulated payload in the W1 phase is critical in spatially and temporally localizing release to the site of ultrasound application. Dynamic micro-PET imaging indicated that the FDG emulsion did not accumulate in the lung, which is critical in minimizing PFC related bioeffects. When delivering a therapeutic agent in the PFC double emulsion, the use of surfactants such as Poloxamer 407 or Poloxamine 908 could assist in decreasing the rate at which the droplets are cleared by the MPS and thereby modulating the release of drug into the general circulation. Given the observed distribution of the FDG loaded into the sonosensitive emulsion, the liver will likely be a critical organ when therapeutic payloads are used.
EXPERIMENTAL
Emulsion preparation
The double emulsion was prepared by modifying previously published methods(31). The primary emulsion (W1/PFC) was formed by dissolving Krytox 157 FSL (CAS# 51798-33-5, DuPont, Wilmington, DE, USA), a perfluoroether with carboxylic acid functionality, and Krytox 157 FSL-polyethylene glycol copolymer in perfluoro-n-pentane (CAS# 678-26-2, Strem Chemicals, Inc., Newburyport, MA, USA) at concentrations of 25 mg/mL and 75 mg/mL, respectively. The PFP phase was then combined with an aqueous solution of FDG at a volumetric ratio of 4:3. FDG was synthesized in-house by the Cyclotron and Radiochemistry Facility at the University of Michigan. The mixture was emulsified, while in an ice bath, via sonication using a microtip (model 450, 20 kHz, Branson, Danbury, CT, USA) operating at 125 W/cm2 for 30 seconds in continuous mode. The resulting primary emulsion was added drop-wise to a 100 mg/mL solution of Poloxamer 188 (Sigma Aldrich, St. Louis, MO, USA) in normal saline (0.9% w/v, Hospira Inc., Lake Forest, IL, USA), which was in an ice bath and being stirred at 1100 rpm for 10 minutes. Poloxamer 188 is a water-soluble, amphiphilic copolymer that stabilizes the PFC/W2 interface, thus enabling the formation of a double emulsion. The coarse double emulsion was then sonicated, as previously described, to reduce droplet size. To minimize the amount of non-emulsified FDG, the double emulsion was washed in triplicate by centrifuging the emulsion at 5000 rpm for 30 seconds, removing the supernatant, and resuspending the pellet (i.e. droplets) in fresh saline. The double emulsion was sized using a Coulter counter (Multisizer III, Beckman Coulter Inc., Brea, CA, USA) with a 50 μm aperture and used immediately for in vivo experiments. To assess the stability of the emulsion in the presence of FDG, the remaining emulsion was stored at room temperature (25°C) and sized, as described previously, 24 hours after formulation.
In vitro FDG release
Release of FDG from the double emulsion was measured at room temperature (25°C) using a Franz diffusion cell (PermeGear, Inc., Hellertown, PA, USA), as described previously(31). Briefly, a cellulose membrane (6–8 kDa molecular weight cutoff, Spectrum Laboratories, Inc., Rancho Dominguez, CA, USA), soaked in normal saline was mounted between the donor and acceptor compartments of the cell. The donor media contained 2 mL FDG emulsion, diluted in either normal saline or human plasma, while the acceptor compartment contained either 7.5 mL normal saline or human plasma (i.e. the same fluid loaded in the donor compartment). Near sink conditions were maintained in the acceptor compartment, which was stirred at 600 rpm, throughout the duration of the experiment. The diffusion area between both compartments was 1.77 cm2. Additionally, an overhead stirrer, operating at 600 rpm, prevented settling of the emulsion on the membrane due to the elevated density of PFP (1.6 g/mL). At 10-minute intervals, aliquots of the acceptor medium were withdrawn and immediately replaced with fresh, normal saline or human plasma. The FDG activity was measured with a dose calibrator (CRC 712M, Capintec, Inc., Ramsey, NJ, USA). The above experiment was repeated for an FDG solution of equal activity as well.
Cell line and tumor model
9L gliosarcoma cells (American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco's Modified Eagle Medium (high glucose) supplemented with 9% (v/v) fetal bovine serum, 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 0.35 mg/mL G418 (Geneticin). Cell culture media and reagents were obtained from Gibco (Invitrogen, Eugene, OR, USA). The cells were grown in a humidified 5% carbon dioxide environment at 37°C. After reaching 80% confluence, 5×106 cells were trypsinized, resuspended in phosphate buffered saline, and injected subcutaneously into the right shoulder of female Fischer 344 rats (n = 12, 200 gram, Harlan Laboratories, Indianapolis, IN, USA). Tumors were allowed to grow for 7–10 days. All animal protocols were approved by the University Committee on Use and Care of Animals.
Dynamic micro-PET imaging
Rats (n = 9) were anesthetized using isoflurane (5% for induction and 1–2% for maintenance) and placed prone within a micro-PET scanner (R4 or P4, Concorde Microsystems, Inc., Knoxville, TN, USA) (64). Body temperature, maintained using a circulating water warming pad, and respiratory rate were monitored during the imaging session. FDG solution (n = 9, 7.4 MBq) was injected via tail vein catheter immediately followed by a 60 minute dynamic PET scan with respiratory gating. The rats were allowed to recover. On the following day, the aforementioned imaging procedure was repeated for the FDG emulsion (n = 6, 1.9 MBq, mass dose of emulsion: 0.08 g PFP per kilogram body weight). A control study was conducted to confirm that the presence of the emulsion alone did not alter the in vivo biodistribution of FDG. Rather than receiving an injection of FDG emulsion, these rats (n = 3) were injected with FDG solution (7.4 MBq) immediately followed by blank (i.e. without FDG) emulsion (mass dose of emulsion: 0.08 g PFP per kilogram body weight). All rats were recovered for tissue sampling (biodistribution) studies on the following day.
Analysis of micro-PET images
After correction for decay, dead time, and random coincidences, PET data were reconstructed using a statistical reconstruction method (ordered-subset expectation maximization). Images were scaled to the SUV defined as the measured radioactivity (in nCi/cm3) normalized by the injected activity and body weight of each rat. Time-activity curves were generated for individual volumes of interest drawn in various organs and tissues using the ASI Pro VM software (Siemens Medical Systems, Malvern, PA, USA). The time-activity curve for blood was generated by analyzing the left ventricular cavity.
MATLAB (The MathWorks, Inc., Natick, MA, USA) was used to calculate the following parameters: AUCSUV, AUCMAX, tMAX, and t1/2.
Direct tissue sampling (biodistribution) studies
The FDG solution (n = 6, 1.9 MBq) or FDG emulsion (n = 6, 1.9 MBq, mass dose of emulsion: 0.08 g PFP per kilogram body weight) was administered intravenously in anesthetized rats. Three minutes after injection, the following tissues were harvested: brain, eyes, heart, lung, liver, pancreas, spleen, adrenal glands, kidneys, adipose tissue, ovaries, uterus, muscle (thigh), bone (femur), blood, and tumor. Radioactivity was counted in a γ-counter (model A5550, Minaxi auto gamma, Packard/Perkin Elmer, Waltham, MA, USA). Decay-corrected results were expressed as percentage of injected radioactivity dose per gram of tissue (%ID/g).
Statistical analysis
Each experimental value is expressed as the mean ± standard deviation and the result of at least three independent measurements. Statistically significant differences between experimental groups was determined using a Student's t-test. A significance level of 0.01 was used for all comparisons.
ACKNOWLEDGMENTS
The authors would like to thank Larry Holt for assistance with micro-PET imaging. This work was supported in part by NIH grant 5R01CA091713 and the University of Michigan-Shanghai Jiao Tong University Collaboration on Biomedical Technologies grant.
REFERENCES
- 1.Ferrara KW, Pollard R, Borden MA. Ultrasound microbubble contrast agents: fundamentals and application to gene and drug delivery. Annual Review of Biomedical Engineering. 2007;9:425–447. doi: 10.1146/annurev.bioeng.8.061505.095852. [DOI] [PubMed] [Google Scholar]
- 2.Miller DL, Averkiou MA, Brayman AA, Everbach EC, Holland CK, Wible JH, Wu J. Bioeffects considerations for diagnostic ultrasound contrast agents. Journal of Ultrasound in Medicine. 2008;27(4):611–632. doi: 10.7863/jum.2008.27.4.611. [DOI] [PubMed] [Google Scholar]
- 3.Pillai R, Marinelli ER, Fan H, Nanjappan P, Song B, von Wronski MA, Cherkaoui S, Tardy I, Pochon S, Schneider M, Nunn AD, Swenson RE. A phospholipid-PEG2000 conjugate of a vascular endothelial growth factor receptor 2 (VEGFR2)-targeting heterodimer peptide for contrast-enhanced ultrasound imaging of angiogenesis. Bioconjugate Chemistry. 2010;21(3):556–562. doi: 10.1021/bc9005688. [DOI] [PubMed] [Google Scholar]
- 4.Bzyl J, Lederle W, Rix A, Grouls C, Tardy I, Pochon S, Siepmann M, Penzkofer T, Schneider M, Kiessling F, Palmowski M. Molecular and functional ultrasound imaging in differently aggressive breast cancer xenografts using two novel ultrasound contrast agents (BR55 and BR38) Eur Radiol. 2011;21(9):1988–1995. doi: 10.1007/s00330-011-2138-y. [DOI] [PubMed] [Google Scholar]
- 5.Datta S, Coussios CC, Ammi AY, Mast TD, de Courten-Myers GM, Holland CK. Ultrasound-enhanced thrombolysis using Definity as a cavitation nucleation agent. Ultrasound in Medicine and Biology. 2008;34(9):1421–1433. doi: 10.1016/j.ultrasmedbio.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watanabe Y, Horie S, Funaki Y, Kikuchi Y, Yamazaki H, Ishii K, Mori S, Vassaux G, Kodama T. Delivery of Na/I Symporter Gene into Skeletal Muscle Using Nanobubbles and Ultrasound: Visualization of Gene Expression by PET. Journal of Nuclear Medicine. 2010;51(6):951–958. doi: 10.2967/jnumed.109.074443. [DOI] [PubMed] [Google Scholar]
- 7.Tartis MS, McCallan J, Lum AFH, LaBell R, Stieger SM, Matsunaga TO, Ferrara KW. Therapeutic effects of paclitaxel-containing ultrasound contrast agents. Ultrasound in Medicine and Biology. 2006;32(11):1771–1780. doi: 10.1016/j.ultrasmedbio.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 8.Cerroni B, Chiessi E, Margheritelli S, Oddo L, Paradossi G. Polymer Shelled Microparticles for a Targeted Doxorubicin Delivery in Cancer Therapy. Biomacromolecules. 2011;12(3):593–601. doi: 10.1021/bm101207k. [DOI] [PubMed] [Google Scholar]
- 9.Yu TH, Fan XL, Xiong SH, Hu K, Wang ZB. Microbubbles assist goat liver ablation by high intensity focused ultrasound. Eur Radiol. 2006;16(7):1557–1563. doi: 10.1007/s00330-006-0176-7. [DOI] [PubMed] [Google Scholar]
- 10.Diaz-Lopez R, Tsapis N, Fattal E. Liquid perfluorocarbons as contrast agents for ultrasonography and 19F-MRI. Pharmaceutical Research. 2010;27(1):1–16. doi: 10.1007/s11095-009-0001-5. [DOI] [PubMed] [Google Scholar]
- 11.Fabiilli ML, Haworth KJ, Sebastian IE, Kripfgans OD, Carson PL, Fowlkes JB. Delivery of chlorambucil using an acoustically-triggered perfluoropentane emulsion. Ultrasound in Medicine and Biology. 2010;36(8):1364–1375. doi: 10.1016/j.ultrasmedbio.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hwang TL, Lin YJ, Chi CH, Huang TH, Fang JY. Development and evaluation of perfluorocarbon nanobubbles for apomorphine delivery. Journal of Pharmaceutical Sciences. 2009;98(10):3735–3747. doi: 10.1002/jps.21687. [DOI] [PubMed] [Google Scholar]
- 13.Rapoport NY, Kennedy AM, Shea JE, Scaife CL, Nam K-H. Controlled and targeted tumor chemotherapy by ultrasound-activated nanoemulsions/microbubbles. Journal of Controlled Release. 2009;138(2):268–276. doi: 10.1016/j.jconrel.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang CH, Kang ST, Lee YH, Luo YL, Huang YF, Yeh CK. Aptamer-conjugated and drug-loaded acoustic droplets for ultrasound theranosis. Biomaterials. 2012;33(6):1939–1947. doi: 10.1016/j.biomaterials.2011.11.036. [DOI] [PubMed] [Google Scholar]
- 15.Zhang M, Fabiilli ML, Haworth KJ, Padilla F, Swanson SD, Kripfgans OD, Carson PL, Fowlkes JB. Acoustic droplet vaporization for enhancement of thermal ablation by high intensity focused ultrasound. Acad Radiol. 2011;18(9):1123–1132. doi: 10.1016/j.acra.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang P, Porter T. An in Vitro Study of a Phase-Shift Nanoemulsion: A Potential Nucleation Agent for Bubble-Enhanced Hifu Tumor Ablation. Ultrasound in Medicine and Biology. 2010;36(11):1856–1866. doi: 10.1016/j.ultrasmedbio.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 17.de Jong N, ten Cate FJ, Vletter WB, Roelandt JRTC. Quantification of Transpulmonary Echocontrast Effects. Ultrasound in Medicine and Biology. 1993;19(4):279–288. doi: 10.1016/0301-5629(93)90100-3. [DOI] [PubMed] [Google Scholar]
- 18.Leakakos T, Schutt EG, Calvin JC, Smith D, Bradley JD, Strnat CA, del Balzo U, Hazard DY, Otto S, Fields TK, Keipert PE, Klein DH, Flaim SF. Pulmonary gas trapping differences among animal species in response to intravenous infusion of perfluorocarbon emulsions. Artificial Cells, Blood Substitutes, and Immobilization Biotechnology. 1994;22(4):1199–1204. doi: 10.3109/10731199409138816. [DOI] [PubMed] [Google Scholar]
- 19.Schutt E, Barber P, Fields T, Flaim S, Horodniak J, Keipert P, Kinner R, Kornburst L, Leakakos T, Pelura T, Weers J, Houmes R, Lachmann B. Proposed mechanism of pulmonary gas trapping (PGT) following intravenous perfluorocarbon emulsion administration. Artificial Cells, Blood Substitutes, and Immobilization Biotechnology. 1994;22(4):1205–1214. doi: 10.3109/10731199409138817. [DOI] [PubMed] [Google Scholar]
- 20.Rapoport N, Nam KH, Gupta R, Gao ZG, Mohan P, Payne A, Todd N, Liu X, Kim T, Shea J, Scaife C, Parker DL, Jeong EK, Kennedy AM. Ultrasound-mediated tumor imaging and nanotherapy using drug loaded, block copolymer stabilized perfluorocarbon nanoemulsions. Journal of Controlled Release. 2011;153(1):4–15. doi: 10.1016/j.jconrel.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willmann JK, Cheng Z, Davis C, Lutz AM, Schipper MK, Nielsen CH, Gambhir SS. Targeted microbubbles for imaging tumor angiogenesis: assessment of whole-body biodistribution with dynamic micro-PET in mice. Radiology. 2008;249(1):212–219. doi: 10.1148/radiol.2491072050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perkins AC, Frier M, Hindle AJ, Blackshaw PE, Bailey SE, Hebden JM, Middleton SM, Wastie ML. Human biodistribution of an ultrasound contrast agent (Quantison(TM)) by radiolabelling and gamma scintigraphy. British Journal of Radiology. 1997;70(834):603–611. doi: 10.1259/bjr.70.834.9227254. [DOI] [PubMed] [Google Scholar]
- 23.Ferrara KW, Borden MA, Zhang H. Lipid-Shelled Vehicles: Engineering for Ultrasound Molecular Imaging and Drug Delivery. Accounts Chem Res. 2009;42(7):881–892. doi: 10.1021/ar8002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tartis MS, Kruse DE, Zheng HR, Zhang H, Kheirolomoom A, Marik J, Ferrara KW. Dynamic microPET imaging of ultrasound contrast agents and lipid delivery. Journal of Controlled Release. 2008;131(3):160–166. doi: 10.1016/j.jconrel.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kok MB, de Vries A, Abdurrachim D, Prompers JJ, Grull H, Nicolay K, Strijkers GJ. Quantitative H-1 MRI, F-19 MRI, and F-19 MRS of cell-internalized perfluorocarbon paramagnetic nanoparticles. Contrast Media Mol I. 2011;6(1):19–27. doi: 10.1002/cmmi.398. [DOI] [PubMed] [Google Scholar]
- 26.Mohan P, Rapoport N. Doxorubicin as a Molecular Nanotheranostic Agent: Effect of Doxorubicin Encapsulation in Micelles or Nanoemulsions on the Ultrasound-Mediated Intracellular Delivery and Nuclear Trafficking. Mol Pharmaceut. 2010;7(6):1959–1973. doi: 10.1021/mp100269f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gorelikov I, Martin AL, Seo M, Matsuura N. Silica-Coated Quantum Dots for Optical Evaluation of Perfluorocarbon Droplet Interactions with Cells. Langmuir. 2011;27(24):15024–15033. doi: 10.1021/la202679p. [DOI] [PubMed] [Google Scholar]
- 28.Anderson CR, Hu XW, Zhang H, Tlaxca J, Decleves AE, Houghtaling R, Sharma K, Lawrence M, Ferrara KW, Rychak JJ. Ultrasound Molecular Imaging of Tumor Angiogenesis With an Integrin Targeted Microbubble Contrast Agent. Investigative Radiology. 2011;46(4):215–224. doi: 10.1097/RLI.0b013e3182034fed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piroth MD, Pinkawa M, Holy R, Klotz J, Nussen S, Stoffels G, Coenen HH, Kaiser HJ, Langen KJ, Eble MJ. Prognostic Value of Early [(18)F]Fluoroethyltyrosine Positron Emission Tomography after Radiochemotherapy in Glioblastoma Multiforme. Int J Radiat Oncol. 2011;80(1):176–184. doi: 10.1016/j.ijrobp.2010.01.055. [DOI] [PubMed] [Google Scholar]
- 30.Basu S. Personalized versus evidence-based medicine with PET-based imaging. Nature Reviews Clinical Oncology. 2010;7(11):665–668. doi: 10.1038/nrclinonc.2010.121. [DOI] [PubMed] [Google Scholar]
- 31.Fabiilli ML, Lee JA, Kripfgans OD, Carson PL, Fowlkes JB. Delivery of water-soluble drugs using acoustically triggered perfluorocarbon double emulsions. Pharm Res. 2010;27(12):2753–2765. doi: 10.1007/s11095-010-0277-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajian JR, Fabiilli ML, Fowlkes JB, Carson PL, Wang X. Drug delivery monitoring by photoacoustic tomography with an ICG encapsulated double emulsion. Opt Express. 2011;19(15):14335–14347. doi: 10.1364/OE.19.014335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Couture O, Faivre M, Pannacci N, Babataheri A, Servois V, Tabeling P, Tanter M. Ultrasound internal tattooing. Medical Physics. 2011;38(2):1116–1123. doi: 10.1118/1.3548068. [DOI] [PubMed] [Google Scholar]
- 34.Apfel RE, Apfel Enterprises, Inc., assignee Activatable Infusable Dispersions Containing Drops of a Superheated Liquid for Methods of Therapy and Diagnosis. United States. 1998 Nov; [Google Scholar]
- 35.Kripfgans OD, Fowlkes JB, Miller DL, Eldevik OP, Carson PL. Acoustic droplet vaporization for therapeutic and diagnostic applications. Ultrasound Med Biol. 2000;26(7):1177–1189. doi: 10.1016/s0301-5629(00)00262-3. [DOI] [PubMed] [Google Scholar]
- 36.Sheeran PS, Luois S, Dayton PA, Matsunaga TO. Formulation and Acoustic Studies of a New Phase-Shift Agent for Diagnostic and Therapeutic Ultrasound. Langmuir. 2011;27(17):10412–10420. doi: 10.1021/la2013705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martz TD, Sheeran PS, Bardin D, Lee AP, Dayton PA. Precision Manufacture of Phase-Change Perfluorocarbon Droplets Using Microfluidics. Ultrasound in Medicine and Biology. 2011;37(11):1952–1957. doi: 10.1016/j.ultrasmedbio.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fabiilli ML, Haworth KJ, Fakhri NH, Kripfgans OD, Carson PL, Fowlkes JB. The role of inertial cavitation in acoustic droplet vaporization. IEEE Trans Ultrason Ferroelectr Freq Control. 2009;56(5):1006–1017. doi: 10.1109/TUFFC.2009.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fang JY, Hung CF, Hua SC, Hwang TL. Acoustically active pefluorocarbon nanoemulsions as drug delivery carriers for camptothecin: drug release and cytoxicity against cancer cells. Ultrasonics. 2009;49(1):39–46. doi: 10.1016/j.ultras.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 40.Rapoport N, Gao Z, Kennedy A. Multifunctional Nanoparticles for Combining Ultrasonic Tumor Imaging and Targeted Chemotherapy. J Natl Cancer I. 2007;99(14):1095–1106. doi: 10.1093/jnci/djm043. [DOI] [PubMed] [Google Scholar]
- 41.Kripfgans OD, Fowlkes JB, Woydt M, Eldevik OP, Carson PL. In vivo Droplet Vaporization for Occlusion Therapy and Phase Aberration Correction. IEEE Transactions on Ultrasonics, Ferroelectrics, and Frequency Control. 2002;49(2):726–738. doi: 10.1109/tuffc.2002.1009331. [DOI] [PubMed] [Google Scholar]
- 42.Kripfgans OD, Orifici CM, Carson PL, Ives KA, Eldevik OP, Fowlkes JB. Acoustic Droplet Vaporization for Temporal and Spatial Control of Tissue Occlusion: a Kidney Study. IEEE Transactions on Ultrasonics, Ferroelectrics, and Frequency Control. 2005;52(7):1101–1110. doi: 10.1109/tuffc.2005.1503996. [DOI] [PubMed] [Google Scholar]
- 43.Zhang M, Fabiilli ML, Haworth KJ, Fowlkes JB, Kripfgans OD, Roberts WW, Ives KA, Carson PL. Initial investigation of acoustic droplet vaporization for occlusion in canine kidney. Ultrasound in Medicine and Biology. 2010;36(10):1691–1703. doi: 10.1016/j.ultrasmedbio.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haworth KJ, Fowlkes JB, Carson PL, Kripfgans OD. Towards Aberration Correction of Transcranial Ultrasound Using Acoustic Droplet Vaporization. Ultrasound Med Biol. 2008;34(3):435–445. doi: 10.1016/j.ultrasmedbio.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bentley PK, Johnson OL, Washington C, Lowe KC. Uptake of Concentrated Perfluorocarbon Emulsions into Rat Lymphoid-Tissues. Journal of Pharmacy and Pharmacology. 1993;45(3):182–185. doi: 10.1111/j.2042-7158.1993.tb05529.x. [DOI] [PubMed] [Google Scholar]
- 46.Satterfield R, Tarter VM, Schumacher DJ, Tran P, Mattrey RF. Comparison of Different Perfluorocarbons as Ultrasound Contrast Agents. Investigative Radiology. 1993;28(4):325–331. doi: 10.1097/00004424-199304000-00013. [DOI] [PubMed] [Google Scholar]
- 47.Riess JG. Oxygen Carriers (“Blood Substitutes”) - Raison d'Etre, Chemistry, and Some Physiology. Chemical Reviews. 2001;101(9):2797–2919. doi: 10.1021/cr970143c. [DOI] [PubMed] [Google Scholar]
- 48.Couture O, Urban A, Bretagne A, Martinez L, Tanter M, Tabeling P. In vivo targeted delivery of large payloads with an ultrasound clinical scanner. Med Phys. 2012;39(8):5229–5237. doi: 10.1118/1.4736822. [DOI] [PubMed] [Google Scholar]
- 49.Lanza GM, Wallace KD, Scott MJ, Cacheris WP, Abendschein DR, Christy DH, Sharkley AM, Miller JG, Gaffney PJ, Wickline SA. A novel site-targeted ultrasonic contrast agent with broad biomedical application. Circulation. 1996;94(12):3334–3340. doi: 10.1161/01.cir.94.12.3334. [DOI] [PubMed] [Google Scholar]
- 50.Ganta S, Paxton JW, Baguley BC, Garg S. Pharmacokinetics and pharmacodynamics of chlorambucil delivered in parenteral emulsion. Int J Pharm. 2008;360(1–2):115–121. doi: 10.1016/j.ijpharm.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 51.Apfel RE. Superheated drop detector. Nuclear Instruments and Methods. 1979;162(1–3):603–608. [Google Scholar]
- 52.d'Errico F. Radiation dosimetry and spectrometry with superheated emulsions. Nuclear Instruments and Methods in Physics Research B. 2001;184:229–254. [Google Scholar]
- 53.Ficheux MF, Bonakdar L, Leal-Calderon F, Bibette J. Some stability criteria for double emulsions. Langmuir. 1998;14(10):2702–2706. [Google Scholar]
- 54.West CML, Jones T, Price P. The potential of positron-emission tomography to study anticancer-drug resistance. Nature Reviews Cancer. 2004;4(6):457–469. doi: 10.1038/nrc1368. [DOI] [PubMed] [Google Scholar]
- 55.Toyama H, Ichise M, Liow JS, Vines DC, Seneca NM, Modell KJ, Seidel J, Green MV, Innis RB. Evaluation of anesthesia effects on [F-18]FDG uptake in mouse brain and heart using small animal PET. Nucl Med Biol. 2004;31(2):251–256. doi: 10.1016/S0969-8051(03)00124-0. [DOI] [PubMed] [Google Scholar]
- 56.Fueger BJ, Czernin J, Hildebrandt I, Tran C, Halpern BS, Stout D, Phelps ME, Weber WA. Impact of animal handling on the results of F-18-FDG PET studies in mice. J Nucl Med. 2006;47(6):999–1006. [PubMed] [Google Scholar]
- 57.Canaud B, Aljama P, Tielemans C, Gasparovic V, Gutierrez A, Locatelli F. Pathochemical toxicity of perfluorocarbon-5070, a liquid test performance fluid previously used in dialyzer manufacturing, confirmed in animal experiment. Journal of the American Society of Nephrology. 2005;16(6):1819–1823. doi: 10.1681/ASN.2004050361. [DOI] [PubMed] [Google Scholar]
- 58.Kutscher HL, Chao P, Deshmukh M, Singh Y, Hu P, Joseph LB, Reimer DC, Stein S, Laskin DL, Sinko PJ. Threshold size for optimal passive pulmonary targeting and retention of rigid microparticles in rats. Journal of Controlled Release. 2010;143(1):31–37. doi: 10.1016/j.jconrel.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schwinn DA, Mcintyre RW, Reves JG. Isoflurane-Induced Vasodilation - Role of the Alpha-Adrenergic Nervous-System. Anesthesia and Analgesia. 1990;71(5):451–459. doi: 10.1213/00000539-199011000-00001. [DOI] [PubMed] [Google Scholar]
- 60.Owens DE, Peppas NA. Opsonization, biodistribution, and pharmaokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 61.Giraudeau C, Djemai B, Ghaly MA, Boumezbeur F, Meriaux S, Robert P, Port M, Robic C, Le Bihan D, Lethimonnier F, Valette J. High sensitivity 19F MRI of a perfluorooctyl bromide emulsion: application to a dynamic biodistribution study and oxygen tension mapping in the mouse liver and spleen. Nmr in Biomedicine. 2012;25(4):654–660. doi: 10.1002/nbm.1781. [DOI] [PubMed] [Google Scholar]
- 62.Troster SD, Muller U, Kreuter J. Modification of the Body Distribution of Poly(Methyl Methacrylate) Nanoparticles in Rats by Coating with Surfactants. Int J Pharm. 1990;61(1–2):85–100. [Google Scholar]
- 63.Vittaz M, Bazile D, Spenlehauer G, Verrecchia T, Veillard M, Puisieux F, Labarre D. Effect of PEO surface density on long-circulating PLA-PEO nanoparticles which are very low complement activators. Biomaterials. 1996;17(16):1575–1581. doi: 10.1016/0142-9612(95)00322-3. [DOI] [PubMed] [Google Scholar]
- 64.Knoess C, Siegel S, Smith A, Newport D, Richerzhagen N, Winkeler A, Jacobs A, Goble RN, Graf R, Wienhard K, Heiss WD. Performance evaluation of the microPET R4 PET scanner for rodents. Eur J Nucl Med Mol Imaging. 2003;30(5):737–747. doi: 10.1007/s00259-002-1052-6. [DOI] [PubMed] [Google Scholar]