

Abstract
A new long chain fatty alcohol acetate identified as 17-hydroxypentacosanyl acetate, (1) together with a new xanthone identified as 1,8-Dihydroxy-3,6-dimethoxy-xanthone-5-O-[α-L-rhamnopyranosyl-(1′′→2′)]-β-D-glucopyranoside (3), as well as two new lignans identified as (+)-Lyoniresinol-3a-O-[α-L-rhamnopyranosyl-(1′′′→6′′)]-β-D-glucopyranoside (4) and (+)-Isolariciresinol-3a-O-[α-L-rhamnopyranosyl-(1′′′→2′′)-α-L-rhamnopyranosyl-(1′′′′→6′′)]-β-D-glucopyranoside (5), in addition to β-sitosterol-3-O-acetate (2) were isolated from the methanolic extract of the aerial parts of Polygonum bellardii growing in Egypt. Their structures were elucidated on the basis of different chemical and spectroscopic evidences. The total extract and its fractions, in addition to compounds (3, 4 and 5) showed significant antioxidant potential by DPPH· scavenging activity technique.
Keywords: Antioxidant, lignans, Polygonaceae, Polyonum bellardii, xanthone
INTRODUCTION
Polygonum bellardii (Polygonaceae) grows in sandy places around Alexandria, Mandara, Abukir, Damietta, Tel-El-Kebir, Assiut, Ekhmim, Dendera, Farafra and Kharge, known locally as Maksus-El-Gariyla, Qardab or Qordob.[1] Many species of the genus Polygonum had been reported to exhibit a variety of interesting biological activities as: Laxative, expectorant, diuretic, tonic, anti-aging, anti-inflammatory, antibacterial, anti-allergic, anthelmintic, astringent, haemostatic and demulcent.[2–5] Moreover, some other species were used in treatment of skin burns, gallstone, hepatitis, osteomyelitis, nephritis, cystitis, suppurative dermatitis, gonorrhea, chronic gastritis, duodenal ulcers, gout, haemorroids and dental diseases.[6–9] A variety of chemical constituents such as flavonoids,[10,11] anthraquinones,[12] sesquiterpenoids,[13] lignans,[14] coumarins[15] and stilbenes[16] have been reported from the genus Polygonum.
Generally Polyphenolic compounds, including phenols, phenolic acids, flavonoids, tannins and lignans have been shown to be potent antioxidants because of their radical scavenging activity and their ability to form complex with heavy metal ions.[17,18] The former chemical and biological information about the genus Polygonum motivated the investigation of Polygonum bellardii All. growing in Egypt for its chemical composition and antioxidant activity.
MATERIAL AND METHODS
General
The UV was measured on Ultrospec 1000, UV/visible Spectrometer, Pharmacia Biotech (England). FAB-MS was measured on JEOL JMS 600 Hz (Japan). IR was measured on Shimadzu Infra-red-470 spectrophotometer (Japan). NMR analysis (1H-NMR, 13C-NMR, HMBC) were measured on JEOL TNM-LA 600 MHz FT- NMR spectrometer (Japan), Varian mercury 400 MHz Spectrometer (Oxford) using TMS as internal standard. Column chromatography was carried on silica gel (70–230, mesh, Merck), Sephadex LH-20 (20–100 μm, Sigma-Aldrich chemicals) and RP-18 (30–50 μm), TLC was carried on aluminium plates covered with a 0.25 mm silica (kieselgel 60 GF254, Merck, Germany). The plates were examined under UV light (at 365 and 254 nm) and visualized by spraying with 20% v/v H2SO4 in EtOH, allowed to dry at room temperature followed by heating at 110–1400C for 1–2 min. The following solvent systems were used for TLC:
I- n-Hexane-EtOAc (9:1 v/v)
II- n-Hexane-EtOAc (8:2 v/v)
III- CHCl3-MeOH-H2O (8: 2:0.2 v/v)
IV- CHCl3-MeOH-H2O (7.5: 2.5:0.3 v/v)
V Toluene-(CH3)2C=O-HCHO (3:6:1 v/v)
VI- MeOH-H2O (6: 4 v/v)
Plant material
The aerial parts of Polygonum bellardii All. were collected during flowering stage in April 2007 from Assiut governorate and kindly identified and authenticated by Prof. Dr. Moamen M. Zarea, Professor of Plant Taxonomy, Department of Botany, Faculty of Science, Assiut University, Assiut, Egypt. A voucher specimen was kept in the Department of Pharmacognosy, Faculty of Pharmacy, Al-Azhar University, Assiut, Egypt.
Authentic sugar materials
D-glucose, L-rhamnose, L-arabinose and L-galactose [obtained from El-Nasr Pharmaceutical and Chemical Co., Egypt (ADWIC) and Sigma-Aldrich chemicals].
Antioxidant materials
2,2-Diphenyl-1-picryl hydrazyl (DPPH), ascorbic acid and quercetin as an antioxidant standard (all were obtained from Sigma-Aldrich Chemicals Co.) and Digital Ultrasonic Cleaner, MTI Corporation, USA for mixing samples.
Extraction and isolation
The air-dried powdered aerial parts (2 Kg) of Polygonum bellardii All. were extracted by maceration and percolation with methanol till complete exhaustion (1:4 ratio) [four times each 8 L; overnight]. The combined methanolic extracts were concentrated under reduced pressure till constant weight to give a syrupy residue (150 g). The extract was subjected to successive solvent fractionation on VLC with n-hexane, chloroform, ethyl acetate, n-butanol and methanol till complete exhaustion in each case to give n-hexane (20 g), chloroform (25 g), ethyl acetate (20 g), n-butanol (10 g) and methanol (50 g).
The chloroform-soluble fraction (10g) was chromatographed on silica gel column chromatography, eluted with n-hexane and n-hexane-ethyl acetate gradients. Fractions 100ml, each were collected. Similar fractions were pooled together, concentrated under reduced pressure to give 6 sub-fractions labeled PCH1-PCH6. A part of sub-fraction PCH3 (500mg), was rechromatographed on silica gel column chromatography and eluted with n-hexane, followed by n-hexane-ethyl acetate gradients. The fractions eluted with n-hexane- ethyl acetate (9:1) afforded compound 1 (35 mg), whereas fractions eluted with n-hexane-ethyl acetate (8:2) afforded compound 2 (30 mg).
The ethyl acetate-soluble fraction (10 g) was chromatographed on silica gel column, eluted with CHCl3 followed by CHCl3-MeOH gradiently; eluates, 100 ml each were collected. Related fractions were grouped together and concentrated under reduced pressure to give 5 sub-fractions labeled PE1-PE5. Fraction PE4 (150mg) was chromatographed on Sephadex LH-20 using CHCl3 -MeOH (1:1). Fractions 8–12 were pooled together, concentrated under reduced pressure and chromatographed on RP- 18 column using MeOH-H2O (6:4), where fractions 5–8 afforded compound 3 (40 mg).
The n-butanol soluble fraction (5 g) was chromatographed on silica gel column chromatography eluted with CHCl3 followed by CHCl3-MeOH gradients; fractions 100 ml, each were collected. Similar fractions were grouped and concentrated under reduced pressure to give 3 sub-fractions labeled PN1-PN3.
Sub-fraction PN2 (200 mg) was chromatographed on Sephadex LH-20 using CHCl3-MeOH (1:1). Fractions 10- 15 were collected, concentrated under reduced pressure and rechromatographed on RP-18 using MeOH-H2O (1:1), afforded compound 4 (50 mg). Furthermore fractions 19–25 rechromatographed on RP-18 using MeOH-H2O (1:1), afforded compound 5 (35 mg).
Compound1 [17-hydroxypentacosanyl acetate]:
Obtained as white amorphous powder from CHCl3, Rf= 0.5 (system, I), +10.3 (MeOH, c.003). The IR spectrum (KBr) showed broad absorption band at 3410 cm-1 (OH stretching), in addition to other bands at 2995 cm-1 (CH stretching), 1722 cm-1 (C = O stretching), 1450 cm-1 (CH2 bending) and 1217 cm-1, 1066 and 989 (C-O stretching). EI mass spectrum showed the molecular ion peak at m/z 426 [M]+(15%), other peaks at m/z 411 [M-CH3]+(3.8%), 408 [M-H2O]+(2.7%), 367 [M-CH3COO]+(10%), 313 [M-CH3(CH2)7]+(2.7%), 283 [M-CH3(CH2)7CHOH]+(3.3%), 269 [M-CH3(CH2)7COO]+(1.4%), 157 [M-CH3(CH2)16 CHOH]+(2.6%), 143 [M-CH3(CH2)16COO]+(63%), 113 [M-CH3(CH2)16COOCHOH]+(58.2%), 59 [M-CH3(CH2)23CHOH]+(4%). Selected 1H-NMR spectral data (CD3OD, 600 MHz): δH 0.94 (3H, t, H3-25), 1.28–1.62 (30 H, m, CH2 residues), 2.30 (2H, m, H2 -2), 2.29 (3H, s, CH3 -C=O), 3.98 (1H, m, H-17), and 4.06 (2H, t, H2 -1). Selected 13C-NMR spectral data (CD3 OD, 150 MHz): δC13.94 (C-25), 23.66 (C-24), 26.66 (C-2), 29.89–30.29 (CH2residue), 31.77 (C-23), 35.03 (C-16 and C-18), 65.14 (C-1), and 69.00 (C-17) in addition to δC 20.11, and 175.61(CH3CO).
Compound 2 [β-sitosterol acetate]:
Obtained as white amorphous powder from CHCl3, Rf = 0.6 (system, II). Positive FAB-MS showed peak at m/z 457 [M+ H]+and 415 [(M+1)-acetyl]+. 1H-NMR spectral data (CD3OD, 600 MHz): δH 0.72 (3H, s, CH3-18), 0.85 (3H, d, J=6.8 Hz, CH3 - 26), 0.88 (3H, d, J=7.5 Hz, CH3 -27), 0.90 (3H, d, J=7.5 Hz, CH3 -29), 1.00 (3H, d, J=6.2 Hz, CH3 -21), 1.05 (3 H, s, CH3 -19), 1.24–2.03 (m, other CH and CH2), 2.05 (3H, s, CO-CH3), 2.32 (1H, br.s, H-4), 3.45 (1 H, m, H-3), and 5.37 (1 H, br s, H-6). 13C-NMR (CD3OD, 150 MHz): δC 37.29 (C-1), 30.62 (C-2), 77.79 (C-3), 39.67 (C- 4), 142.86 (C-5), 122.76 (C-6), 32.92 (C-7), 32.92 (C-8), 49.42 (C-9), 37.29 (C-10), 21.63 (C-11), 39.67 (C-12), 42.13 (C- 13), 58.34 (C-14), 25.92 (C-15), 30.26 (C-16), 57.56 (C- 17), 12.23 (C-18), 19.32 (C-19), 37.29 (C-20), 19.32 (C- 21), 34.98 (C- 22), 25.92 (C-23), 47.43 (C-24), 30.26 (C- 25), 20.07 (C-26), 21.63 (C-27), 24.02 (C-28), 14.36 (C- 29), 20.07 (CO-CH3) and 175.20 (CO-CH3).
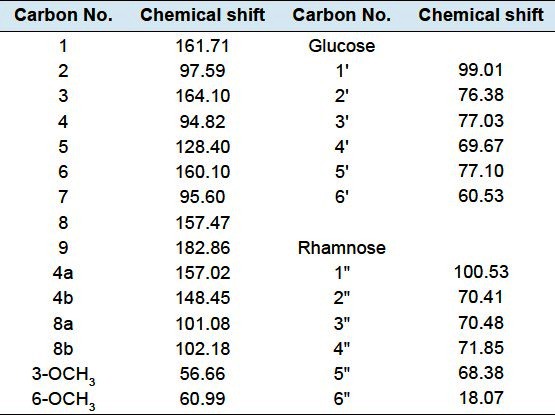
Compound 3 1,8-Dihydroxy-3,6-dimethoxyxanthone-5-O-[α-L-rhamnopyranosyl-(1”→2′)]-β-D-glucopyranoside]:
Obtained as yellowish needles from MeOH (m.p. 177–180), Rf = 0.40 (system, III), -9.8 (MeOH, c.001). Positive FAB-MS showed peak at m/z 613 [M+1]+, 467 [(M+1)-rhamnose]+and 305 [(M+1)-(rhamnose + glucose)]+. 1H-NMR spectral data (DMSO-d6, 600 MHz) was listed in Table 1. 13C-NMR spectral data (DMSO-d6, 150 MHz) was listed in Table 2.
Table 1.
1H-NMR spectral data of compound 3 (DMSO-δ6, 600 MHz)

Table 2.
13C-NMR spectral data of compound 3 (DMSO-δ6, 150 MHz)
Compound 4 [Lyoniresinol-3a-O-[α-L-rhamnopyranosyl-(1”′→6”)]-β-D-glucopyranoside]:
Obtained as a white amorphous powder from MeOH, Rf = 0.40 (system, IV), +9.6 (MeOH, c.05). Positive FAB-MS showed peak at m/z 729 [M+1]+and other peaks at 583 [(M+1)-rhamnose]+and 421 [(M+1)-(glucose + rhamnose)]+. The 1H-NMR spectral data (DMSO-d6, 400 MHz) was listed in Table 3. 13C-NMR spectral data (DMSO-d6, 100 MHz) was listed in Table 4.
Table 3.
1H-NMR spectral data of compound 4 and 5 (DMSO-d6, 400 and 600 MHz)
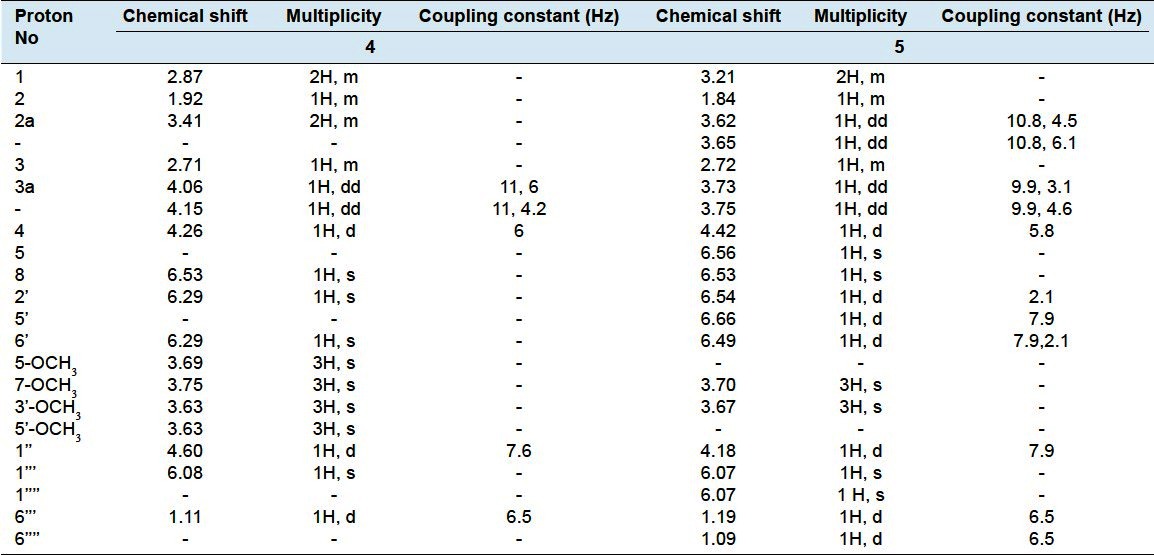
Table 4.
13C-NMR spectral data of compounds 4 and 5 (DMSO-δ6, 100 and 150 MHz)

Compound 5 [(+)-Isolariciresinol-3a-O-[α-L-rhamnopyranosyl-(1”′→2”)-α-L-rhamnopyranosyl-(1””→6”)]-β-D-glucopyranoside]:
Obtained as white powder from MeOH, Rf = 0.35 (system, IV), +11 (MeOH, c. 02). Positive FAB-MS showed peak at m/z 815 [M+1]+and other peaks at 669 [(M+1)- rhamnose]+, 521 [(M+1)-two rhamnose units]+, 361 [(M+1)- (glucose + two rhamnose units)]+. The 1H-NMR spectral data (CD3OD, 600 MHz) was listed in Table 3 and 13C NMR spectral data (CD3OD, 150 MHz) was listed in Table 4.
DPPH radical-scavenging assay
Firstly, free radical scavenging activity of the total methanolic extract, its fractions in addition to compounds 3, 4 and 5 isolated from the aerial parts of Polygonum bellardii All. was carried out against stable DPPH· with a rapid TLC screening method using 0.2% DPPH· in methanol. Thirty minutes after spraying, the active compounds appear as yellow spots against purple background.[19]
In a second experiment, DPPH· scavenging activity was measured by spectrophotometric method.[20] Briefly, 1ml of a wide range of concentrations (25–200 μg/ml) of test sample (in methanol) was added to 1 ml of 200 μM of methanolic solution of DPPH·. The mixture was mixed by Digital Ultrasonic Cleaner (sonicator) for 1 min and then left to stand at room temperature for 30 min in dark. When DPPH· reacts with an antioxidant compound, which can donate hydrogen, it is reduced and the changes in colour (from deep-violet to light-yellow) were measured at 517nm on a UV/visible light spectrophotometer. Absorption of blank sample containing the same amount of methanol and DPPH· solution was prepared and measured daily. The experiment was carried out in triplicate, using ascorbic acid and quercetin as a positive control standards.[21] The percentage reduction of the DPPH· was calculated by the following formula: %Inhibition (scavenging) = [(AB-AA)/AB]×100 Where: AB-absorption of blank sample (t = 0 min) and AA-absorption of tested extract solution (t = 30 min).
RESULTS AND DISCUSSION
Compound 1
The low resolution EI-MS showed the molecular ion peak [M]+ at m/z 426, together with IR, 1H-NMR and 13C-NMR spectral data confirmed the molecular formula to be C27H54O3. Careful investigation of 1H-NMR and 13C-NMR spectral data of compound 1 demonstrated the presence of long chain aliphatic ester moiety. EI-MS showed a peak at 408 [M-H2O]+(2.7%) indicated the presence of hydroxyl group, confirmed from IR band at 3410. Promising abundant peaks at m/z 143 [M-CH3(CH2)16COO]+(63%) and 113 [M-CH3(CH2)16COOCHOH]+(58.2%) indicated the presence of hydroxyl group at C-17. Additionally, it revealed the presence of an ester methyl moiety at m/z 59 (4%). The 1H-NMR showed a triplet methyl at δH 0.94 (3H, t) attributed to terminal methyl at C-25 together with another methyl signal at δH 2.63 (3H, s) belonging to methyl protons of the acetyl group. It also showed a signal at δH 3.98 (1H, m) assigned to oxygenated methine group. Moreover, it revealed the presence of a triplet signal at δH 4.06 (2H, t, H-1) and a multiplet signal at δH 2.30 (2H, m, H-2). Moreover, a cluster of CH2 signals at δH 1.28–1.6 (30 H, m) indicative for long chain aliphatic moiety, which was obvious from 13C-NMR signals at δC 29.89–30.29.[22,23] The 13C-NMR spectrum displayed two signals at δC 175.61 and 20.11, assigned to acetyl group. The oxygenated methine carbon appeared at δC 69.00, while the terminal methyl appeared at δC 13.94. Therefore, compound 1 was assigned as17-hydroxypentacosanyl acetate,[22,23] a new natural product [Figure 1–3, Tables 1–4].
Figure 1.
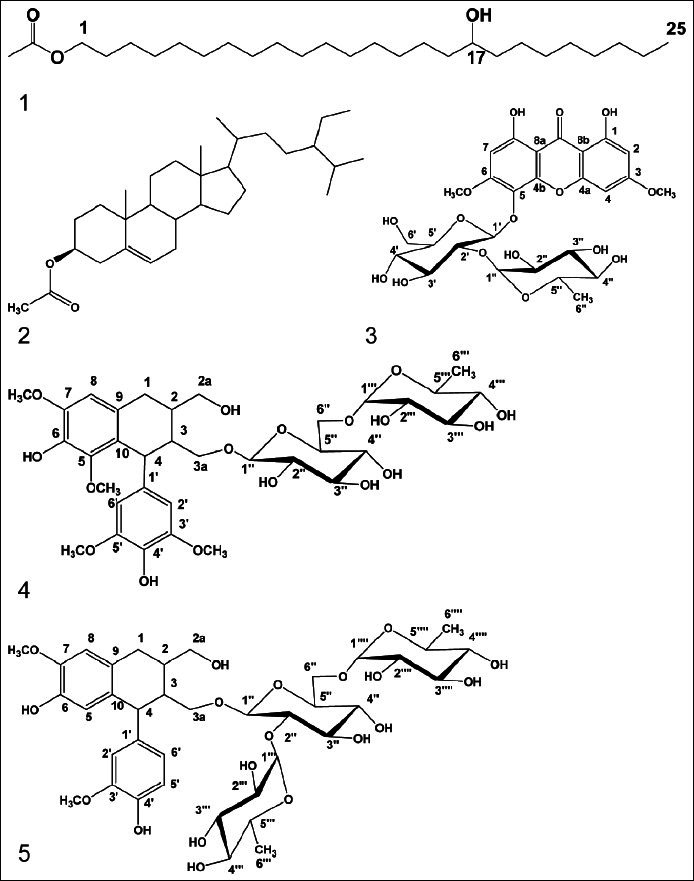
Structures of the isolated compounds 1–5
Figure 3.
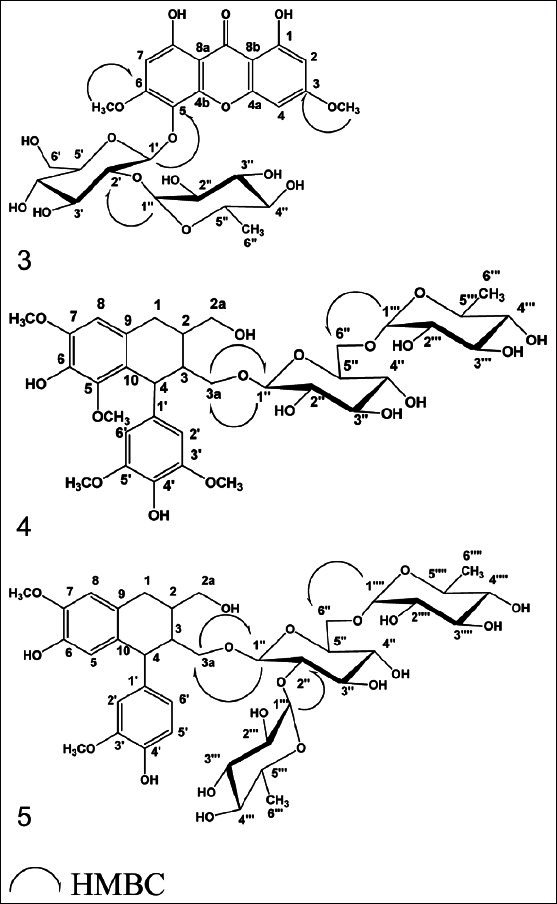
Significant HMBC correlations of compounds 3–5
Figure 2.

Significant fragments of compound 1
Compound 2
Chemical investigation of compound 2 indicated its steroidal nature.[24] 1H-NMR spectrum of compound 2 showed six methyl signals at δH 0.72, 0.85, 0.88, 0.90, 1.00 and 1.05 each (3H, s) assigned to CH3-18, 26, 27, 29, 21 and 19 respectively in addition to an extra downfield shifted oxygenated methyl signal at δH 2.05 (3H, s) allocated to acetyl group. It also showed signals at δH 2.32 (1H, br. s, H-4), 3.45 (1 H, m, H-3) and 5.37 (IH, br s, H-6). The 1H-NMR together with 13C-NMR data concluded the existence of acetylated β-sitosterol moiety, this was confirmed from 13C-NMR signals at δC 20.07 and 175.35 assigned to acetate moiety. The localization of acetyl group at C-3 was obvious from the downfield shift of C-3 at δC 77.79 upon comparison with non-substituted β-sitosterol.[25] From the previous data and upon comparison with literature,[26,27] compound 2 was identified as β-sitosterol-3-O-acetate, previously isolated from Prunella vulgaris[26] and firstly reported from the genus Polygonum.
Compound 3
The 1H and 13C-NMR measurements of compound 3 indicated the presence of xanthone glycoside moiety.[28] Positive FAB-MS spectrum showed the quasi-molecular ion peak at m/z 613 [M+1]+, together with 1H-, 13C-NMR spectral data confirmed the molecular formula to be C27H32O16. 1H-NMR spectrum of compound 3 showed two downfield shifted singlet signals at δH 11.72, 11.87 each (1H, s) ascribed to chelated 1,8-OH groups respectively. The singlet signal at δH 6.37 and broad singlet signals at δH 6.55 and 6.67 were assigned to isolated aromatic protons H-7, H-2 and H-4 respectively,[29–32] two intense signals appeared at δH 3.91, 3.77 each (3H, s) indicating two aromatic methoxy groups. Moreover, the 1H-NMR showed signals at δH 5.35 (1H d, J = 7.4 Hz) and 5.17 (1H, s) assigned to couple of anomeric sugar protons, indicative for the existence of bioside moiety. The previous 1H-NMR data together with diagnostic 13C-NMR signals listed in Table 2 were conclusive for the presence of pentasubstituted 1,8-dihydroxy bioside moiety. The site of sugar attachment at C-5 was obvious from 13C-NMR downfield shifted signals at δC 128.4 allocated to C-5 upon comparison with C-5 unsubstituted 1,8 dihydroxyxanthone series.[28–32] 13C-NMR displayed pair of diagnostic signals at δC 99.01, 100.53 attributed to two anomeric sugar carbons.[28–32] The aforementioned data associated with 13C-NMR prominent signals at δC 99.01, 76.38, 77.03, 69.67, 77.10 and 60.53 deduced the occurrence of C-2 substituted glucose moiety. The position of the intraglycosidic linkage was approved from 13C-NMR downfield shifted signal at δC 76.38 belonging to C-2 of glucose.[28–32] Other 13C-NMR signals at δC 100.53, 70.41, 70.48, 71.85, 68.38 and 18.07 in addition to characteristic doublet methyl group at δH 1.19 (3H, d, J=6 Hz) in 1H-NMR were ascribed to rhamnosyl moiety.[28–32] The partial and complete acid hydrolysis of compound 3 in parallel with investigation of the hydrolysate by paper chromatography (system IV) revealed the presence of glucose and rhamnose sugars. On the other hand, HMBC measurements showed a significant correlation between the anomeric proton of glucose unit H-1′ (δH 5.35) and C-5 (δC 128.40) of the aglycone part, supported our hypothesis about position of attachment of the sugar part to C-5 of the xanthone aglycone. It also showed an important correlation between anomeric proton of rhamnosyl moiety H-1″ (dH 5.17) and C-2′ (δC 76.38) of glucose moiety, confirming the site of attachment of rhamnose to glucose at C-2′. Furthermore the HMBC experiment showed cross linkage between protons of two methoxy groups at C-3 (δH 3.77) and C-6 (δH 3.91) and carbons C-3 δC 164.10) and C-6 (δC 160.10) respectively, supporting the presence of 3,6-dimethoxylated xanthone moiety. From the previously mentioned chromatographic, spectral and results of acid hydrolysis data, compound 3 was identified as 1,8-Dihydroxy-3,6-dimethoxy-xanthone-5-O-[α-L-rhamnopyranosyl-(1″→2′)]-β-D-glucopyranosid, a new natural product.
Compound 4
Its positive FAB-MS showed the quasi-molecular ion peak at m/z 729 [M+1]+, together with 1H-, 13C-NMR spectral data confirmed the molecular formula to be C34H48O17. The detailed analysis of 1H and 13C-NMR spectra of compound 4 deduced the occurrence of glycosylated (+)-lyoniresinol lignan moiety;[33,37] However the 1H-NMR spectrum of compound 4 displayed signals at δH 4.15 (1H, dd, J=11, 6 Hz), 4.06 (1H, dd, J=11, 4.2 Hz) assigned for primary alcoholic methylene protons H-3a, in addition to signal at δH 2.87 (2H, m) assigned to methylene protons, H-1. The signals appeared at δH 3.69 (3H, s), 3.75 (3H, s) and 3.63 (6H, s) could be assigned to four methoxy groups attached to C-5, C-7 and C-3´, C-5´ respectively.[33–37] Moreover, 1H-NMR showed the oxygenated methylene protons (H-2a) as a multiplet signal at δH 3.41 (2H, m). The aliphatic methine protons H-2, H-3 and H-4 appeared at δH 1.92 (1H, br.s), 2.71 (1H, m) and 4.26 (1H, d, J=6 Hz) respectively while the aromatic protons appeared at δH 6.53, (1H, s, H-8) and 6.29 (2H, s, H-2′ and H-6′). The 13C- NMR spectrum of compound 4 [Table 4] illustrated the presence of 34 carbon atoms, of them 12 carbons belonging to sugar moiety and the rest 22 carbons indicated an aryl-tetralin-type lignin.[33–37] Concerning the sugar moiety, 1H-NMR spectrum showed the presence of two anomeric protons, one of them appeared at δH 4.60 (1H, d, J = 7.6 Hz), the other proton noticed at δH 6.08 (1H, s). The 1H-NMR data in conjunction with 13C-NMR signals at δC 103.87, 100.86, indicating the existence of two anomeric carbons, in addition to the other signals at δC 73.41, 76.80, 69.04, 77.29 and 64.04 pointed out the incidence of C-6″ substituted glucose. This was further supported from C-6″ downfield shifted signal at δC 64.04 in comparison with non-substituted glucose moiety.[33–37] The appearance of a characteristic doublet at δH 1.11 (3H, δ, J= 6.5 Hz) in addition to other carbon signals at δC 69.74, 70.86, 70.99, 69.55 and 17.99, indicated the presence of rhamnose moiety attached to C-6″ of glucose. The sugar linkage at C-3a was determined mainly by the comparison of the 1H, 13C-NMR data of compound 4 with (+)-lyoniresinol, where two protons at C-3a (δH 4.06, 4.15) of compound 4 showed obvious downfield shift signal upon comparison with corresponding protons at C-3a (δH 3.48) of (+)-lyoniresinol (about 0.6 ppm) and C-3a shifted from δC 64.20 to 70.75 when compared with (+)-lyoniresinol.[33,38] The optical rotation of Compound 4 is +9.6 (MeOH, c.05) compared with (+)-lyoniresinol with optical rotation +13.3 (MeOH, c.32),[38] indicated that, compound 4 is (+)-lyoniresinol derivative. Investigation of the HMBC spectrum revealed the presence of significant correlations between; methylene protons H-3a (δH 4.15, 4.06) of the aglycone and C-1″ (δC 103.87) of glucose moiety, confirming the position of sugar linkage at C-3a. The HMBC spectrum showed diagnostic correlation between H-1″′ (δH 6.08) of rhamnose and C-6″ (δC 64.04) of glucose, confirming the attachment of rhamnose to C-6″ of glucose unit. From the above chemical, chromatographic and spectral data,[33–38] compound 4 was identified as (+)-Lyoniresinol-3a-O-[α-L-rhamnopyranosyl-(1″′→6″)]-β-D-glucopyranoside,[33–38] a new natural product.
Compound 5
The positive FAB-MS spectrum showed the quasi-molecular ion peak at m/z 815 [M+1]+, together with 1H-, 13C-NMR spectral data confirmed the molecular formula to be C38H54O19. The 1H-NMR together with 13C- NMR spectral data illustrated the presence of 38-carbon atoms, among them, 18 carbons for two aromatic rings and six aliphatic carbon exhibiting the existence of an aryl-tetralin-type lignan, could be identified as (+)-isolariciresinol.[34–37] Remaining 18 carbons concluded the presence of substituted glucose moiety. The 1H-NMR of compound 5 displayed signals at δH 3.62 (1H, dd, J = 10.8, 4.5) and 3.65 (1H, dd, J = 10.8, 6.1), H-2a; δH 3.21 (2H, m), H-1; δH 3.70 and 3.67 each (3H, s), two methoxy groups at C-7 and C-3´ respectively and δH 3.73 (1H, dd, J= 9.9, 3 Hz), 3.75 (1H, dd, J= 9.9, 4.6 Hz), H-3a. Moreover, it revealed the presence of the following signals: At δH 1.84 (1H, m, H-2), 2.72 (1H, br.s, H-3), 4.42 (1H, d, J= 5.8 Hz, H-4), 6.56 (1H, s, H-5), 6.54 (1H, s, H-8), 6.49 (1H, d, J=7.9, 2.1 Hz, H-6′), 6.66 (1H, d, J=7.9 Hz, H-5′) and 6.54 (1H, d, J=2.1 Hz, H-2′). The anomeric protons appeared at δH 4.18 (1H, d, J= 7.9 Hz) and 6.07 (2H, s). The 1H-NMR data in association with 13C-NMR signals at δC 102.24, 101.92 and 101.22, indicating the occurrence of three anomeric carbons, in addition to the other 13C-NMR signals at δC 74.91, 77.91, 68.01, 78.23 and 65.36 declared the presence of C-2′′ and C-6″ substituted glucose moiety,[37–40] This was further supported from C-2″, C-6″ downfield shift signals at δC 74.91 and 65.36 accordingly upon comparison with non-substituted glucose moiety.[38] The two doublet signals at δH 1.09 and 1.19 each (3H, d, J= 6.5 Hz), in addition to other 1H and 13C-NMR sugar signals [Tables 3 and 4] indicated the existence of two rhamnose moieties attached to C-2″ and C-6″ of glucose. The linkage of sugar was configured at C-3a mainly by the comparison of the 1H and 13C-NMR data of compound 5, with those reported for (+)-isolariciresinol, however the methylene protons H-3a (δH 3.73, 3.75) was obviously downfield shifted.[39,40] The optical rotation of Compound 5 is +11 (MeOH, c 0.05) compared with (+)-isolariciresinol +34 (MeOH, c 0.10),[40] indicated that, compound 5 is (+)-isolariciresinol derivative. HMBC correlations showed significant cross linkage between the protons at C-3a (δH 3.73, 3.75) of the aglycone and anomeric carbon of glucose moiety C-1″ (δC 102.24), confirming the attachment of sugar part at C-3a. The inter-glycosidic linkages between glucose and two rhamnose units were daignosed from HMBC correlations between the two anomeric protons of rhamnose units H-1″′, H-1″″ (δH 6.07) and C-2″ (δC 74.91) together with C-6′′ (δC 65.36) of glucose moiety which confirmed the site of attachment of rhamnose units to C-2″ and 6″ of glucose. From the above chemical, chromatographic and spectral data,[33–40] it could be concluded that compound 5 was identified as (+)-Isolariciresinol-3a-O-[α-L-rhamnopyranosyl-(1″′→2″)-α-L-rhamnopyranosyl-(1″″→6″)]-β-D-glucopyranoside, a new natural product.
Results of radical scavenging activity
The obtained results revealed that compounds 4, 5 in addition to ethyl acetate, n-butanol and methanol extracts displayed the strongest radical scavenging activity, while the weakest radical scavenging activity was observed with n-hexane extract. The radical scavenging activities of the extracts were correlated to the presence of high concentrations of flavonoids and phenolic compounds in the extracts which were isolated from Polygonum bellardii in our previous work.[10] The pure compounds showed lower activity than that of crude extracts (ethyl acetate and n-butanol) may be due to synergism (synchronization) of high flavonoid content in the plant (18.52 mg/kg) and other phenolic constituents present in crude extracts of the plant [Table 5, Figure 4].
Table 5.
Antioxidant activity of total extract, fractions and isolated compounds (3–5) from the aerial parts of Polygonum bellardii All

Figure 4.
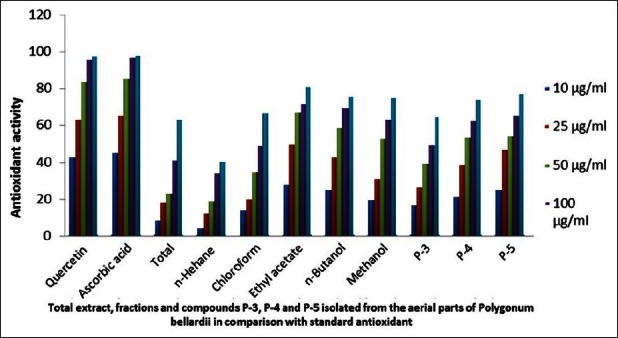
Antioxidant activity of the total extract, fractions and compounds 3, 4 and 5 isolated from the aerial parts of Polygonum bellardii All
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Muschler R. A Manual Flora of Egypt. Vol. 1. New York: S. H Service Agency, Inc.; 1970. pp. 255–68. [Google Scholar]
- 2.Mittal D. Hepatoprotective effects of Polygonum bistorta and active principles on albino rats intoxicated with carbon tetrachloride and paracetamol. Toxicol Lett. 2009;1895:557–61. [Google Scholar]
- 3.Demiray S, Pintado ME, Castro PM. Evaluation of phenolic profiles and antioxidant activities of Turkish medicinal plants: Tilia argentea, Crataegi folium leaves and Polygonum bistorta roots. World Acad Sci Engg Technol. 2009;54:312–7. [Google Scholar]
- 4.Ogweno-Midiwo J, Yenesew A, Juma BF, Derese S, Ayoo IA, Aluoch AO, et al. Bioactive compounds from some kenyan ethnomedicinal plants: Myrsinaceae, Polygonaceae, and psiadia punctulata. Phytochem Rev. 2002;1:311–23. [Google Scholar]
- 5.Sun XB, Zhao PH, Xu YJ. Chemical constituents from the roots of Polygonum bistorta. Chem Nat Compounds. 2007;43:563–6. [Google Scholar]
- 6.Wang K, Zhang Y, Yang C. Antioxidant phenolic compounds from rhizomes of Polygonum paleaceum. J Ethnopharmacol. 2005;96:483–7. doi: 10.1016/j.jep.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 7.Ban SH, Kwon YR, Pndit S, Lee YS, Yi HK, Joen JG. Effects of a bio-assay guided fraction from Polygonum cuspidatum root on the viability, acid production and glucosyltransferase of mutant Streptococci. Fitoterapia. 2010;81:30–4. doi: 10.1016/j.fitote.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 8.Lv L, Gu X, Tang J, Ho CH. Antioxidant activity of stilbene glycoside from Polygonum multiflorum Thunb. In vivo. Food Chem. 2007;104:1678–81. [Google Scholar]
- 9.Xiao K, Xuan L, Xu Y, Bai D. Stilbene glycoside sulfates from Polygonum cuspidatum. J Nat Prod. 2000;63:1373–6. doi: 10.1021/np000086+. [DOI] [PubMed] [Google Scholar]
- 10.Mohamed MH, Ibraheim ZZ, Abd El-Mawla AM, Abd El-Kader AM. A new flavonol glycoside from Polygonum bellardii All. growing in Egypt. Bull Pharma Sci Assiut Univ. 2006;28:262–70. [Google Scholar]
- 11.López SN, Sierra MG, Gattuso SJ, Furlàn RL, Zacchino S A. An unusual homoisoflavanone and a structurally-related dihydrochalcone from Polygonum ferrugineum (Polygonaceae) Phytochemistry. 2006;67:2152–8. doi: 10.1016/j.phytochem.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 12.Zhang F, Chen W, Sun L. LC-VWD-MS Determination of three anthraquinones and one stilbene in the quality control of crude and prepared roots of Polygonum multiflorum Thunb. Chromatographia. 2008;67:869–74. [Google Scholar]
- 13.Datta BK, Rahman MM, Gray AI, Sayed AH, Auzi AA, Sarker SD. Polygosumic acid, a new cadinane sesquiterpene from Polygonum viscosum, inhibits the growth of drug-resistant Escherichia coli and Staphylococcus aureus (MRSA) in vitro. J Nat Med. 2007;61:391–6. [Google Scholar]
- 14.Xiao K, Xuan L, Xu Y, Bai D, Zhong D. Constituents from Polygonum cuspidatum. Chem Pharma Bull. 2002;50:605–8. doi: 10.1248/cpb.50.605. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Yu M, Meng D, Li Z, Zhang L. A new chromone glycoside from Polygonum capitatum. Fitoterapia. 2007;78:506–9. doi: 10.1016/j.fitote.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 16.Jiang Z, Xu J, Long M, Tu Z, Yang G, He G. Tetrahydroxystilbene-2-O-β-D-glucoside (THSG) induces melanogenesis in B16 cells by MAP kinase activation and tyrosinase upregulation. Life Sci. 2009;85:345–50. doi: 10.1016/j.lfs.2009.05.022. [DOI] [PubMed] [Google Scholar]
- 17.Pan Y, Zhang X, Wang H, Liang Y, Zhu J, Li H, et al. Antioxidant potential of ethanolic extract of Polygonum cuspidatum and application in peanut oil. Food Chemistry. 2007;105:1518–24. [Google Scholar]
- 18.Hsin-Tang L, Sui-Lin N, Ya-Yin H, She-Ching W. Potential Antioxidant Components and Characteristics of Fresh Polygonum multiflorum. J Food Drug Anal. 2010;18:120–7. [Google Scholar]
- 19.Yrjönen T, Peiwu L, Summanen J, Anu-Hopia A, Vuorela H. Free radical-scavening activity of phenolics by reversed-phase TLC. J Am Oil Chem Soc. 2003;80:9–14. [Google Scholar]
- 20.Qureshi MN, Kuchekar BS, Logade NA, Haleem MA. In-vitro Antioxidant and in-vivo Hepatoprotective activity of Leucas ciliata leaves. Rec Nat Prod. 2010;4:124–30. [Google Scholar]
- 21.Fan P, Terrier L, Hay AE, Marston A, Hostettmann K. Antioxidant and enzyme inhibition activities and chemical profiles of Polygonum sachalinensis F. Schmidt ex Maxim (Polygonaceae) Fitoterapia. 2010;81:124–31. doi: 10.1016/j.fitote.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 22.Pande A, Shukla YN, Tripathi AK. Lipid constituents from Stellaria media. Phytochemistry. 1995;39:709–11. [Google Scholar]
- 23.Silverstein RM, Bassler GC, Morrill TC. Spectrometric Identification of Organic Compounds. 5th. New York, Chichester, Brisbane, Toronto and Singapore: John Willy and Sons, Inc; 1991. pp. 27–9. [Google Scholar]
- 24.Schmidt J. Organic Chemistry. 8th. Edinburgh and London: Oliver and Boyd; pp. 318–673. [Google Scholar]
- 25.Asakawa Y, Toyota M, Harrison LJ. Isotachin A and isotachin B, two sulphur-containing acrylates from the liverwort Isotachis japonica. Phytochemistry. 1985;24:1505–8. [Google Scholar]
- 26.Kojima H, Sato N, Hatano A, Ogura H. Sterol glucosides from Prunella vulgaris. Phytochemistry. 1990;29:2351–5. [Google Scholar]
- 27.Ibraheim ZZ, Ahmed AS, Ramadan MA. Lipids and triterpenes from Maerua crassifolia growing in Egypt. Saudi Pharma J. 2008;16:69–74. [Google Scholar]
- 28.Vieira LM, Kijjoa A. Naturally-Occurring Xanthones: Recent Developments. Curr Med Chem. 2005;12:2413–46. doi: 10.2174/092986705774370682. [DOI] [PubMed] [Google Scholar]
- 29.Elshanawany AM, Mohamed GA, Nafady AM, Ibrahim SR, Radwan MM, Ross SA. A new xanthone from the roots of Centaurium spicatum. Phytochem Lett. 2011;4:126–8. [Google Scholar]
- 30.Xue HQ, Ma XM, Wu SX, Xing XL, Wang HQ. Xanthones from Gentianopsis paludosa. Chem Nat Compounds. 2011;46:979–81. [Google Scholar]
- 31.Jiang Y, Tu PF. Xanthone O-glycosides from Polygala tenuifolia. Phytochemistry. 2002;60:813–6. doi: 10.1016/s0031-9422(02)00184-x. [DOI] [PubMed] [Google Scholar]
- 32.Nugroho A, Choi JK, Park JH, Lee KT, Cha BC, Park HJ. Two New flavonol glycosides from Lamium amplexicaule L. and their in vitro. Free Radical Scavenging and Tyrosinase Inhibitory Activities, Planta Medica. 2009;75:364–6. doi: 10.1055/s-0028-1112216. [DOI] [PubMed] [Google Scholar]
- 33.Tripetch K, Mohamed SK, Ryoji K, Kazuo Y, Chayan P, Yoshikazu H. Lignan glucosides from Acanthus ilicifolius. Phytochemistry. 2001;56:369–72. doi: 10.1016/s0031-9422(00)00362-9. [DOI] [PubMed] [Google Scholar]
- 34.Tripetch K, Ryoji K, Kazuo Y. Lignan and phenylpropanoid glycosides from fernandoa adenophylla. Phytochemistry. 2001;57:1245–8. doi: 10.1016/s0031-9422(01)00212-6. [DOI] [PubMed] [Google Scholar]
- 35.Piyanuch T, Rutt S, Rapepol B, Robert V. Antioxidant lignan glucosides from Strychnos vanprukii. Fitoterapia. 2004;75:623–8. doi: 10.1016/j.fitote.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 36.Tripetch K, Phannipha C, Ryoji K, Hideaki O, Kazuo Y. Lignan and megastigmane glycosides from Sauropus androgynus. Phytochemistry. 2003;63:985–8. doi: 10.1016/s0031-9422(03)00219-x. [DOI] [PubMed] [Google Scholar]
- 37.Tian-Tian N, Huan-Xin Z, Hong B. Chemical Constituents of Pittosporum glabratum. Chinese J Nat Med. 2011;9:180–4. [Google Scholar]
- 38.Tung YT, Cheng KC, Ho ST, Chen YL, Wu TL, Hung KC, et al. Comparison and Characterization of the Antioxidant Potential of 3 Wild Grapes-Vitis thunbergii, V. flexuosa, and V kelungeusis. J Food Sci. 2011;76:701–6. doi: 10.1111/j.1750-3841.2011.02178.x. [DOI] [PubMed] [Google Scholar]
- 39.Yean MH, Kim JS, Lee JH, Kang SS. Lignans from Lonicerae caulis. Nat Prod Sci. 2010;16:15–9. [Google Scholar]
- 40.Jutiviboonsuk A, Zhang H, Tan GT, Ma C, Hung NV, Cuong NM, et al. Bioactive constituents from roots of Bursera tonkinensis. Phytochemistry. 2005;66:2745–51. doi: 10.1016/j.phytochem.2005.09.025. [DOI] [PubMed] [Google Scholar]