

Abstract
Vitronectin (VN) is a multi-functional protein involved in extracellular matrix (ECM)-cell binding through integrin receptors on the cell surface, which is an important environmental process for maintaining biological homeostasis. We investigated how VN affects the survival of endothelial cells after radiation damage. VN attenuated radiation-induced expression of p21, an inhibitor of cell cycle progression, and selectively inhibited Erk- and p38 MAPK-dependent p21 induction after radiation exposure through regulation of the activity of GSK-3β. VN also reduced the cleavage of caspase-3, thereby inhibiting radiation-induced apoptotic cell death. These results suggest that VN has important roles in cell survival after radiation damage.
Keywords: Vitronectin, Integrin, p21, Radiation, MAPK
Highlights
▸ Vitronectin prevented apoptosis of endothelial cells after radiation damage. ▸ Treatment of endothelial cells with vibronectin abrogated G1 arrest. ▸ Vitronectin inhibited p21 expression through prevention of ERK and p38 MAPK pathways. ▸ Radiation-induced inactivation of GSK-3β was suppressed by vibronectin.
1. Introduction
After accidental exposure to a high dose of ionizing radiation (IR), therapeutic strategies for patients with acute radiation syndrome (ARS) remain a major problem. At relatively high doses, gastrointestinal and vascular syndromes emerge in a dose-dependent manner, which lead to multi-organ dysfunction [1]. Moreover, damage to the target organ is related to systemic inflammatory response mainly caused by the release of cytokines from damaged vascular endothelial cells [1]. Vascular endothelial cells restrict the passage of inflammatory cells and circulating molecules into underling tissues. Following endothelial cell death after radiation exposure, para-cellular permeability is increased, which leads to unrestricted passage of circulating molecules and inflammatory cells and eventually triggers uncontrollable inflammation and further tissue damage [2]. Therefore, it is important to improve survival and to maintain the function of IR-damaged vascular endothelial cells in order to control and mitigate ARS.
A recent report showed that IR enhanced the expression of integrin αvβ3 in human umbilical vein endothelial cells (HUVECs), and HUVECs were sensitized to IR by blocking this receptor [3]. Vitronectin (VN) recognizes the integrin αvβ3 receptor on the cell surface, resulting in receptor clustering for the subsequent initiation of intracellular signaling pathways [4,5]. Therefore, it is reasonable to postulate that vascular endothelial cells demand binding of VN by its integrin receptor in order to sustain its own survival signaling after radiation damage.
VN is a cell-adhesive glycoprotein found in the extracellular matrix (ECM) of various tissues and in circulating blood, and VN is implicated in various pathophysiological processes including inflammation, cell migration, angiogenesis, and regulation of plasminogen activation [4]. More than 95% VN in plasma exists as a latent form (monomeric/dimeric form) and others in the ECM as multimeric forms [4]. The function and activity of VN strongly depend on its binding partners, such as plasminogen activator inhibitor 1 (PAI-1), heparin, or cell surface receptors including integrin receptors [4]. However, it remains unclear how abundant latent VN (concentration: 200–400 μg/mL) in plasma impacts on the fate of vascular endothelial cells after IR damage. Therefore, in this study, we investigated the effects and mechanisms of latent VN in plasma against IR-damaged HUVECs.
2. Materials and methods
2.1. Reagents
Human purified plasma VN protein was purchased from MILLIPORE (CC808, Lot NG1888899). Antibodies against GAPDH (#5174), GSK-3β (#9315), phosphoglycogen-synthase kinase-3β (GSK-3β) at Ser9 (#9336), p44/42 (extracellular signal-regulated kinase; ERK) kinase (#9102), phospho-ERK at Thr202/Tyr204 (#9101), p38 mitogen activated protein kinase (MAPK) (#9212), phospho-p38 MAPK at Thr180/Tyr182 (#9211), phospho-stress activated protein kinase/c-Jun-N-terminal kinase (SAPK/JNK) at Thr183/Tyr185 (#9251), AKT (#9272), AKT1 (#2938), AKT2 (#3063), phospho-AKT at Ser473 (#4051), and poly (ADP-ribose) polymerase (PARP) (#9532) were purchased from Cell Signaling Technology. An antibody against SAPK/JNK (sc-571) was purchased from Santa Cruz Technology, an antibody against p21 (556431) was purchased from BD Bioscience, and an antibody against integrin αv/CD51 was purchased from EPITOMICS. Inhibitors of PI3K (LY294002), MEK1 (PD98059), p38 (SB203580), and JNK (JNK inhibitor II, Anthra[1,9-cd]pyrazol-6(2H)-one) were purchased from CALBIOCHEM. An inhibitor of GSK3-β (LiCl) was purchased from Wako.
2.2. Cell lines
HUVECs were purchased from the Health Science Research Resource Bank and maintained in MCDB107 (COSUMO BIO Co., Ltd.) supplemented with 15% heat-inactivated FBS, penicillin/streptomycin (Invitrogen), 10 ng/mL bFGF (Sigma), and 50 μg/mL heparin. Cells were incubated in a humidified atmosphere at 37 °C with 5% CO2. Col-I coated dishes (11-018-002, IWAKI) were used for cell culture.
2.3. γ-Ray irradiation
Cells were irradiated using a Cesium-137 (Cs137) gammator (model M Gammator, Irradiation Machinery), at a dose rate of 8.0 Gy/min on a rotating platform.
2.4. Cell cycle analysis
Cells were irradiated and cultured with or without VN (10 μg/mL) for 28 h. The harvested cells were treated with PBS containing 0.1% Triton X-100 (Wako) for 5 min on ice, and stained with propidium iodide (PI; 50 μg/mL, Sigma). An analysis of the cell cycle distribution was performed using a fluorescent cell analyzer (FACSCalibur, Becton Dickinson).
2.5. SDS–PAGE and western blotting analysis
Harvested cells were lysed in SDS sample buffer (300 mM Tris–HCl, pH 6.8, 10% SDS, 1% 2-mercaptoethanol, 1% glycerol) with sonication, and then heated for 7 min at 95 °C. Proteins were separated by SDS–PAGE and transferred onto PVDF membranes (Advantec Toyo). The membrane was reacted with each primary antibody in Tris-buffered saline (10 mM Tris–HCl, pH 7.4, 100 mM NaCl, 0.1% Tween-20) supplemented with 2% blocking agent (GE Healthcare) for 1 h at room temperature after blocking of the membrane. After the primary antibody reaction, membranes were labeled with each corresponding HRP-conjugated antibody (GE Healthcare) and then detected using ECL Prime western blotting substrate (GE Healthcare).
2.6. MAPK, AKT and GSK-3β inhibition assay
HUVECs with incubated with inhibitors; LY294002 (20 μM), PD98059 (50 μM), SB203580 (20 μM), JNK II (200 nM) and/or LiCl (20 mM), for 2 h at 37 °C before irradiation. Then, the cells were irradiated (8 Gy), and cultured for a further 28 h. Harvested cells were subjected to SDS–PAGE and immunoblotting analysis in accordance with the procedure described above.
2.7. Statistical analysis
The significance of differences between the control and experimental groups was determined using Student's t-test depending on the data distribution. Statistical analysis was performed using Excel 2003 software (Microsoft) with add-in software Statcel 2.
3. Results
3.1. Exogenously added latent vitronectin prevents γ-ray-induced cell death in HUVECs
Radiation-enhanced expression of integrin αv was detected in HUVECs by western blotting analysis (Fig. 1(A)), suggesting that cellular signaling from integrin affects the survival of radiation-damaged cells. Therefore, we examined whether VN, one of the ligands of integrin αvβ3, affects the survival of radiation-damaged HUVECs. As shown in Fig. 1(B), exogenously added latent VN increased the survival ratio of radiation-damaged HUVECs in a dose-dependent manner, which was also observed by recombinant VN treatments (Supplementary Fig. S2(A)). The addition of VN to HUVECs 10 min after irradiation inhibited the production of cleaved active-caspase3 and also inhibited the cleavage of a downstream substrate of caspase3, PARP (Fig. 1(C)). These results indicated that latent VN can prevent radiation-induced apoptotic cell death in HUVECs.
Fig. 1.
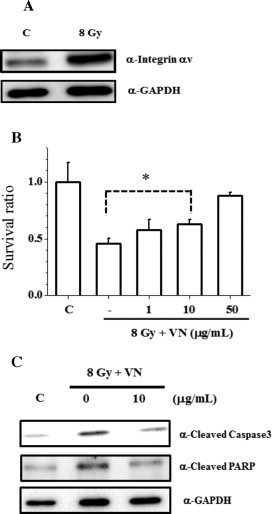
VN protects against ionizing irradiation-induced cell death in HUVECs. (A) Radiation-induced integrin αv expression. Cells were cultured for 28 h after 8 Gy irradiation. Subsequently, cells in each condition were harvested and whole-cell extracts were subjected to immunoblotting analysis with the indicated antibodies. C, unirradiated control. (B) Cells were exposed to irradiation at a dose of 8 Gy. VN was added to the cell culture 10 min after irradiation, and the cells were continued to be cultured for 28 h. Subsequently, the cells in each condition were harvested, and the number of viable cells was determined by the trypan blue exclusion test using a hemocytometer. *p < 0.05 by Student's t-test. (C) After irradiation, cells were treated with VN, and subsequently harvested as in (B). Whole-cell extracts were subjected to immunoblotting analysis with the indicated antibodies.
3.2. Vitronectin suppresses cell cycle arrest by inhibiting γ-ray-induced up-regulation of cell cycle inhibitor p21 in HUVECs
In general, radiation induces DNA damage and subsequent cell cycle arrest before activation of apoptotic cell death. Therefore, we next examined the effect of VN on cell cycle distribution of irradiated HUVECs by flow cytometric analysis. As shown in Fig. 2(A), radiation exposure induced cell cycle arrest at G1 and G2/M phase. However, VN treatment abolished G1 phase arrest, which was supported by a 1.3-fold increase in the G2/M population compared with radiation treatment alone (Fig. 2(A)). VN treatment also decreased the sub-G1 phase population by one-third, an indicator of apoptotic DNA fragmentation (Fig. 2(A)). These results again support the conclusion that VN treatment prevents radiation-induced apoptotic cell death in HUVECs. Because p21 is an inhibitor of the G1-S phase transition and its expression is induced by DNA damage, we determined whether VN affects DNA damage-induced p21 expression. As shown in Fig. 2(B), VN treatment decreased radiation-induced expression of p21 protein. Similarly, recombinant VN also suppressed p21 expression (Supplementary Fig. S2(B) and (C)). Although the levels of p53, a major transactivator of p21, also increased in response to DNA damage, p53 protein levels were unchanged by VN treatment (data not shown). These results suggest that other mechanisms may accommodate the regulation of p21 expression by VN treatment (Fig. 2(B)).
Fig. 2.

VN attenuates radiation-induced G1 phase cell cycle arrest through decreasing the expression level of the cell cycle inhibitor protein p21. Cells were exposed to irradiation at a dose of 8 Gy. VN was added to the cell culture 10 min after irradiation, and cells culturing was continued for 28 h. (A) Cells in each condition were subjected to cell cycle analysis using PI staining in accordance with the procedure as described in Section 2. (B) Whole-cell extracts were prepared from HUVECs under the indicated conditions and were subjected to immunoblotting analysis with the indicated antibodies. The relative amounts of p21 protein were quantified and shown as graphs.
3.3. Vitronectin suppresses γ-ray-induced up-regulation of p21 through prevention of activation of ERK and p38 MAP kinase pathways in HUVECs
A number of reports indicate that signal transduction, such as the MAPK pathways, is involved in the regulation of p21 expression, although the involvement and effect of p21 expression depend on each cell type [6]. Therefore, we examined the involvement of the PI3K/AKT, ERK, p38 MAPK, and SAPK/JNK pathways in the radiation-induced p21 expression in HUVECs. For this purpose, we used chemical inhibitors, LY294002 for PI3K/AKT, PD98059 for ERK, SB203580 for p38 MAPK, and JNK inhibitor II for SAPK/JNK pathways. Except for the JNK inhibitor II, the other inhibitors, including LY294002, PD98059, and SB203580, prevented the radiation-induced expression of p21 in HUVECs (Fig. 3(A)). When pairs of inhibitors, including LY294002, PD98059, and SB203580, were added, the inhibitory effect was enhanced. Furthermore, treatment with all three inhibitors inhibited the expression of p21 much more strongly. Therefore, these results suggest the involvement of the PI3K/AKT, ERK, and p38 MAPK pathways in the radiation-induced p21 expression in HUVECs. We subsequently examined whether VN treatment affected the radiation-induced activation of the protein kinases in these signal transduction pathways. VN treatment preferentially inhibited radiation-induced phosphorylation of ERK and p38 MAPK whereas phosphorylation of AKT and JNK was maintained after VN treatment (Fig. 3(B)). Moreover, although previous studies suggest that AKT1 and AKT2 have distinct functions in DNA damage response [7], VN treatment did not change the cellular amount of AKT1 or AKT2 (Fig. 3(C)). Therefore, the above results indicate that VN treatment prevents radiation-induced p21 expression through inhibiting the activation of both ERK and p38 MAPK pathways in HUVECs.
Fig. 3.
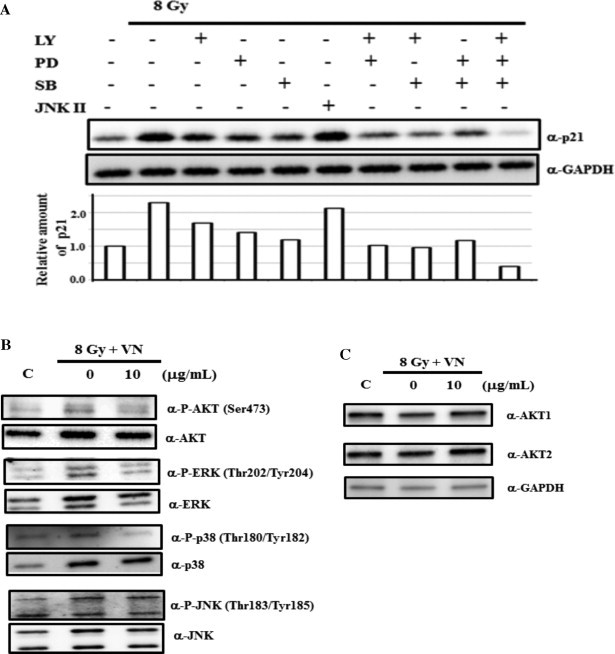
VN inhibits p21 expression through the ERK and p38MAPK pathways. (A) HUVECs were pre-treated with the indicated inhibitors for 2 h before irradiation, and cell culture was continued for 28 h in the presence of the indicated inhibitors, as shown in Section 2. Whole-cell extracts were subjected to immunoblotting analysis with the indicated antibodies. (B, C) Cells were exposed to irradiation at a dose of 8 Gy. VN was added to the cell culture 10 min after irradiation, and the cell culture was continued for 28 h. Whole-cell extracts were subjected to immunoblotting analysis with the indicated antibodies.
3.4. Vitronectin suppresses γ-ray-induced inactivation of GSK-3β in HUVECs
Several studies indicate that GSK-3β is involved in the regulation of p21 expression in response to genotoxic stresses. Therefore, we initially examined the effect of VN treatment on the phosphorylation state of GSK-3β at Ser9, an indicator of its inactive form. Consistent with the previous study [8], radiation exposure increased the phosphorylation of GSK-3β at Ser9 in HUVECs (Fig. 4(A)). VN treatment suppressed the radiation-induced phosphorylation of GSK-3β at Ser9, which was correlated with a reduction in p21 protein levels. Because our initial results indicated the involvement of PI3K/AKT, ERK, and p38 MAPK pathways in radiation-induced expression of p21, we examined the relationship between these protein kinases and the radiation-induced phosphorylation of GSK-3β. Treatment with PD98059 or SB203580, but not LY294002, markedly abolished the phosphorylation of GSK-3β (Fig. 4(B)). On the other hand, when cells were pre-treated with LiCl, an inhibitor of GSK3β, before irradiation, phosphorylation of GSK-3β at Ser9 was further increased compared with cells without the inhibitor (Fig. 4(C)). Moreover, the expression levels of p21 were also increased (Fig. 4(C)). Collectively, our results suggest that VN treatment inhibits radiation-induced expression of p21 through regulation of the activity of GSK-3β in HUVECs.
Fig. 4.
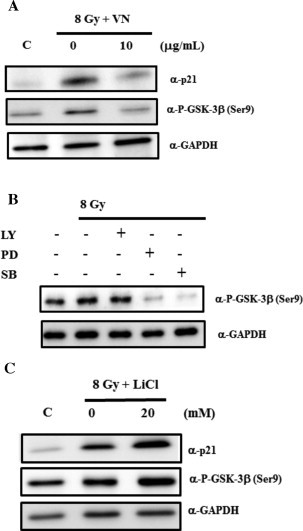
VN inhibits p21 expression thorough the inactivation of GSK-3β following the ERK and p38MAPK pathways. (A) Cells were exposed to irradiation at a dose of 8 Gy. VN was added to the cell culture 10 min after irradiation, and cell culture was continued for 28 h. Whole-cell extracts were subjected to immunoblotting analysis with the indicated antibodies. (B, C) HUVECs were pre-treated with the indicated inhibitors for 2 h before irradiation, and cell culture was continued for 28 h in the presence of the indicated inhibitors, as shown in Section 2. Whole-cell extracts were subjected to immunoblotting analysis with the indicated antibodies.
4. Discussion
In this study, we demonstrated that latent VN inhibits radiation-induced p21 expression and protects against apoptotic cell death in HUVECs via the suppression of ERK and p38 MAPK signal pathways. Furthermore, VN protected against radiation-induced phosphorylation of GSK-3β, and inhibition of ERK or p38 MAPK using chemical inhibitors similarly abolished the radiation-induced phosphorylation of GSK-3β. Therefore, our results strongly suggest that the reduced activation of both ERK and p38 MAPK prevented radiation-induced phosphorylation of GSK-3β in HUVECs. To our best knowledge, this is the first report demonstrating that latent VN improved the survival of vascular endothelial cells by inhibiting radiation-induced apoptotic cell death.
We demonstrated that latent VN reduced p21 expression through the prevention of phosphorylation of GSK-3β in radiation-damaged HUVECs. This observation is strongly supported by Tan et al. [8]. They revealed that increased levels of the dephosphorylated form of GSK-3β at Ser9 might reduce the expression levels of p21 by inhibiting a transactivator of p21, c-Jun, or by inducing the proteasomal degradation of p21 [8].
In studying the intracellular signaling pathway of VN, both ERK and p38 MAPK participated in the anti-apoptotic process in our HUVEC model. A recent report also showed that VN inhibits neutrophil apoptosis [9]. In this report, the anti-apoptotic effect against neutrophil apoptosis was regulated through the PI3K/AKT and ERK pathways but not the p38 MAPK pathway. Thus, the intracellular signaling of the anti-apoptotic ability of VN may depend on each cell-type or apoptotic inducer.
VN can exist in two forms with latent (monomer/dimer) or multimers status [4]. Previous studies revealed that fixed multimetric VN had anti-apoptotic ability against genotoxic stimuli [10,11]. We used latent VN in our experiment because more than 95% of VN in plasma is in the latent form [4]. We assumed that an experiment using latent VN may be more suitable for examining and reflecting anti-apoptotic ability of VN in vivo against IR-damaged endothelium. However, there have been no previous reports evaluating the anti-apoptotic activity of latent VN.
The major biological functions and characteristics of VN may be determined by the binding ligands and biochemical modifications to the protein itself [4]. The integrin receptor is one of the ligands of VN and is important to the induction of survival signaling after IR damage [3,4]. We revealed that the expression levels of this VN ligand, integrin αv, were enhanced in IR-damaged HUVECs. Although we did not examine other VN ligands, e.g. PAI-1, a previous report revealed that PAI-1 levels were also increased after IR-damage [12], which was considered interfere with VN-integrin binding [13]. Therefore, it is possible that quantitative changes in VN ligands could affect the biological functions of VN in ARS.
With regard to the biochemical modifications of VN itself, the phosphorylation of VN at Ser378 by cAMP-dependent protein kinase A (PKA) leads to reduced PAI-1 binding, whereas phosphorylation of VN at Ser362 by protein kinase C (PKC) induces persistent PAI-1 binding [14]. Moreover, the phosphorylation of VN at Thr50 and Thr57 by casein kinase 2 (CK2) leads to enhanced integrin αvβ3 binding [14]. IR-damaged cells may release various enzymes, including PKA, PKC or CK2, which could modify biochemical characteristics of VN. Thus, owing to its multifaceted abilities, the regulatory systems of VN may be very complicated in vivo. In our limited in vitro study, the latent form of VN can protect against apoptotic cell death in HUVECs after IR-damage. However, it is important to also understand these complicated situations in vivo, and to clarify the precise mechanisms about how to modify VN.
In conclusion, we revealed that latent VN can attenuate cell cycle arrest and prevent cell death after IR-damage. Although most plasma VN exists as a latent form, even this form clearly has the potential to sustain survival signaling depending on the demands of surrounding vascular endothelial cells after radiation damage.
Acknowledgments
This work was supported in part by a grant of NIRS and also partly by Grants-in-Aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Appendix A. Supplementary material
Purity of recombinant human vitronectin. Each recombinant human vitronectin (rVN) expressed in HEK293 (A) and N-terminal GST-tagged rVN expressed in E. coli (B) was subjected to Coomassie Brilliant Blue (CBB) staining (left) and immunoblotting analysis (right).
rVN improved survival and inhibited p21 expression in radiation-damaged HUVECs. (A) Cells were exposed to irradiation at a dose of 8 Gy. Each rVN was added to the cell culture 10 min after irradiation, and the cells were continued to be cultured for 28 h. Subsequently, the cells in each condition were harvested, and the number of viable cells was determined by the trypan blue exclusion test using a hemocytometer. (B) After irradiation, cells were treated with each rVN, and subsequently harvested as in (A). Whole-cell extracts were subjected to immunoblotting analysis with the indicated antibodies. (C) HUVECs were treated with rVN as in (A), and cells in each condition were immunostained: the cells were stained for p21 (green) and nucleus using DAPI.
References
- 1.Herodin F., Drouet M. Cytokine-based treatment of accidentally irradiated victims and new approaches. Exp. Hematol. 2005;33:1071–1080. doi: 10.1016/j.exphem.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Pai S., Khakoo A.Y., Zhao J., Jimenez F., Gerber M.H., Harting M., Redell J.B., Grill R., Matsuo Y., Guha S., Cox C.S., Reitz M.S., Holcomb J.B., Dash P.K. Human mesenchymal stem cells inhibit vascular permeability by modulating vascular endothelial cadherin/β-catenin signaling. Stem Cells Dev. 2011;20:89–101. doi: 10.1089/scd.2010.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Albert J.M., Cao C., Geng L., Leavitt L., Hallahan D.E., Lu B. Integrin alpha v beta 3 antagonist Cilengitide enhances efficacy of radiotherapy in endothelial cell and non-small-cell lung cancer models. Int. J. Radiat. Oncol. Biol. Phys. 2006;65:1536–1543. doi: 10.1016/j.ijrobp.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 4.Preissner K.T., Reuning U. Vitronectin in vascular context: facets of a multitalented matricellular protein. Semin. Thromb. Hemost. 2011;37:408–424. doi: 10.1055/s-0031-1276590. [DOI] [PubMed] [Google Scholar]
- 5.Jin Y.J., Park I., Hong I.K., Byun H.J., Choi J., Kim Y.M., Lee H. Fibronectin and vitronectin induce AP-1-mediated matrix metalloprotease-9 expression through integrin α5β1/αvβ3-dependent Akt, ERK and JNK signaling pathways in human umbilical vein endothelial cells. Cell Signal. 2011;23:125–134. doi: 10.1016/j.cellsig.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Dent P., Yacoub A., Fisher P.B., Hagan M.P., Grant S. MAPK pathways in radiation responses. Oncogene. 2003;22:5885–5896. doi: 10.1038/sj.onc.1206701. [DOI] [PubMed] [Google Scholar]
- 7.Heron-Milhavet L., Franckhauser C., Rana V., Berthenet C., Fisher D., Hemmings B.A., Fernandez A., Lamb N.J. Only Akt1 is required for proliferation, while Akt2 promotes cell cycle exit through p21 binding. Mol. Cell Biol. 2006;26:8267–8280. doi: 10.1128/MCB.00201-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan J., Geng L., Yazlovitskaya E.M., Hallahan D.E. Protein kinase B/Akt-dependent phosphorylation of glycogen synthase kinase-3 β in irradiated vascular endothelium. Cancer Res. 2006;66:2320–2327. doi: 10.1158/0008-5472.CAN-05-2700. [DOI] [PubMed] [Google Scholar]
- 9.Bae H.B., Zmijewski J.W., Deshane J.S., Zhi D., Thompson L.C., Peterson C.B., Chaplin D.D., Abraham E. Vitronectin inhibits neutrophil apoptosis through activation of integrin-associated signaling pathways. Am. J. Respir. Cell Mol. Biol. 2012;46:790–796. doi: 10.1165/rcmb.2011-0187OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uhm J.H., Dooley N.P., Kyritsis A.P., Rao J.S., Gladson C.L. Vitronectin, a glioma-derived extracellular matrix protein, protects tumor cells from apoptotic death. Clin. Cancer Res. 1999;5:1587–1594. [PubMed] [Google Scholar]
- 11.Wadsworth S.J., Freyer A.M., Corteling R.L., Hall I.P. Biosynthesized matrix provides a key role for survival signaling in bronchial epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2004;286:L596–603. doi: 10.1152/ajplung.00217.2003. [DOI] [PubMed] [Google Scholar]
- 12.Abderrahmani R., Francois A., Buard V., Tarlet G., Blirando K., Hneino M., Vaurijoux A., Benderitter M., Sabourin J.C., Milliat F. PAI-1-dependent endothelial cell death determines severity of radiation-induced intestinal injury. PLoS One. 2012;7:e35740. doi: 10.1371/journal.pone.0035740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stefansson S., Lawrence D.A. The serpin PAI-1 inhibits cell migration by blocking integrin alpha V beta 3 binding to vitronectin. Nature. 1996;383:441–443. doi: 10.1038/383441a0. [DOI] [PubMed] [Google Scholar]
- 14.Schvartz I., Seger D., Shaltiel S. Vitronectin. Int. J. Biochem. Cell Biol. 1999;31:539–544. doi: 10.1016/s1357-2725(99)00005-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Purity of recombinant human vitronectin. Each recombinant human vitronectin (rVN) expressed in HEK293 (A) and N-terminal GST-tagged rVN expressed in E. coli (B) was subjected to Coomassie Brilliant Blue (CBB) staining (left) and immunoblotting analysis (right).
rVN improved survival and inhibited p21 expression in radiation-damaged HUVECs. (A) Cells were exposed to irradiation at a dose of 8 Gy. Each rVN was added to the cell culture 10 min after irradiation, and the cells were continued to be cultured for 28 h. Subsequently, the cells in each condition were harvested, and the number of viable cells was determined by the trypan blue exclusion test using a hemocytometer. (B) After irradiation, cells were treated with each rVN, and subsequently harvested as in (A). Whole-cell extracts were subjected to immunoblotting analysis with the indicated antibodies. (C) HUVECs were treated with rVN as in (A), and cells in each condition were immunostained: the cells were stained for p21 (green) and nucleus using DAPI.