

Abstract
We characterize the allylic epoxyalcohols and their trihydroxy hydrolysis products generated from 9R- and 9S-hydroperoxy-octadecenoic acid (HPODE) under non-enzymatic conditions, reaction with hematin and subsequent acid hydrolysis, and enzymatic conditions, incubation with Beta vulgaris containing a hydroperoxide isomerase and epoxide hydrolase. The products were resolved by HPLC and the regio and stereo-chemistry of the transformations were determined through a combination of 1H NMR and GC-MS analysis of dimethoxypropane derivatives. Four trihydroxy isomers were identified upon mild acid hydrolysis of 9S,10S-trans-epoxy-11E-13S-hydroxyoctadecenoate: 9S,10R,13S, 9S,12R,13S, 9S,10S,13S and 9S,12S,13S-trihydroxy-octadecenoic acids, in the ratio 40:26:22:12. We also identified a prominent -ketol rearrangement product from the hydrolysis as mainly the 9-hydroxy-10E-13-oxo isomer. Short incubation (5 min) of 9R- and 9S-HPODE with Beta vulgaris extract yielded the 9R- and 9S-hydroxy-10E-12R,13S-cis-epoxy products respectively. Longer incubation (60 min) gave one specific hydrolysis product via epoxide hydrolase, the 9R/S,12S,13S-trihydroxyoctadecenoate. These studies provide a practical approach for the isolation and characterization of allylic epoxy alcohol and trihydroxy products using a combination of HPLC, GC-MS and 1H NMR.
Keywords: 9-HPODE, trihydroxyoctadecenoic acid, linoleic acid, epoxyalcohol synthase, epoxide hydrolase, hematin
1. Introduction
The oxygenation at C9 of linoleic acid is the gateway to a diverse collection of oxylipin products (Andreou et al., 2009). C9 oxygenation in plants is catalyzed by 9-lipoxygenases (9-LOX), well-represented among the LOX genes (Feussner and Wasternack, 2002). Linoleate 9-lipoxygenases are also known in prokaryotes (Andreou et al., 2008; Gao et al., 2010; Zheng et al., 2008), and are a catalytic activity of arachidonate 12R-LOX in higher animals (Meruvu et al., 2005; Siebert et al., 2001; Zheng et al., 2011). Among the many possible transformations of the resulting 9-hydroperoxy-octadecenoic acid (9-HPODE), in keeping with other fatty acid hydroperoxides are rearrangements to epoxy-hydroxy derivatives (fatty acid epoxyalcohols), with subsequent hydrolysis producing a collection of trihydroxy-octadecenoates.
The non-enzymatic transformation of fatty acid hydroperoxides to epoxyalcohols is catalyzed by heme or transition metals, resulting in a mixture of isomers, as analyzed earlier utilizing 13-HPODE (Dix and Marnett, 1985; Gardner et al., 1974; Hamberg, 1975). The present study aims to address the heme transformations with 9-HPODE as starting material. Enzymatic conversion results in regio- and stereo-specific formation of epoxyalcohols. Lipoxygenases can also catalyze epoxyalcohol synthesis (Garssen et al., 1976; Nigam et al., 2004; Pace-Asciak et al., 1995; Yu et al., 2003), the transformation being favored under anaerobic conditions (Zheng and Brash, 2010). Heme and lipoxygenase catalysis involves formation of alkoxyl and epoxyallylic radical intermediates, whereas oxygen transfer from the hydroperoxide with epoxidation of a remote double bond is effected by other enzymes. These include peroxygenase (Blée et al., 1993; Hamberg and Hamberg, 1996), catalase-related hemoproteins (Gao et al., 2009; Niisuke et al., 2009), as well as putative P450-like enzymes that remain to be characterized, for example in the fish fungus Saprolegnia parasitica (Hamberg et al., 1986), potato leaves (Hamberg, 1999), and beetroot (Hamberg and Olsson, 2011).
Trihydroxy hydrolysis products are readily formed from fatty acid epoxyalcohols, and in the case of potential 9-HPODE-derived products, these triols are reported in such diverse settings as; an in-vivo pathogen response in potato leaves (Göbel et al., 2002; Hamberg, 1999), in brewing, where their formation contributes to the bitter flavor of beer (Garbe et al., 2005; Hamberg, 1991b), as products in mammalian blood vessels and leukocytes (Claeys et al., 1985; Funk and Powell, 1985), and in preserved meat as a marker of lipid peroxidation (Püssa et al., 2009). The adjuvant activity of 9S,12S,13S-trihydroxyoctadecenoic acid (commonly known as pinellic acid) and its isomers in the administration of flu vaccine has also been reported (Shirahata et al., 2003). In addition, our interests in the transformations of linoleate and its esters through LOX activity in mammalian epidermis (Zheng et al., 2011) has focused our attention on the preparation and characterization of 9-HPODE-derived epoxyalcohols and the resulting triols. There are effective methods for their analysis in the literature, although the separations cannot be related to typical HPLC methodology, which was one impetus for our conducting the present study. Hamberg made assignments based on GLC and TLC mobilities coupled with mass spectrometry and chemical degradation (oxidative ozonolysis, followed by GLC analysis of the fragments) (Hamberg, 1991a). Shirahata et al (Shirahata et al., 2006) prepared all possible stereo isomers of pinellic acid by total synthesis and compared their spectral properties by NMR. The use of an acetonide derivative to capture the vicinal 12,13-diol in the 9,12,13-triol is part of this procedure and has been applied in other studies to identify single naturally occurring fatty acid triols by NMR (Bruno et al., 1992; Cardellina and Moore, 1980). We make use of this procedure in our NMR assignments herein.
Accordingly, the objectives of the current study were: i) To develop a practical method for production and purification of allylic epoxyalcohols via hematin treatment of 9-hydroperoxy-linoleic acid. ii) To characterize the HPLC separation of the trihydroxy-octadecenoates resulting from hydrolysis of the allylic alcohols. iii) To utilize a GC-MS and NMR approach for the assignment of the trihydroxy-octadecenoate regio- and stereo-chemistry. iv) To compare the enzymatic synthesis, isomeric distribution and chirality of epoxyalcohols and triols from 9R- and 9S-hydroperoxy-linoleic acid in Beta vulgaris, a source of epoxyalcohol synthase (Hamberg and Olsson, 2011), and v) To characterize specific delta-ketols as products from the mild acid treatment of allylic trans-epoxyalcohols.
2. Experimental
2.1 Generation of 9R-HPODE and 9S-HPODE substrates
2.1.1 9R-HPODE
Linoleic acid (10 mg) in 0.5 ml ethanol was stirred into 200 ml 50 mM Tris pH 7.5, 150 mM NaCl, 20 mM CHAPS with a strong oxygen stream on the surface of the solution. Anabaena LOX enzyme (a linoleate 9R-LOX, (Zheng et al., 2008)) was added as 150 μl of a 20 mg/ml solution; reaction was complete in 2 min as evidenced by UV analysis of the conjugated diene chromophore of the product. The solution was then acidified to pH 4.0 and extracted with ethyl acetate, washed twice with half volume of water, taken to dryness and redissolved in a small volume of methanol for storage prior to HPLC. The 9R-HPODE was purified by semi-preparative SP-HPLC using a Beckman Ultrasphere silica 10 μm column, with isocratic hexane/ IPA/acetic acid (100:2:0.1), a flow rate of 4 ml/min, with UV monitoring at 235 nm.
2.1.2 9S-HPODE
9S-HPODE was prepared using the linoleate 9S-LOX in potato (Galliard and Phillips, 1971). Yukon Gold potatoes were diced and blended with 255 ml water. To eliminate starch, the solution was then stirred with 250 μl Tween 80, 320 μl Viscozyme L (Sigma) and 160 μl Amyloglucosidase (from Aspergillus niger 300 units/ml, Sigma) for one hour at room temperature. After a low speed centrifugation (500 RPM, 10 min) the supernatant was poured through 2 layers of cheesecloth into a graduated cylinder. Sodium acetate (1M) pH 5.5 was then added to 10% volume. The resulting enzyme solution was then suspended in a water bath at 15°C. Linoleic acid in ethanol (1 g) was added drop wise with a strong stream of oxygen blowing on the surface of the solution and the reaction stirred for 90 min. The resulting solution was acidified to pH 4.0 and extracted with dichloromethane (DCM). The DCM was then washed twice with half volume of H2O and taken to dryness. The 9S-HPODE was purified using SP-HPLC with a Beckman Ultrasphere silica 10 μm column with an isocratic mobile phase of hexane/IPA/acetic acid (100:2:0.1).
2.2 Reaction of 9S-HPODE with hematin
A hematin solution (5 mg/ml) was prepared by grinding the crystals to a fine powder and stirring with 0.1M K2HPO4 adjusted to pH 11.0 with NaOH. 9S-HPODE (20 mg in 0.5 ml ethanol) was mixed with 0.1 M K2HPO4 (20 ml, pH 8.5), 1 ml of the hematin solution was added and the reaction left for 15 min at room temperature. The sample was cooled on ice, two volumes of ice-cold DCM were added, then a predetermined mix of 1M KH2PO4 (5 ml) and 1 M HCl solution, sufficient to bring the pH to ~ 4.5, followed immediately by vigorous mixing of the phases. The aqueous layer was removed and the DCM washed twice with ice-cold water. The DCM was transferred to a fresh vial, dried under N2, and the sample stored at −20°C in DCM:MeOH (2:1, v/v) prior to initial purification on an open-bed silica Bond-Elut cartridge.
2.2.1 Silica Cartridge purification of extract
The extract from the hematin reaction was pre-dissolved in ethyl acetate (2 ml) then hexane (8 ml) was added and the sample immediately applied to a 1g Bond-Elut silica cartridge preconditioned with 25% ethyl acetate in hexane. A further 10 ml rinse of the sample vial with ethyl acetate (25%) in hexane was added and followed by a further 10 ml 25% ethyl acetate. The products were then eluted with 100% ethyl acetate (10 ml), and the sample was dried under N2 and stored for analysis in DCM:MeOH, 2:1.
2.2.2 HPLC purification of hematin products
The range of products generated from the hematin reaction were analyzed on an Agilent 1100 series HPLC using a Thomson Advantage Silica 150Å 5μ column (250 × 4.6 mm) an isocratic solvent of hexane:IPA:acetic acid (100:2:0.02), a flow rate of 1 ml/min, with diode array detection at wavelengths of 205, 220, 235 and 270 nm.
2.3 HPLC separation of trihydroxy-octadecenoate products from allylic epoxyalcohol hydrolysis
2.3.1 Acid hydrolysis of 9S,10S-trans-epoxy-11E-13S-hydroxyoctadecenoate
To the allylic epoxide dissolved in acetonitrile (5 ml), 1% aqueous acetic acid (20 ml) was added and mixed thoroughly. After standing at room temperature for 30 min, the hydrolysis products were extracted in a C18 Bond Elute cartridge, washed with two volumes of H2O and eluted with MeOH.
2.3.2 Purification of trihydroxy-octadecenoate products
Initial RP-HPLC analysis employed a Waters Symmetry C18 column (250 × 4.6 mm) eluted isocratically with methanol/water/acetic acid (80/20/0.01) at 1 ml/min. The 205 nm-absorbing peaks in the triol region of the chromatogram at ~ 4 min retention time were pooled and subsequently resolved using a Chiralpak AD column (250 × 4.6 mm) with an isocratic mobile phase of hexane/methanol/ethanol/acetic acid (86.5/4.5/9/0.1) run at 1 ml/min. The peak designated as II was further separated using a Chiralpak OD-H column (250 × 4.6 mm) with a mobile phase of hexane/ethanol/acetic acid, (97.5/2.5/0.1) run at 1 ml/min.
2.4 Assignment of regio- and stereo-chemistry of trihydroxy-octadecenoates via 1H NMR and GC-MS analysis
Methyl esters were prepared by dissolving the products in MeOH (typically 10 μl), followed by addition of several drops of ethereal diazomethane (~ 100 μl); after mixing and standing for 30 s on ice the sample is taken to dryness under a stream of nitrogen. Methyl esters of the triols derived from hydrolysis of methyl 9S,10S-trans-epoxy-11E-13S-hydroxyoctadecenoate were purified as a group by SP-HPLC using a Thomson Advantage silica column (25 × 0.46 cm), a solvent of hexane/IPA (90:10, by vol.) run at 1 ml/min, with UV detection at 205 nm and a retention time of 9–10 min. Acetonide derivatives were prepared by addition of DMP:acetone (1:1) containing 1 mM p-toluene sulfonic acid to the dry methyl ester derivatives (reaction is complete within 5 min at room temperature); the acid was then neutralized using an equal volume of triethylamine (1.5 mM) in DCM and the sample taken to dryness. Individual methyl ester DMP derivatives were purified by HPLC using a Thomson Scientific silica column (25 × 0.46 cm), a solvent of hexane/IPA (97:3, v/v), and a flow rate of 1 ml/min with UV detection at 205 nm; the methyl ester DMP derivatives eluted with retention times close to 5 min. Resolution of mixtures of triol methyl ester DMP derivatives was achieved using a solvent of hexane/IPA (100:1, by vol.).
GC-MS analyses were run using an Agilent/J&W DB-5MS column (25m × 0.2mm, 0.33μm film) in a ThermoFinnigan DSQ instrument operated in EI mode (ion source temperature 250°C, electron energy 70 eV) set to full scans from m/z 50 to 650.
1H NMR spectra of the DMP derivatized trihydroxy methyl esters were recorded in C6D6 (150 μl solvent in a 3 mm tube) at 25°C using a Bruker 600 MHz instrument equipped with a 5mm Z-gradient TCI Cryo-probe and using pulse program zg with parameter settings including a pulse length 8.50 μs, time domain 32768, and a relaxation time of 2 s.
2.5 Enzymatic synthesis of epoxyalcohols and triols from 9S/R-HPODE with Beta vulgaris extract
A single beet was peeled and diced, 100 g was added to ice cold 0.1 M potassium phosphate pH 6.7 (600 ml) and blended using a Polytron homogenizer for 3 × 30 s or until a smooth consistency was obtained. The solution was filtered through 2 layers of gauze and used directly for incubation with HPODE. Metabolizing activity was monitored by disappearance of the UV absorbance at 235 nm using 130 μl of homogenate in 870 μl buffer and 50 μM HPODE substrate as described (Hamberg and Olsson, 2011). For larger scale incubations, 4 mg of either 9R- or 9S-HPODE in 200 μl methanol was added to the beetroot homogenate (26 ml) with stirring at room temperature for 1 h. The incubations were then acidified to pH 3.0 with 1N HCl and extracted with 2 volumes of ethyl acetate. The ethyl acetate was washed twice with half a volume of water, taken to dryness, and the recovered products resuspended in isopropanol/chloroform (1:2). The sample was then loaded onto a Waters Sep-Pak Plus NH2 cartridge. The reaction products were eluted in 15 mL ethyl acetate/acetic acid (98:2). HPLC purification was then performed using a Waters Symmetry C18 column with an isocratic 1 ml/min flow of methanol/water/acetic acid (80:20:0.01). Generation of the epoxyalcohol product was carried out in a similar way with the following deviations; (1) reaction time was reduced to 5 min to prevent conversion by epoxide hydrolase, (2) the unstable epoxyalcohol products were extracted with diethyl ether eliminating the requirement for the Sep-Pak chromatography.
2.5.1 Assignment of regio- and stereo-chemistry
1H NMR and GC-MS analysis was carried out as section 2.4 for triol analysis. Deuterated benzene was used as solvent (locked on the 7.15 ppm signal of residual C6H6), except for underivatized triols and their methyl esters, recorded in deuterated methanol (locked on 3.34 ppm). Epoxyalcohol methyl esters were converted to the trimethylsilyl derivative by treatment with BSTFA for 1 h (or overnight) at room temperature.
For CD analysis, approximately 50 μg of epoxyalcohol methyl ester was converted to the 9-O-benzoate ester (1 μl benzoyl chloride, ~1 mg dimethylaminopyridine in 30 μl pyridine, 15 min at room temperature) and repurified by RP-HPLC (MeOH/H2O, 95:5 solvent, flow rate 1 ml/min, retention time approximately 7 min) as described (Schneider et al., 1997). The samples were redissolved in acetonitrile and the concentration adjusted to give 1 AU at the 226 nm UV chromophore. CD spectra (200 – 300 nm) were recorded using a Jasco J-700 spectropolarimeter (3 scans of each sample averaged, scan rate 30 nm/min).
3. Results
3.1 Generation and isolation of epoxyalcohols from hematin treatment of 9-HPODE
Figure 1A illustrates the RP-HPLC separation of the hematin products from 9S-HPODE. These include four 9,10-epoxy-13-hydroxy isomers appearing late in the chromatogram, of which the more prominent are the two trans epoxide isomers, identified earlier as 9S,10S-trans-epoxy-13S-hydroxy followed by the 9S,10S,13R diastereomer (Zheng et al., 2011). On either side of the first of these are the two less abundant 9,10-cis-epoxide isomers labeled as peaks 6 and 8, the stereochemistry of which can be deduced from earlier studies as the 9S,10R-cis-epoxy-13R-hydroxy diastereomer followed by the 9S,10R,13S (Gao et al., 2009). We also identified two 9S,10S-trans-epoxy-11-hydroxy diastereomers (Figure 1A, peaks 4 and 5): the 11S-erythro isomer elutes earlier at ~9.3 min, and the 11R-threo-hydroxy isomer co-elutes with the first cis-epoxide at 11.0 min (see Supplementary Tables S9 and S10 for 1H NMR assignments); this is consistent with previous findings that the erythro diastereomers are the less polar on SP-HPLC or TLC (Corey and Mehrotra, 1983; Corey et al., 1983; Dix and Marnett, 1985; Gardner and Kleiman, 1981; Vasiljeva et al., 1993).
Figure 1. Isolation of epoxyalcohols and trihydroxyoctadecenoic acids.

(A) SP-HPLC analysis of products from reaction of 9S-HPODE with hematin. The four allylic epoxy alcohols are the smaller 9,10-cis-epoxy-11E-13-hydroxy products (peak 6, 9S,10R,13R, and peak 8, 9S,10R,13S), and the more prominent trans epoxides (peak 7, 9S,10S,13S, and peak 9, 9S,10S,13R). Other products appearing on the chromatogram include the following: (1) 9-keto-octadec-11E,12Z-dienoic acid, (2) 9S,10S-epoxy-13-oxo-octadec-11E-enoic acid, (3) 9S-HODE, (4) 9S,10S-trans-epoxy-11S-erythro-hydroxy-octadec-12Z-enoic acid, and (5) 9S,10S-trans-epoxy-11R-threo-hydroxy-octadec-12Z-enoic acid. Column: Thomson Advantage Silica 150Å 5μ column, 25 × 0.46 cm; solvent: hexane/IPA/HAc 100/2/0.02 (by vol.), flow rate 1 ml/min. (B) RP-HPLC analysis after mild acid hydrolysis of the 9S,10S-trans-epoxy-13S-hydroxy isomer with unresolved triols (4 min), -ketol (5.5 min), and unreacted epoxyalcohol (6.7 min). Column: Waters Symmetry C18 column 250 × 4.6mm; solvent: methanol/water/acetic acid (80/20/0.01) with UV detection at 205nm. (C) Further separation of the collected triols using a Chiralpak AD column produced three peaks, designated I, II and III. Solvent: hexane/methanol/ethanol/acetic acid (86.5/4.5/9/0.1), 1 ml/min, UV detection at 205nm. (D) Resolution of II was achieved using a Chiralpak OD-H column yielding IIa and IIb. Solvent: hexane/ethanol/acetic acid, (97.5/2.5/01), 1 ml/min, UV detection at 205nm.
3.2 Separation and characterization of trihydroxyoctadecenoate isomers from the acid hydrolysis of 9S,10S-trans-epoxy-11E, 13S-hydroxyoctadecenoate
To prepare a series of trihydroxy standards for analytical work, we resolved the products of mild acid hydrolysis of 9S,10S-trans-epoxy-11E-13S-hydroxyoctadecenoate. After epoxide ring opening, attack of water on either face of the molecule at C-10 or C12 is expected, giving rise to two 9S,10(R/S),13S- and two 9S,12(R/S),13S-triols. Using an RP-HPLC system in which the epoxyalcohol starting material elutes at 7 min, a -ketol by-product (Section 3.4) elutes at ~5.5 min, the four trihydroxy isomers co-elute around 4 minutes (Figure 1B). Although the four triols are diastereomers, not enantiomers, use of a Chiralpak AD chiral column proved most helpful for their separation, and using a combination of successive chromatographic steps the four were resolved: Chiralpak AD separation of the collected trihydroxy isomers allows separation of 3 of the 4 trihydroxy products (labeled I, II and III) (Figure 1C). Peak II is further separated to peaks IIa and IIb using the Chiralpak OD-H column (Figure 1D). Each peak was collected and analyzed individually.
3.2.1 GC-MS analysis of triol isomers
The four trihydroxyoctadecanoate methyl esters from acid hydrolysis of 9S,10S-trans-epoxy-13S-hydroxyoctadecenoate were treated with dimethoxypropane/toluene sulphonic acid, thus coupling the vicinal diol as the dimethoxypropane (DMP) acetonide derivative, then silylated on the free hydroxyl with BSTFA and analyzed by GC-MS. This clearly identified triols I and IIb as having a 13-OTMS group (prominent m/z 173), and IIa and III as 9-OTMS (m/z 259):
TriOH Peak I and IIb
The mass spectrum of the methyl ester, Me3Si, DMP derivative of peaks I and IIb showed ions at m/z 441 (M+ - 15; loss of CH3), 295 (C1-C13 – 90; loss of Me3SiOH), 199 (C11-C18), 173 (C13-C18), 103 (Me3SiO+ =CH2), 99 (C13-C18 – Me3Si+) and 73 (Me3Si++). These fragment ions indicate a 9,10,13 trihydroxyoctadecenoate (Figure 2A and C).
Figure 2. Regio isomer assignment of 9S-HPODE-derived triols.

EI mass spectra of the triols as the methyl ester, dimethoxypropane, trimethylsilyl ether derivative (M+• 456). A prominent m/z 173 ion indicates a 13-OTMS structure and is diagnostic of a 9,10,13-triol (I and IIb, panels A and C) with other characteristic ions at m/z 327, 295, 270, 228, 227 and 212. A prominent ion at m/z 259 indicates a 9-OTMS and is diagnostic of a 9,12,13-triol (IIa and III, panels B and D) with other characteristic ions at m/z 356, 298, 241 and 209.
TriOH IIa and III
Analyzed as the methyl ester, DMP, Me3Si ether derivatives the following ions were detected; m/z 441 (M+ - 15; loss of CH3), 356 (M+ - 100, rearrangement followed by loss of OHC-(CH2)4-CH3), 298 (C1-C13 – Me3Si+ – CH3), 259 (C1-C9), 209 (C9-C18 – Me3SiOH), 103 (Me3SiO+ =CH2), 73 (Me3Si+). The m/z 259 ion representing α-cleavage next to the 9-OTMS group is diagnostic for the 9,12,13 trihydroxyoctadecenoate (Figure 2B and D).
3.2.2 1H NMR of DMP derivative of products from acid hydrolysis of 9S,10S-trans-epoxy-11E, 13S-hydroxyoctadecenoate
Table I details the diagnostic chemical shift, multiplicity and coupling constants for all 4 trihydroxy isomers formed (See also Supplementary tables S1–S4). From the GC-MS analysis, I and IIb are 9,10,13-trihydroxy, and IIa and III are the 9,12,13-trihydroxy epimers. As these products are generated from 9S,10S-epoxy-13S-hydroxy, the difference in the NMR data is due to the chirality of either the 10-hydroxy or 12-hydroxy. The results are consistent with the unknown hydroxy groups in I and III having the same relative configuration (I, J9,10 = 7.5 Hz and III, J12,13 = 7.62 Hz with both the H10 and H12 chemical shifts at 4.06 ppm). The chemical shifts of the acetonide methyl groups (I and III – 1.47 and 1.45 ppm) indicate the syn-diol configuration (Bruno et al., 1992; Cardellina and Moore, 1980). Trihydroxy IIa and IIb show the opposite chirality of the unconfirmed hydroxyl (IIa, J12,13 = 6.84 Hz and IIb, J9,10 = 6.72 Hz with both the H10 and H12 chemical shifts at 4.41 ppm). The acetonide methyl groups in this case indicate an anti-diol (IIa and IIb – 1.56 and 1.375 ppm). All products have a trans double bond (J10,11 / 11,12 = 15.5 Hz).
Table I.
Diagnostic MS and 1H NMR characteristics of triol hydrolysis products of 9S,10S-epoxy-13S-hydroxy-octadec-11E-enoic acid
| Hydrolysis Products | Epoxyalcohol precursor:9S,10S,12S |
Product stereochemistry | GC-MS (DMP)-MeTMS diagnostic ions | 1H-NMR DMP geminal (OH) proton | 1H-NMR DMP-methyl (ppm) |
|---|---|---|---|---|---|
| I |
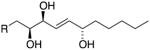
|
9S,10S,13S 9,10-syn (threo) |
m/z 173 | H10 – 4.06 ppm J9,10 = 7.7 Hz J10,11 = 7.5 Hz |
1.45 1.47 |
| IIa |
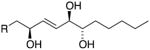
|
9S,12R,13S 12,13-anti (erythro) |
m/z 209 & 259 | H12 – 4.42 ppm J12,13 = 6.8 Hz J11,12 = 6.8 Hz |
1.38 1.56 |
| IIb |
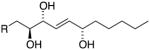
|
9S,10R,13S 9,10-anti (erythro) |
m/z 173 | H10 – 4.42 ppm J9,10 = 7.0 Hz J10,11 = 6.7 Hz |
1.38 1.56 |
| III |
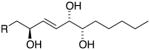
|
9S,12S,13S 12,13-syn (threo) |
m/z 209 & 259 | H12 – 4.06 ppm J12,13 = 7.6 Hz J11,12 = 7.6 Hz |
1.45 1.47 |
| -ketol |
|
9-hydroxy, 13-keto | m/z 241 & 99 |
Diagnostic signal H12 – doublet at 2.73 ppm J11,12 = 7.0Hz |
|
R = HO2C(CH2)7−
Peak III co-eluted with pinellic acid, (the beetroot product from 9S-HPODE (Hamberg and Olsson, 2011), see the following Section 3.3) in the Chiralpak AD separation and also shares the same 1H NMR and GC-MS data, confirming its structure as pinellic acid, 9S,12S,13S-trihydroxyoctadec-10E-enoic acid. Triol I shares the same 1H NMR profile as III (Table I) and can therefore be assigned as 9S,10S,13S-trihydroxyoctadec-11E-enoic acid. The stereochemistry of IIa, the second 9,12,13-triol, has an 1H NMR spectrum consistent with an anti-diol and is therefore 9S,12R,13S-trihydroxyoctadec-10E-enoic acid. The second 9,10,13-triol, IIb, also shares this anti-diol and can be assigned as 9S,10R,13S-trihydroxyoctadec-11E-enoic acid.
3.2.3 SP-HPLC resolution of the triols as the methyl ester DMP derivative
As suggested by the results above, chromatographic separation of the four triols from the 9S,10S,13S epoxyalcohol is quite challenging as they are poorly resolved in typical RP-HPLC and SP-HPLC systems. In a late development in this work, we found that the triols are widely resolved by SP-HPLC of the methyl ester DMP derivative (Figure 3A).
Figure 3. SP-HPLC resolution of triols as the methyl ester DMP derivative.

(A) The four triol hydrolysis products of methyl 9S,10S-epoxy-13S-hydroxyoctadec-11E-enoate were purified as a group by SP-HPLC, derivatized to the acetonide, and analyzed using a Thomson Advantage Silica 150Å 5μ column (25 × 0.46 cm) with a solvent of hexane/IPA 100/1 (by vol.) at a flow rate 1 ml/min, with UV detection at 205 nm. The relative proportions of the four triols are indicated as percentages. (B) The same four products were chromatographed with the triols (methyl ester DMP derivative) from the second major epoxyalcohol of the hematin reaction, the 13R diastereomer, 9S,10S-epoxy-13R-hydroxyoctadec-11E-enoate (the shaded peaks). Two of the new triols coelute in the peak marked with an asterisk; these two are easily resolved by SP-HPLC of the methyl ester derivative using the same Thomson silica column with a solvent of hexane/IPA 90/10 (by vol.), retention times 8.8 and 11.5 min (data not shown).
To further test the capabilities of this method, we prepared the four diastereomeric triols from acid hydrolysis of the 9S,10S,13R-epoxyalcohol (the second of the two major epoxyalcohols from the hematin reaction with 9S-HPODE). These are separated into an additional three new peaks in this system (Figure 3B). As indicated in the figure legend, the first of these new peaks (marked with an asterisk) contains two triols which can be resolved by SP-HPLC of the methyl ester derivative.
3.3 Enzymatic conversion of 9S- and 9R-HPODE by beetroot extract to epoxyalcohols and triols
Hamberg and Olsson reported the efficient enzymatic transformation of 9S-HPODE by a filtered homogenate of Beta vulgaris (beetroot) to 9S-hydroxy-12R,13S-cis-epoxy-octadec-10E-enoic acid and upon longer incubation its enzymatic hydrolysis to 9S,12S,13S-trihydroxy-octadec-10E-enoate (pinellic acid); 13S-HPODE was not metabolized (Hamberg and Olsson, 2011). In extending these experiments we found that 13R-HPODE is also not metabolized, while 9R-HPODE was converted with only a slightly lower rate of reaction compared to 9S-HPODE. The 9S-HPODE enantiomer gave products as reported (Hamberg and Olsson, 2011), a single epoxyalcohol product after 5 min incubation (data not shown), and one prominent triol peak following its hydrolysis during 60 min incubation (RP-HPLC, peak at 4 min retention time, Figure 4A with GC-MS, Figure 4B and 1H NMR, Table II and Supplementary table S5, confirming the structure). The transformations of 9R-HPODE produced one main epoxyalcohol with two minor isomers upon short-term incubation (5 min), Figure 4C. Incubation for 60 min yielded their enzymatic hydrolysis products with the polarity of triols (Fig. 4E), resolved on a Chiralpak AD column into one predominant trihydroxy isomer with two minor products (Fig. 4E, inset).
Figure 4. Isolation and structural characterization of products from the incubation of 9R- and 9S-HPODE with Beta vulgaris extract.

(A) RP-HPLC (as per Section 2.3.2) of products from 60 min incubation of 9S-HPODE: trihydroxy product(s) elute at 4 min with unreacted 9-HPODE substrate and 9-HODE eluting at 15 and 17.5 min respectively. Inset: separation using a Chiralpak AD column confirms the generation of one major trihydroxy isomer. (B) EI mass spectrum of the methyl ester DMP TMS ether derivative with a diagnostic ion at m/z 259 indicating a 9-OTMS group and a 9,12,13-trihydroxyoctadecenoate structure. (C) RP-HPLC of the epoxyalcohol (retention time, 6 min) from a 5 min incubation of 9R-HPODE with Beta vulgaris. (D) EI mass spectrum (methyl ester TMS ether derivative) of the 9-hydroxy-12,13-epoxyoctadecenoic acid product. (E) RP-HPLC of products from the 60 min incubation of 9R-HPODE with two triol peaks eluting at 4 min. Inset: the triols were resolved using a Chiralpak AD column as major (17 min) and minor (24 min) isomers. (F) EI spectrum of the major isomer from 9R-HPODE, a 9,12,13-trihydroxyoctadecenoate, analyzed as in (B).
Table II.
Diagnostic MS and 1H NMR characteristics of 9S/R-hydroperoxy-10E, 12Z-octadecadienoic acid and Beta vulgaris incubation products
| Incubation Products | Hydroperoxide precursor: 9R |
Product stereochemistry | GC-MS (DMP)-MeTMS diagnostic ions | 1H-NMR DMP geminal (OH) proton cis-epoxide | 1H-NMR DMP-methyl (ppm) |
|---|---|---|---|---|---|
| 5 minute incubation |
|
9R-hydroxy,12R,13S-epoxy | m/z 259 | H12 – 3.25 ppm J12,13 = 4.4 Hz |
|
| 60 minute incubation |
|
9R,12S,13S 12,13-syn (threo) |
m/z 209 & 259 | H12 – 4.06 ppm J12,13 = 7.3 Hz J11,12 = 7.3 Hz |
1.46 1.48 |
|
| |||||
|
Hydroperoxide precursor: 9S |
|||||
|
| |||||
| 5 minute incubation |
|
9S-hydroxy,12R,13S-epoxy | m/z 259 |
cis-epoxide H12 – 3.25 ppm J12,13 = 4.4 Hz |
|
| 60 minute incubation |
|
9S,12S,13S 12,13-syn (threo) |
m/z 209 & 259 | H12 – 4.06 ppm J12,13 = 7.1 Hz J11,12 = 7.3 Hz |
1.46 1.47 |
R = HO2C(CH2)7−
3.3.1 Identification of the main 9R-HPODE-derived epoxyalcohol formed in beetroot
The epoxyalcohol produced by 9R-HPODE incubation with beetroot was isolated as per methods as a single main product eluting at 6 minutes on RP-HPLC (Figure 4C). The 1H-NMR spectrum clearly established the presence of a trans double bond with allylic cis-epoxide (J = 4 Hz) and carbon-bearing hydroxyl on either side (Table II, Supplementary Table S6). However, due to the symmetry of structure in this type of linoleate derivative, the NMR does not establish whether the epoxyalcohol is 9,10-epoxy-11E-13-hydroxy or the reversed 9-hydroxy-10E-12,13-epoxy.
The mass spectrum of the methyl ester TMS ether derivative (Figure 4D) showed ions at m/z 398 (M+), 327 (C1-C13), 297, 259 (C1-C9), 237 (327-Me3SiOH), 199 (C1-C10), 173 (C13-C18), 103 (Me3SiO+ =CH2), 73 (Me3Si+). The presence of ions at both m/z 173 ion (C13-C18, typical of 13-OTMS) and m/z 259 (C1-C9, typical of 9-OTMS) also fails to resolve the issue. Indeed, Gardner and Kleiman (Gardner and Kleiman, 1981) have shown that both m/z 173 and 259 are prominent ions in the mass spectra of the methyl ester TMS ether derivative of 9-hydroxy-12,13-epoxyoctadecenoates.
Based on its biosynthesis in beetroot and by analogy to the metabolism of 9S-HPODE (Hamberg and Olsson, 2011), an oxygen transfer mechanism converting 9R-HPODE to 9R-hydroxy-12,13-cis-epoxyoctadecenoate is anticipated. This was confirmed by comparison of the beetroot 9R-HPODE-derived epoxyalcohol with the two 9,10-cis-epoxy-13-hydroxy diastereomers obtained from hematin treatment of 9-HPODE (section 3.1), and the corresponding two from 13S-HPODE (9-hydroxy-10E-12,13-cis-epoxy). By RP-HPLC, three resolved from the beetroot product and one of 13S-HPODE cis-epoxyalcohols co-chromatographed (data not shown). This one must have the same regio- and stereochemistry as beetroot 9R-HPODE product or be the enantiomer. Finally, we assigned this 13S-HPODE-derived cis-epoxyalcohol with the 9R-hydroxy configuration by CD analysis (Gonnella et al., 1982; Schneider et al., 1997) (Figure 5), establishing it as identical to the beetroot product which therefore has the structure, 9R-hydroxy-12R,13S-cis-epoxyoctadec-10E-enoic acid (Scheme 1).
Figure 5. CD analysis of the hydroxyl configuration in 9-hydroxy-12,13-cis-epoxyalcohols.

CD spectra were recorded using a Jasco-700 spectropolarimeter on the 9-O-benzoate esters of the 9-hydroxy-10E-12,13-cis-epoxy-octadecenoate methyl esters in acetonitrile solution with the concentration adjusted to give 1.0 AU at 227 nm in the UV (methods in section 2.5.1). Interaction of the chromophores of the 9-O-benzoate ester and the 10E bond produces either a positive first Cotton effect (top) associated with a positive interaction (+) in the Newman projection, and allowing designation of the 9S configuration, or the negative Cotton effect, negative Newman projection, designating 9R .
Scheme 1.

Summary of trihdroxy product formation from 9S- and 9R-HPODE
3.3.2 Identification of the main 9R-HPODE-derived triols formed in beetroot
The major triols from the 9S- and 9R-HPODE incubations with beetroot show indistinguishable mass spectra (Figure 4B, 4F), indicating they are the same regio isomers. Analyzed as the methyl ester, DMP, TMS ether derivatives the following ions were detected; m/z 441 (M+ - 15; loss of CH3), 356 (M+ - 100, rearrangement followed by loss of OHC-(CH2)4-CH3), 298 (C1-C13 – Me3Si+ – CH3), 259 (C1-C9), 209 (C9-C18 – Me3SiOH), 103 (Me3SiO+ =CH2), 73 (Me3Si+). The presence of the ion at m/z 259, diagnostic of the 9-OTMS group, identifies these products as 9,12,13 trihydroxy (Table II).
Table II includes a summary of the diagnostic 1H NMR data for the major 9S- and 9R-HPODE-derived triols as the methyl ester DMP derivative (with full details given in Supplementary Tables S5 and S7). The original 9R/13S or 9S/13S configurations in the parent epoxyalcohols should be retained, and therefore the unresolved issue is the chirality at C-12. The chemical shifts of the C12 proton and coupling constant for H12,13 are almost identical (4.06 ppm, J12,13 = 7.1 or 7.3 Hz), indicating the same relative configuration in the vicinal diol: the substituents are trans to each other on the acetonide ring and so the hydroxyls must be in the syn configuration and are 12S,13S. The chemical shifts of the acetonide methyl groups also support this conclusion (Bruno et al., 1992; Cardellina and Moore, 1980). This reversal of configuration at C-12 upon hydrolysis of the epoxyalcohols is expected through the actions of epoxide hydrolase (Arand et al., 2005). Finally, the coupling constants for both these products show the double bond is trans (J10,11 = 15.5 Hz). Therefore, the 9S-HPODE-derived triol, pinellic acid, is 9S,12S,13S as already reported (Hamberg and Olsson, 2011), and the main triol from 9R-HPODE is 9R,12S,13S-trihydroxy-10E-octadecenoic acid (Scheme 1).
3.4 Identification of a specific -ketol product from mild acid hydrolysis of 9S, 10S-trans-epoxy-11E, 13S-hydroxyoctadecenoate
Upon RP-HPLC analysis of acid-treated 9S,10S-trans-epoxy-11E-13S-hydroxy-octadecenoate, we consistently observed a peak, more polar than the parent epoxyalcohol yet considerably less polar than the triol hydrolysis products, and exhibiting only end absorbance in the UV (Figure 1B). After further purification as the methyl ester (Chiralpak AD column), the main component was identified by NMR as a -ketol. The significant chemical shifts, multiplicity and coupling constants for the -ketol product are detailed in Table I (Supplementary Table S8). Diagnostic features of the spectrum include the doublet signal for the CH2 at 2.7ppm (J11,12 = 7.02 Hz), with coupling only to the one trans double bond (J =15.5 Hz), with no additional coupling due to the adjacent ketone and a second CH2 triplet representing H14 near H2 (Supplementary Table S8). Due to the symmetry typical of many linoleate-derived products in the NMR, it was not possible to distinguish the 9-oxo-11E-13-hydroxyoctadecenoate from the 9-hydroxy-10E-13-oxo isomer. In this case, however, GC-MS analyses were definitive. The methyl ester, methoxime, TMS ether derivative gave a pair of closely-eluting peaks for the syn and anti isomers on GC with slightly differing ion abundances on MS (Figure 6A and 6B); both peaks gave ions at m/z 429 (M+•) 396 (M+-CH3O), 356 (C1-C13), 285 (C1-C11), 270 (C9-C18), 168 (C10-C18), 73 (Me3Si+). GC-MS of the methyl ester, TMS derivative yielded one main peak showing two main ions at m/z 241 (C9-C18) and 99 (C13-C18) (Table I, Figure 6C and 6D) (Gardner and Kleiman, 1979). These results, together with the NMR data, identify the δ-ketol as 9-hydroxy-13-oxo-octadec-10E-enoic acid.
Figure 6. Identification of a δ-ketol.

(A) Total ion current chromatogram of the methyl ester, methoxime, TMS ether derivative of the -ketol formed during acid hydrolysis of 9S,10S-trans-epoxy,13S-hydroxy-octadec-11E-enoic acid showing the closely eluting syn and anti isomers at ~9.8 min. (B) EI mass spectrum of the first-eluting isomer with diagnostic ions at m/z 168 and 285. (C) Total ion current chromatogram of the methyl ester, TMS ether derivative. (D) The mass spectrum of the main peak has diagnostic ions at m/z 99 and 241 (Gardner and Kleiman 1979).
4. Discussion
Utility of hematin method
In the hematin treatment of fatty acid hydroperoxides under the conditions we describe, the allylic epoxyalcohols are recovered as major products when suitable precautions are taken to avoid their facile hydrolysis. To avoid hydrolysis of these highly acid-labile allylic epoxyalcohols, the samples are kept on ice during acid extraction, the organic solvent is added prior to acidification (and thus is ready for mixing the instant after acid is added), and the acidification itself uses a pre-determined quantity of 1M KH2PO4 and HCl to produce the desired pH, immediately followed by organic extraction; subsequent washing of the organic phase with water prior to evaporation is another essential precaution. Although the products chromatograph satisfactorily in RP-HPLC solvent containing 0.01% glacial acetic acid, prior formation of the methyl esters (diazomethane) and elimination of the acetic acid improves the subsequent recovery of intact allylic epoxyalcohol products from the solvent.
Identification of hydrolysis products
In linoleate-derived triols arising from intermediate allylic epoxyalcohols, the structural symmetry of the 9,10,13- and 9,12,13-trihydroxy products confounds their definitive assignment solely by NMR. On the other hand, these regioisomers are readily distinguished by GC-MS of the DMP derivative (Figure 2, Table I and Supplementary tables S1–S4). Epoxide ring opening under mild acid hydrolysis induces formation of an intermediate allylic carbocation with the hydroxyl configurations at 9 and 13 fixed with respect to the substrate. When the structure of the epoxyalcohol is known, therefore, the remaining issue of stereochemistry in the resulting triols is the configuration of the hydroxy at C10 or C12. In 1H-NMR of the DMP derivative, the chemical shift of the allylic hydroxyl in the vicinal diol is diagnostic of the syn or anti configuration. Thus, the chemical shift of the allylic hydroxyl in the syn vicinal diols (products I and III, Table I) is 0.4 ppm upfield in comparison to anti (products IIa, IIb), which is sufficient to define the complete stereochemistry.
The chemical shifts of the geminal methyl groups on the acetonide ring in the DMP derivatives also provide structural information, confirming the chirality of the unknown hydroxyl (cf. (Bruno et al., 1992; Cardellina and Moore, 1980; Shirahata et al., 2006)). When the 9,10 or 12,13 hydroxyls are syn, the chemical shifts of the acetonide methyl groups are almost superimposable, only ~0.02 ppm apart. When the vicinal diol hydroxyls are anti, the methyl groups are in a cis configuration and the chemical shifts differ by ~0.2 ppm. Thus, for products I and III (Table I), the acetonide methyls have chemical shifts of 1.45 and 1.47 ppm, whereas they are separated in IIa and IIb with values of 1.38 and 1.56 ppm.
Four additional triols with the 9S,10(R or S),13R and 9S,12(R or S),13R configurations are available by acid hydrolysis of the second trans-epoxyalcohol from 9S-HPODE (9S,10S-epoxy-11E-13R-hydroxy). We show that these can be resolved by SP-HPLC of the methyl ester DMP derivative (Figure 3B). Although not addressed in this study, the two trans-epoxyalcohols derived from the enantiomeric hydroperoxide, 9R-HPODE, will give rise to eight triol enantiomers to the products resolved here. As an alternative to the reported degradative methods of chiral analysis via ozonolysis (Hamberg, 1991, Garbe et al, 2005), chiral HPLC column methods could be explored as a means to their assignment.
Proportions of the hydrolysis products
Hamberg has reported the relative proportions of the four products of mild acid hydrolysis from a linoleate-derived 9,10-cis-epoxy-13-hydroxy epoxyalcohol: 61% and 15% attack on the allylic epoxy carbon (favoring reversal of configuration), 15% and 8% on the carbon α to the hydroxyl (favoring formation of the erythro vicinal diol) (Hamberg, 1991a). He found similar proportions, 61:18:15:6, in hydrolysis of the 12,13-cis-epoxy-9-hydroxy precursor of pinellic acid (Hamberg and Olsson, 2011). We found different proportions for mild acid hydrolysis of an analogous trans-epoxy product, methyl 9S,10S-trans-epoxy-11E-13S-hydroxy-octadecenoate. Our best estimate comes from resolution of the four diastereomers as the methyl ester DMP derivative showing a ratio of 22:26:40:12 for products I, IIa, IIb, and III respectively (Figure 3A). A point of agreement with hydrolysis of the cis-epoxyalcohols is that the highest abundance triol is formed via attack on the allylic epoxy carbon with reversal of configuration (product IIb in our case), and the lowest is the threo vicinal diol δ to the original epoxide (product III in our case).
Stereospecificity of the beetroot epoxyalcohol synthase
Here we showed that 9R-HPODE, enantiomeric to the 9S-HPODE studied by Hamberg and Olsson is also a good substrate for the beetroot enzymes (Hamberg and Olsson, 2011) (whereas neither of the 13-HPODE enantiomers react). The initial major product from the 9R-hydroperoxide is the analogous 9R-hydroxy-12R,13S-cis-epoxide, similarly formed by epoxidation at 12,13. Subsequent hydrolysis gave a major product formed via epoxide hydrolase. Recently it was shown that cis epoxides present in epoxyeicosatrienoic acids (EETs) are converted to the syn-diol by soluble epoxide hydrolase present in rat red blood cells; conversely the trans epoxides became anti-diols (Jiang et al., 2008). These results are in line with our findings. It is logical, therefore, that the main product from the enzymatic hydrolysis of 9R-hydroxy-12R, 13S-trans-epoxide is indeed the 9R,12S,13S-triol.
To assign the stereoconfigurations in the primary beetroot epoxyalcohol product from 9R-HPODE, NMR was used to establish the hydroxy-trans double bond-cis-epoxy partial structure. Again, because of the symmetry in linoleate-derived structures, NMR did not reveal the orientation of this unit in the fatty acid. Similarly, GC-MS is not definitive because the usual “diagnostic” ions for 9-OTMS (m/z 259) or 13-OTMS (m/z 173) were both present in the EI mass spectrum of the methyl ester TMS derivative (cf. (Gardner and Kleiman, 1981)). We assigned the configuration by (i) comparison of its HPLC mobility to each of the potential cis-epoxy diastereomers prepared by hematin treatment of 9-HPODE (two 9,10-cis-epoxy-11E-13-hydroxy diastereomers) and 13S-HPODE (two 9-hydroxy-10E-12R, 13S-cis-epoxides). The 9R-HPODE-derived beetroot product co-chromatographed on RP-HPLC only with one of the two 12R,13S-epoxides; (ii) their 9-hydroxy configuration was then established by CD analysis (Gonnella et al., 1982), proving that the beetroot product is 9R-hydroxy-10E-12R,13S-cis-epoxy-octadec-10E-enoic acid. Although we did not pursue the matter, another potential application of the CD method is assignment of the stereochemistry of the free hydroxyl in the DMP derivative of the triols.
Mechanism of transformation to delta-ketol
We observed that mild acid hydrolysis of the trans-epoxyalcohol, 9S,10S-epoxy-11E-13S-hydroxy-octadecenoate, formed not only the distinctly more polar triols, but also a peak of intermediate polarity on RP-HPLC (Fig 1B) that we identified by NMR and GC-MS as mainly one δ-ketol. In the 1970s, Gardner reported formation of two isomeric δ-ketols from treatment of either 9-HPODE or 13-HPODE with FeCl3/cysteine (Gardner and Kleiman, 1979), but there has been little attention to such products since. Gardner described that a single epoxyalcohol intermediate treated with a Lewis acid (BF3-etherate) gave rise to similar quantities of two isomeric ketols, 9-hydroxy-10E-13-oxo and 9-oxo-11E-13-hydroxy. We found a 10-fold relative abundance of the 9-hydroxy-13-oxo isomer over the 9-oxo-13-hydroxy. Whereas Gardner and Kleiman implicated a symmetrical carbocation intermediate to account for the appearance of apparently similar amounts of two diastereomers, our results suggest asymmetry in the reaction intermediate, giving the bias towards predominantly one outcome (Scheme 2). Although delta-ketols have not appeared in the recent literature, given the similarity in the EI mass spectra of the methyl ester TMS ether derivatives of certain epoxyalcohols and delta-ketols (Gardner and Kleiman, 1979), they are a candidate that might provide an alternative explanation for the appearance of unlikely epoxyalcohol isomers in the analysis of human skin fatty acids (Zheng et al., 2011). Even more probable, we suspect a delta-ketol(s) accounts for the peak of intermediate polarity illustrated in the RP-HPLC profile in our acid hydrolysis of the arachidonate-derived epoxyalcohol, 8R-hydroxy-11R,12R-trans-epoxy-eicosatrienoic acid (Yu et al., 2003); in that case LC-MS analysis confirmed that the intermediate polarity product(s) has the same molecular weight as the starting epoxyalcohol and potential δ-ketol product.
Scheme 2.

Proposed δ-ketol formation during acid hydrolysis of 9,10-trans-epoxy-11E-13-hydroxy-C18:1
Supplementary Material
Highlights.
Allylic epoxy alcohols were isolated from 9-HPODE reaction with hematin.
HPLC analysis of trihydroxy hydrolysis products, assignment by GC-MS, CD, and 1H NMR.
9R-and 9 S-HPODE metabolism by epoxyalcohol synthase and epoxide hydrolase.
Identification of a d-ketol rearrangement product of 9,10-trans-epoxy alcohol.
Acknowledgments
Financial support was provided by European Union FP7, Marie Curie Outgoing Fellowship (C.P.T.), by NIAMS grant AR-51968 (to A.R.B.) and a voucher for use of the mass spectrometry resource through the Vanderbilt-Ingram Cancer Center Support grant (P30 CA068485). We would also like to thank Drs. Wade Calcutt and Donald Stec for their assistance with GC-MS and 1H NMR respectively, and Nicholas M. Adams and Dr. David Wright for the CD analysis.
Abbreviations
- CD
circular dichroism
- COSY
correlation spectroscopy
- DCM
dichloromethane
- DMP
dimethoxypropane
- GC-MS
gas chromatography-mass spectrometry
- H(P)ODE
hydro(pero)xyoctadecadienoic acid
- IPA
isopropyl alcohol
- LOX
lipoxygenase
- RP-HPLC
reversed-phase high pressure liquid chromatography
- SP-HPLC
straight-phase high pressure liquid chromatography
- TMS
trimethylsilyl
- UV
ultraviolet
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andreou A, Brodhun F, Feussner I. Biosynthesis of oxylipins in non-mammals. Prog Lipid Res. 2009;48:148–170. doi: 10.1016/j.plipres.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Andreou AZ, Vanko M, Bezakova L, Feussner I. Properties of a mini 9R-lipoxygenase from Nostoc sp PCC 7120 and its mutant forms. Phytochemistry. 2008;69:1832–1837. doi: 10.1016/j.phytochem.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Arand M, Cronin A, Adamska M, Oesch F. Epoxide hydrolases: structure, function, mechanism, and assay. Methods Enzymol. 2005;400:569–588. doi: 10.1016/S0076-6879(05)00032-7. [DOI] [PubMed] [Google Scholar]
- Blée E, Wilcox AL, Marnett LJ, Schuber F. Mechanism of reaction of fatty acid hydroperoxides with soybean peroxygenase. J Biol Chem. 1993;268:1708–1715. [PubMed] [Google Scholar]
- Bruno I, Dauria MV, Iorizzi M, Minale L, Riccio R. Marine eicosanoids: Occurrence of 8,11,12-trihydroxylated eicosanoic acids in starfishes. Experientia. 1992;48:114–115. [Google Scholar]
- Cardellina JH, Moore RE. Malyngic acid, a new fatty acid from Lyngbya majuscula. Tetrahedron. 1980;36:993–996. [Google Scholar]
- Claeys M, Kivits GA, Christ-Hazelhof E, Nugteren DH. Metabolic profile of linoleic acid in porcine leukocytes through the lipoxygenase pathway. Biochim Biophys Acta. 1985;837:35–51. doi: 10.1016/0005-2760(85)90083-9. [DOI] [PubMed] [Google Scholar]
- Corey EJ, Mehrotra MM. Stereochemistry of the lipoxygenase-catalyzed allylic hydroperoxide to oxiranylcarbinol rearrangement. Tetrahedron Lett. 1983;45:4921–4922. [Google Scholar]
- Corey EJ, Mehrotra MM, Cashman JR. New synthetic routes to leukotrienes and other arachidonate derived epoxy eicosatetraenoic acids (EPETE's) - Exclusion of the hydroxy epoxide pathway for leukotriene biosynthesis. Tetrahedron Lett. 1983;24:4917–4920. [Google Scholar]
- Dix TA, Marnett LJ. Conversion of linoleic acid hydroperoxide to hydroxy, keto, epoxyhydroxy, and trihydroxy fatty acids by hematin. J Biol Chem. 1985;260:5351–5357. [PubMed] [Google Scholar]
- Feussner I, Wasternack C. The lipoxygenase pathway. Annu Rev Plant Biol. 2002;53:275–297. doi: 10.1146/annurev.arplant.53.100301.135248. [DOI] [PubMed] [Google Scholar]
- Funk CD, Powell WS. Release of prostaglandins and monohydroxy and trihydroxy metabolites of linoleic and arachidonic acids by adult and fetal aortae and ductus arteriosus. J Biol Chem. 1985;260:7481–7488. [PubMed] [Google Scholar]
- Galliard T, Phillips DR. Lipoxygenase from potato tubers. Partial purification and properties of an enzyme that specifically oxygenates the 9-position of linoleic acid. Biochem J. 1971;124:431–438. doi: 10.1042/bj1240431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Boeglin WE, Brash AR. Omega-3 fatty acids are oxygenated at the n-7 carbon by the lipoxygenase domain of a fusion protein in the cyanobacterium Acaryochloris marina. Biochim Biophys Acta. 2010;1801:58–63. doi: 10.1016/j.bbalip.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Boeglin WE, Zheng Y, Schneider C, Brash AR. Evidence for an ionic intermediate in the transformation of fatty acid hydroperoxide by a catalase-related allene oxide synthase from the cyanobacterium Acaryochloris marina. J Biol Chem. 2009;284:22087–22098. doi: 10.1074/jbc.M109.013151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbe LA, Hübke H, Tressl R. Enantioselective formation pathway of a trihydroxy fatty acid during mashing. J Am Soc Brew Chem. 2005;63:157–162. [Google Scholar]
- Gardner HW, Kleiman R. Lack of regioselectivity in formation of oxohydroxyoctadecenoic acids from the 9-hydroperoxide or 13-hydroperoxide of linoleic acid. Lipids. 1979;14:848–851. [Google Scholar]
- Gardner HW, Kleiman R. Degradation of linoleic acid hydroperoxides by a catalyst as a model for similar biochemical reactions. 2 Specificity in cysteine FeCl3 formation of fatty acid epoxides. Biochim Biophys Acta. 1981;665:113–125. doi: 10.1016/0005-2760(81)90239-3. [DOI] [PubMed] [Google Scholar]
- Gardner HW, Kleiman R, Weisleder D. Homolytic decomposition of linoleic acid hydroperoxide - Identification of fatty acid products. Lipids. 1974;9:696–706. [Google Scholar]
- Garssen GJ, Veldink GA, Vliegenthart JFG, Boldingh J. The formation of threo-11-hydroxy-trans-12: 13-epoxy-9-cis-octadecenoic acid by enzymic isomerisation of 13-L-hydroperoxy-9-cis, 11-trans-octadecadienoic acid by soybean lipoxygenase-1. Eur J Biochem. 1976;62:33–36. doi: 10.1111/j.1432-1033.1976.tb10094.x. [DOI] [PubMed] [Google Scholar]
- Göbel C, Feussner I, Hamberg M, Rosahl S. Oxylipin profiling in pathogen-infected potato leaves. Biochim Biophys Acta. 2002;1584:55–64. doi: 10.1016/s1388-1981(02)00268-8. [DOI] [PubMed] [Google Scholar]
- Gonnella NC, Nakanishi K, Martin VS, Sharpless BK. General method for determining absolute configurations of acyclic allylic alcohols. J Am Chem Soc. 1982;104:3775–3776. [Google Scholar]
- Hamberg M. Decomposition of unsaturated fatty acid hydroperoxides by hemoglobin - Structures of major products of 13L-hydroperoxy-9,11-octadecadienoic acid. Lipids. 1975;10:87–92. doi: 10.1007/BF02532161. [DOI] [PubMed] [Google Scholar]
- Hamberg M. Regiochemical and stereochemical analysis of trihydroxyoctadecenoic acids derived from linoleic acid 9-hydroperoxide and 13-hydroperoxide. Lipids. 1991a;26:407–415. doi: 10.1007/BF02536065. [DOI] [PubMed] [Google Scholar]
- Hamberg M. Trihydroxyoctadecenoic acids in beer: qualitative and quantitative analysis. J Agric Food Chem. 1991b;39:1568–1572. [Google Scholar]
- Hamberg M. An epoxy alcohol synthase pathway in higher plants: biosynthesis of antifungal trihydroxy oxylipins in leaves of potato. Lipids. 1999;34:1131–1142. doi: 10.1007/s11745-999-0464-7. [DOI] [PubMed] [Google Scholar]
- Hamberg M, Hamberg G. Peroxygenase-catalyzed fatty acid epoxidation in cereal seeds (sequential oxidation of linoleic acid into 9(S),12(S),13(S)-trihydroxy-10(E)-octadecenoic acid) Plant Physiol. 1996;110:807–815. doi: 10.1104/pp.110.3.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamberg M, Herman RP, Jacobsson U. Stereochemistry of two epoxy alcohols from Saprolegnia parasitica. Biochim Biophys Acta. 1986;879:410–418. [Google Scholar]
- Hamberg M, Olsson U. Efficient and specific conversion of 9-lipoxygenase hydroperoxides in the beetroot. Formation of pinellic acid Lipids. 2011;46:873–878. doi: 10.1007/s11745-011-3592-7. [DOI] [PubMed] [Google Scholar]
- Jiang H, Zhu AG, Mamczur M, Morisseau C, Hammock BD, Falck JR, McGiff JC. Hydrolysis of cis- and trans-epoxyeicosatrienoic acids by rat red blood cells. J Pharmacol Exp Ther. 2008;326:330–337. doi: 10.1124/jpet.107.134858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meruvu S, Walther M, Ivanov I, Hammarstrom S, Furstenberger G, Krieg P, Reddanna P, Kuhn H. Sequence determinants for the reaction specificity of murine (12R)-lipoxygenase: Targeted substrate modification and site-directed mutagenesis. J Biol Chem. 2005;280:36633–36641. doi: 10.1074/jbc.M508260200. [DOI] [PubMed] [Google Scholar]
- Nigam S, Patabhiraman S, Ciccoli R, Ishdorj G, Schwarz K, Petrucev B, Kuhn H, Haeggstrom JZ. The rat leukocyte-type 12-lipoxygenase exhibits an intrinsic hepoxilin A3 synthase activity. J Biol Chem. 2004;279:29023–29030. doi: 10.1074/jbc.M307576200. [DOI] [PubMed] [Google Scholar]
- Niisuke K, Boeglin WE, Murray JJ, Schneider C, Brash AR. Biosynthesis of a linoleic acid allylic epoxide: mechanistic comparison with its chemical synthesis and leukotriene A biosynthesis. J Lipid Res. 2009;50:1448–1455. doi: 10.1194/jlr.M900025-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace-Asciak CR, Reynaud D, Demin PM. Hepoxilins: a review on their enzymatic formation, metabolism and chemical synthesis. Lipids. 1995;30:107–114. doi: 10.1007/BF02538262. [DOI] [PubMed] [Google Scholar]
- Püssa T, Raudsepp P, Toomik P, Pallin R, Maeorg U, Kuusik S, Soidla R, Rei M. A study of oxidation products of free polyunsaturated fatty acids in mechanically deboned meat. J Food Comp Anal. 2009;22:307–314. [Google Scholar]
- Schneider C, Schreier P, Humpf HU. Exciton-coupled circular dichroism (ECCD) in acyclic hydroxylated dienes: a sensitive method for the direct stereochemical assignment of lipoxygenase products. Chirality. 1997;9:563–567. [Google Scholar]
- Shirahata T, Sunazuka T, Yoshida K, Yamamoto D, Harigaya Y, Kuwajima I, Nagai T, Kiyohara H, Yamada H, Omura S. Total synthesis, elucidation of absolute stereochemistry, and adjuvant activity of trihydroxy fatty acids. Tetrahedron. 2006;62:9483–9496. [Google Scholar]
- Shirahata T, Sunazuka T, Yoshida K, Yamamoto D, Harigaya Y, Nagai T, Kiyohara H, Yamada H, Kuwajima I, Omura S. Total synthesis and adjuvant activity of all stereoisomers of pinellic acid. Bioorg Med Chem Lett. 2003;13:937–941. doi: 10.1016/s0960-894x(02)01069-7. [DOI] [PubMed] [Google Scholar]
- Siebert M, Krieg P, Lehmann WD, Marks F, Fürstenberger G. Enzymic characterization of epidermis-derived 12-lipoxygenase isoenzymes. Biochem J. 2001;355:97–104. doi: 10.1042/0264-6021:3550097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiljeva LL, Manukina TA, Demin PM, Lapitskaja MA, Pivnitsky KK. Synthetic study of hepoxilins. 3 Synthesis, properties, and identification of epimeric hepoxilins (−)-(10R)-B3 and (+)-(10S)-B3. Tetrahedron. 1993;49:4099–4106. [Google Scholar]
- Yu Z, Schneider C, Boeglin WE, Marnett LJ, Brash AR. The lipoxygenase gene ALOXE3 implicated in skin differentiation encodes a hydroperoxide isomerase. Proc Natl Acad Sci USA. 2003;100:9162–9167. doi: 10.1073/pnas.1633612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Boeglin WE, Schneider C, Brash AR. A 49-kDa mini-lipoxygenase from Anabaena sp PCC 7120 retains catalytically complete functionality. J Biol Chem. 2008;283:5138–5147. doi: 10.1074/jbc.M705780200. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Brash AR. On the role of molecular oxygen in lipoxygenase activation: comparison and contrast of epidermal lipoxygenase-3 with soybean lipoxygenase-1. J Biol Chem. 2010;285:39876–39887. doi: 10.1074/jbc.M110.180794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Yin H, Boeglin WE, Elias PM, Crumrine D, Beier DR, Brash AR. Lipoxygenases mediate the effect of essential fatty acid in skin barrier formation: A proposed role in releasing omega-hydroxyceramide for construction of the corneocyte lipid envelope. J Biol Chem. 2011;286:24046–24056. doi: 10.1074/jbc.M111.251496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.