

Abstract
Our objectives were to determine if porcine serum could be enriched with selenium (Se) by feeding pigs with high concentrations of dietary Se and if the Se-biofortified serum inhibited proliferation of 3 types of human cancer cells. In Expt. 1, growing pigs (8 wk old, n = 3) were fed 0.02 or 3.0 mg Se/kg (as sodium selenite) for 16 wk and produced serum with 0.5 and 5.4 μmol/L Se, respectively. In Expt. 2, growing pigs (5 wk old, n = 6) were fed 0.3 or 1.0 mg Se/kg (as Se-enriched yeast) for 6 wk and produced serum with 2.6 and 6.2 μmol/L Se, respectively. After the Se-biofortified porcine sera were added at 16% in RPMI 1640 to treat NCI-H446, DU145, and HTC116 cells for 144 h, they decreased (P < 0.05) the viability of the 3 types of human cancer cells by promoting apoptosis, compared with their controls. This effect was replicated only by adding the appropriate amount of methylseleninic acid to the control serum and was mediated by a downregulation of 8 cell cycle arrest genes and an upregulation of 7 apoptotic genes. Along with 6 previously reported selenoprotein genes, selenoprotein T (Selt), selenoprotein M (Selm), selenoprotein H (Selh), selenoprotein K (Selk), and selenoprotein N (Sepn1) were revealed to be strongly associated with the cell death-related signaling induced by the Se-enriched porcine serum. In conclusion, porcine serum could be biofortified with Se to effectively inhibit the proliferation of 3 types of human cancer cells and the action synchronized with a matrix of coordinated functional expression of multiple selenoprotein genes.
Introduction
Cancer is a major public health threat and afflicts millions of people in the world. Approximately 30% of women and 50% of men in the United States may develop cancer within their lifetime (1). Lung cancer represents the leading cause of cancer mortality, followed by prostate and colon cancers (2). The Nutritional Prevention of Cancer Trial showed that daily supplementation of 200 μg selenium (Se) as Se-enriched yeast decreased mortalities of lung, colon, and prostate cancers in senior citizens (3). Other animal and human studies have also demonstrated a similar anticancer potential of supranutritional levels of Se (4, 5). Because ingesting high levels of Se from currently used sources may be associated with increased risks of diabetes and hyperlipidemia in humans (6, 7), it is important to explore novel or safe sources of Se that possess more anticancer capacity with fewer health concerns.
Selenoproteins and nonprotein-bound Se metabolites in the liver of mice fed supranutritional Se were reported to reduce the risks of colon cancer (5). As a vital organ for Se metabolism (8), the liver may produce anticancer selenocompounds and distribute them into target organs via the circulation. Those active selenocompounds are presumably present in the serum. In certain parts of the world, serum from food animals such as pigs is used as foods or food ingredients. Although pigs are an excellent model for human nutrition and medicine (9), the effects of Se-enriched serum from pigs or any other food-producing species on treating human cancer cells have not been reported. Various forms of inorganic and organic Se compounds have been used in studying the anticancer role and mechanism of Se (5, 10). These include sodium selenite, sodium selenate, seleno-dl-cystine (SeCys)8, seleno-dl-methionine (SeMet), Se-methylseleno-l-cysteine (SeMC), and methylseleninic acid (MSA) (10, 11). Comparatively, MSA is one of the most effective forms of Se in inhibiting cancer cell proliferation (11). In general, anticancer functions of various Se compounds are mediated by regulating major cell cycle arrest and apoptosis signaling proteins (12, 13). The former include cyclin-dependent kinase 1 (Cdc2), cell division cycle 25 homolog A (Cdc25A), cyclin-dependent kinase 2 (Cdk2), cyclin-dependent kinase 4 (Cdk4), cyclin A, cyclin D1, transcription factor Dp-1 (DP1), growth arrest and DNA damage-inducible gene 153 (GDAA153). The latter include RAC-beta serine/threonine-protein kinase (AKT2), B-cell lymphoma 2 (Bcl-2), Caspase3, cFos-binding protein (c-Jun), forkhead box O1 (FoxO1), cyclin-dependent kinase inhibitor 2A (p19), cyclin-dependent kinase inhibitor 1 (p21), mitogen-activated protein kinase p38 (p38), and tumor suppressor protein 53 (p53) (12–17). However, there is no information on the effectiveness and underlying mechanism of Se-biofortified porcine serum compared with other well-tested Se sources in treating or inhibiting the proliferation of human cancer cells.
Although a total of 25 selenoproteins (18) was identified in the human genome, the involvement and relative importance of these proteins in the anticancer actions of Se, in particular in inhibiting the recurrence of cancer cells, have remained unclear (5, 19). Many past studies were focused on single or only a few selenoproteins (5, 20–22) in the formation of primary cancer (5, 22) and were conducted with the perception that the expression of most, if not all, selenoproteins in normal cells was saturated at Se levels much lower than the anticancer doses (5). Little information is available on the responses of the whole selenogenome or multiple selenoprotein genes in the formed cancer cells to exposure of high levels of Se. Because recent animal studies have demonstrated regulations of high Se intake (up to 3 mg Se/kg diet) on the expressions of deiodinase, iodothyronine, type I (Dio1), cytosolic glutathione peroxidase (Gpx1), gastro-intestinal glutathione peroxidase (Gpx2), plasma glutathione peroxidase (Gpx3), phospholipidhydroperoxide glutathione peroxidase (Gpx4), selenoprotein H (Selh), selenoprotein K (Selk), selenoprotein M (Selm), selenoprotein O (Selo), selenoprotein S (Sels), selenoprotein T (Selt), selenoprotein V (Selv), 15 kDa selenoprotein (Sep15), selenoprotein N (Sepn1), Sepp1, selenoprotein W (Sepw1), thioredoxin reductase-1 (Txnrd1), and thioredoxin reductase-2 (Txnrd2) (23–25), it is fascinating to examine if high Se serum induces cell cycle arrest and apoptosis via regulating individual or groups of selenoprotein genes in human cancer cells. Therefore, our objectives were to determine if: 1) porcine serum could be biofortified with Se by feeding pigs with supranutritional levels of sodium selenite and Se-enriched yeast; 2) the Se-biofortified porcine serum was more effective than the commonly used selenocompounds in inhibiting proliferation of human lung, prostate, and colon cancer cells; and 3) the high-Se porcine serum inhibited the human cancer cells by activating cell cycle arrest and apoptosis signaling via regulation of the functional expression of selected selenoprotein genes.
Materials and Methods
Pigs, diets, and serum preparations.
The pig protocol for Expt. 1 was approved by Sichuan Agricultural University, Chengdu, China. Six Duroc × Landrance × Yorkshire growing pigs (8 wk old) were fed a Se-deficient, corn-soybean basal diet (0.02 mg Se/kg; 23) or the diet plus 3.0 mg Se/kg as sodium selenite (Kermel) for 16 wk. The pig protocol for Expt. 2 was approved by the Cornell University Institutional Animal Care and Use Committee. Twelve Yorkshire × Hampshire × Landrace growing pigs (5 wk old) were fed a similar corn-soybean meal diet (26) supplemented with Se at 0.3 and 1.0 mg /kg as Se-enriched yeast (ADM Alliance Nutrition) for 6 wk. In both experiments, pigs were given free access to feed and water and were housed in temperature (22°C)- and light (12 h)-controlled pens. The serum Se enrichment protocol in Expt. 2, including dietary Se supplemental level and feeding duration, was based on the serum Se responses of pigs in Expt. 1.
At the end of feeding, blood was collected from the anterior vena cava of pigs feed-deprived overnight for 8 h. The serum was prepared by centrifugation at 1000 × g for 15 min at 4°C and was filtered twice through a 0.22-μm membrane for sterilization and stored at −20°C before use. Serum from pigs fed the Se-deficient basal diet or 3.0 mg Se/kg in Expt. 1 was designated as control and Se-Na, respectively. Serum from pigs fed 0.3 and 1.0 mg Se/kg in Expt. 2 was designated as control and Se-Y, respectively. The concentrations of total Se, GPX3 activity, lactate dehydrogenase (LDH) activity, and selenoprotein P (Sepp1) in the prepared serum samples were determined as previously described (23, 27, 28). Separation of nonprotein-bound Se and protein-bound Se in pig serum was conducted as described by Beilstein and Whanger (29).
Cell culture and viability assay.
The human small cell lung cancer cell line NCI-H446 used in Exp. 1 was obtained from Shanghai Institute of Cell Biology, Chinese Academy of Sciences. The human prostate cancer cell line DU145 and human colon cancer cell line HCT116 used in Exp. 2 were generously donated by Dr. David Shalloway, Department of Molecular Biology and Genetics of Cornell University. Before the Se-enriched serum treatments, cells were grown in RPMI 1640 medium supplemented with 100 iu penicillin, 100 μg streptomycin/mL, and 10% FBS. After FBS-free cell suspension was prepared and cells were seeded at 1× 104 cells/well in 24-well plates, the selected porcine serum was added (at 16%) to the medium for various tests. Our preliminary experiment indicated that 16% pig serum could replace 10% FBS in the media to maintain growth and morphology of the selected 3 human cancer cell lines. All cell culture plates were maintained in a humidified incubator containing 5% CO2 and 95% air at 37°C. The medium was changed every 48 h. After 144 h of incubation, cell viability was determined using the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide assay (10). Cell counting was conducted using a hemacytometer and viable cells were determined by the trypan blue exclusion method.
In Expt. 1, the baseline Se concentration in the RPMI 1640 media was 0.08 μmol/L. The viability of human small cell lung cancer cells (NCI-H446) was determined with or without the Se concentration in the control serum equalized with that of Se-Na by adding the proper amount of sodium selenite. The apoptotic rate of treated cells was assayed using a FITC Annexin V Apoptosis Detection kit II (BD Bioscience) by a flow cytometer (BD FACSCalibur). The LDH activity in the medium supernatant was determined as described above.
In Expt. 2, the baseline Se concentration in the RPMI 1640 media was undetectable. The viability of human prostate cancer cell line DU145 and human colon cancer cell line HCT116 was determined as described above after the Se concentration in the control serum was equalized with that of Se-Y by adding the proper amounts of SeCys, SeMet, SeMC, sodium selenite, sodium selenate, or MSA. To exclude nonspecific toxicity associated with the Se dose used, an amount of Se equal to that in Se-Y was added into the regular RPMI 1640 media as sodium selenite, SeMet, or MSA to treat DU145, HCT116, and HepG2 cells.
Real-time qPCR and Western-blot analyses.
The cancer cells were collected after treatments with Se-Na or Se-Y along with the respective control serum with balanced (equalized) Se concentrations for 144 h. Total RNA was prepared from fresh cells by using Trizol (Invitrogen) and following the manufacturer’s instructions. The mRNA levels of pertaining genes in cell cycle arrest, apoptosis, and selenoprotein genes were determined by qPCR (7900 HT; Applied Biosystems). The genes assayed and primer sequences used for each gene are presented in Supplemental Table 1. The 2−ddCt method was used for the quantification with hypoxanthine-guanine phosphoribosyltransferase (HPRT) as a reference gene, and the relative abundance was normalized to the control (as 1). The validity of HPRT as the reference was verified by its amplification plots and dissociation curves of all samples from the 2 cell lines (Supplemental Fig. 1). Protein concentration determinations and Western-blot analyses of the pertaining samples were performed as previously described (24). The primary antibodies used for each gene are presented in Supplemental Table 2.
Statistical analyses.
Data generated from Expt. 1 were analyzed using Student’s t test. Data generated from Expt. 2 with ≥3 treatments were analyzed by 1-way ANOVA to test the main effects. The Bonferroni t test was followed for multiple mean comparisons if there was a main effect. The correlation between the expression profiles of selenoprotein genes and cell cycle arrest/apoptosis genes was analyzed using the stepwise regression program of PROC REG. Significance was set at P < 0.05. All analyses were conducted using SAS 8.2 (SAS Institute).
Results
Expt. 1
Pig study.
Feeding pigs with 3 mg Se/kg as sodium selenite elevated (P < 0.05) the total serum Se concentration, nonprotein and protein-bound Se, relative percentage of nonprotein-bound Se, and GPX3 activity compared with the control diet (Table 1). The total Se concentration in the control and Se-Na was 0.5 and 5.4 μmol/L, respectively. Meanwhile, the Se supplementation decreased (P < 0.05) the serum LDH activity and relative percentage of protein-bound Se. Dietary Se concentrations did not show any effect on growth performance or apparent pig health (data not shown).
TABLE 1.
Effects of supranutritional dietary Se on biochemical profiles of pig serum in Expt. 1 and 21
| Control2 | Biofortified3 | |
| Expt. 1 | ||
| Total Se, μmol/L | 0.5 ± 0.2 | 5.4 ± 0.6* |
| NBS, μmol/L | 0.02 ± 0.01 | 1.5 ± 0.1* |
| PBS, μmol/L | 0.48 ± 0.1 | 4.2 ± 0.6* |
| NBS, % | 4.0 ± 2.8 | 24.6 ± 1.3* |
| PBS, % | 96.0 ± 2.8 | 75.4 ± 5.9* |
| Protein, g/L | 35.5 ± 3.3 | 36.1 ± 3.4 |
| GPX3, units/mL | 8.1 ± 2.8 | 28.8 ± 4.5* |
| LDH, units/mL | 184 ± 8.9 | 102 ± 10.8* |
| Expt. 2 | ||
| Total Se, μmol/L | 2.6 ± 0.2 | 6.2 ± 0.8* |
| GPX3, units/mL | 37.9 ± 0.9 | 35.6 ± 1.6 |
| Sepp1,4 % | 100 ± 7.2 | 157 ± 23.1* |
Values are means ± SEs, n = 3–6. Means with * are different from the controls, P < 0.05. GPX3, plasma glutathione peroxidase; LDH, lactate dehydrogenase; NBS, nonprotein-bound Se; PBS, protein-bound Se; Sepp1, selenoprotein P.
In Expt. 1, the control diet contained 0.02 mg Se/kg; in Expt. 2, the control diet was supplemented with 0.3 mg Se/kg.
In Expt. 1, the biofortified diet was supplemented with 3.0 mg Se/kg as sodium selenite; in Expt. 2, the biofortified diet was supplemented with 1.0 mg Se/kg as Se-enriched yeast.
Values are relative density from the protein band.
Cell study.
After 144 h of incubation, the cells treated with Se-Na had lower (P < 0.05) viable cell counts (25%; Fig. 1A) and cell viability (22%; Fig. 1B), along with increased (P < 0.05) LDH activity released into the media (Fig. 1C), compared with pigs treated with the control serum. The difference (P < 0.05) in cell viability between the 2 types of serum still remained (Fig. 1D) even after their Se concentrations were matched by adding the appropriate amount of sodium selenite to the control serum. Although typical quadrantal diagram of cells was shown (Fig. 2A,B), the cells treated with Se-Na had a greater (P < 0.05) apoptotic rate than those treated with the Se-equalized control serum (Fig. 2C). Compared with the control group, the Se-Na–treated cells had a substantial decrease (P < 0.05) in the Bcl-2 mRNA level and increases (P < 0.05) in p53 and p38 mRNA levels (Fig. 2D). However, there was no difference in Caspase3 mRNA level between the 2 treatments.
FIGURE 1.

Effects of the Se-biofortified porcine serum (Se-Na) treatment on viable cell numbers (A), cell viability (B), medium LDH activity (C), and cell viability (D) of the human small cell lung cancer cells (NCI-H446) in Expt. 1. The control serum was not supplemented with extrinsic Se in A–C, but was supplemented with sodium selenite to match the Se concentration with that of Se-Na in D. Data are means ± SEs, n = 6. *Different from control, P < 0.05. LDH, lactate dehydrogenase; Se-Na, serum from pigs fed 3.0 mg Se/kg in Expt. 1.
FIGURE 2.

Effects of the Se-biofortified porcine serum (Se-Na) treatment on the quadrantal diagrams (A,B), apoptotic rate (C), and relative mRNA abundance of Bcl-2, p53, p38, and Caspase3 (D) of the human small cell lung cancer cells (NCI-H446) in Expt. 1. The early and late apoptotic cells are located in the lower right and upper right quadrants of A and B, respectively. In all panels, the control porcine serum was supplemented with sodium selenite to match the Se concentration with that of Se-Na. Data in C and D are means ± SEs, n = 4. *Different from control, P < 0.05. Bcl-2, B-cell lymphoma 2; p38, mitogen-activated protein kinase p38; p53, tumor suppressor protein 53; Se-Na, serum from pigs fed 3.0 mg Se/kg in Expt. 1.
Expt. 2
Pig study.
Feeding pigs with 3 mg Se/kg as Se-enriched yeast elevated (P < 0.05) the total serum Se concentration from 2.6 μmol/L of the control to 6.2 μmol/L (Table 1). Whereas the serum Sepp1 concentration was greater (P < 0.05) in the Se-Y than in the control, GPX3 activity did not differ between the 2 types of sera. Growth performance or health condition of pigs was not affected by dietary Se concentrations (data not shown).
Cell viability.
Compared with those treated with the control serum, the Se-Y–treated DU145 and HCT116 cells exhibited lower (P < 0.05) cell viability (Fig. 3A,B). After proper amounts of various Se compounds were added to the control serum to match the Se concentration of the Se-Y serum, only the MSA balanced serum resulted in viability decreases (P < 0.05) similar to those of Se-Y in the DU145 (31 vs. 29%) and HCT116 (25 vs. 20%) cell lines. None of the SeCys, SeMet, SeMC, sodium selenite, or sodium selenate balanced-sera affected cell viability in either cancer cell line compared with the control serum. Adding the same amount of Se as in Se-Y in the form of sodium selenite or organic forms other than MSA to the RPMI 1640 or DMEM media caused no change in the viability of DU145 and HCT116 cells (Supplemental Fig. 2) or HepG2 cells (Supplemental Fig. 3) compared with the respective medium controls.
FIGURE 3.
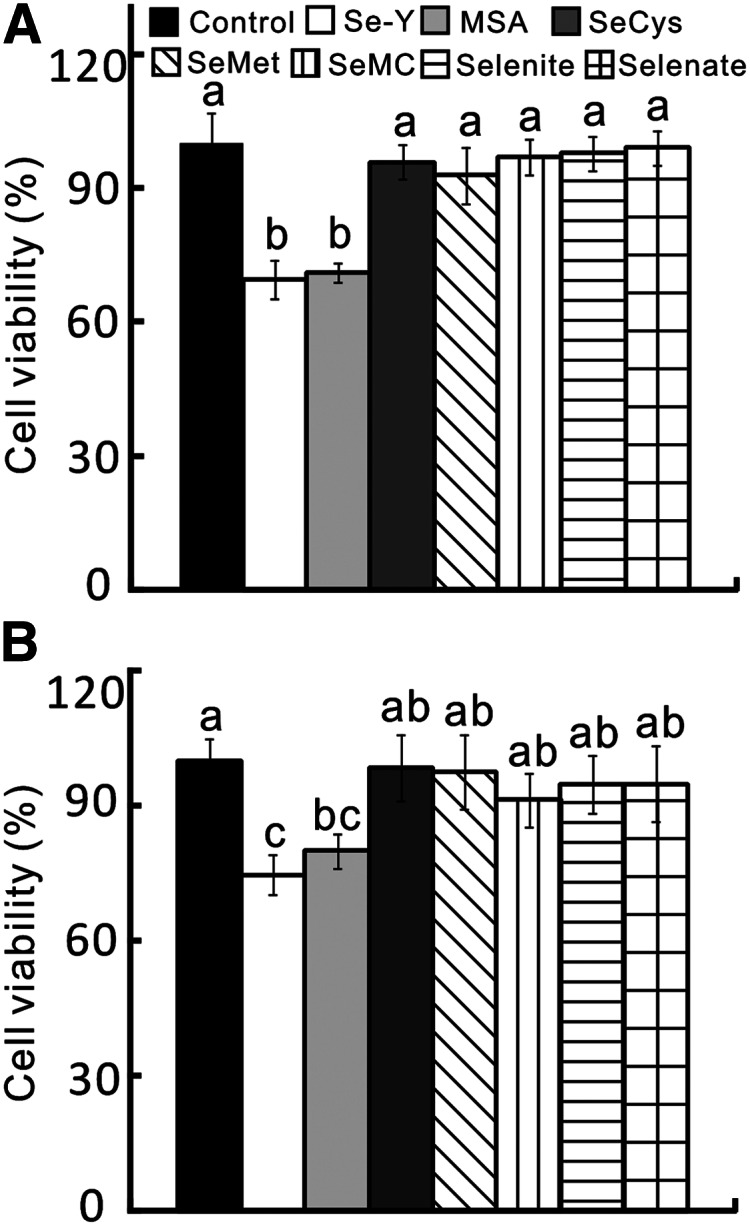
Effects of Se-biofortified porcine serum (Se-Y) treatment on cell viability of human prostate cancer cells (DU145; A) and colon cancer cells (HTC116; B) in Expt. 2 compared with the control porcine serum or that serum was added with different Se compounds to match the Se concentration in Se-Y. Data are means ± SEs, n = 5. Bars without a common letter differ, P < 0.05. MSA, methylseleninic acid; SeCys, seleno-dl-cystine; SeMC, Se-methylseleno-l-cysteine; SeMet, seleno-dl-methionine; Se-Y, serum from pigs fed 1.0 mg Se/kg in Expt. 2.
Cell cycle arrest and apoptosis gene expression.
Among the 17 cell cycle arrest and apoptosis genes assayed, 10 genes in the DU145 cell line (Fig. 4A) and 9 genes in the HCT116 cell line (Fig. 4B) were affected (P < 0.05) by the high-Se serum treatments. In the DU145 cell line, both Se-Y– and MSA-balanced serum resulted in similar or consistent decreases (P < 0.05) in mRNA levels of the cell death genes DP1, Cyclin A, Cdk2, and Cdk4 as well as increases (P < 0.05) in mRNA levels of p53, Caspase3, and c-Jun compared with the control serum and/or those balanced with SeMet and selenite. However, only Se-Y decreased (P < 0.05) mRNA levels of Cdc25A and increased GADD153 and FoxO1 mRNA levels compared with the control serum. In addition, the selenite-balanced serum increased (P < 0.05) the mRNA levels of p53, Caspase3, and FoxO1, whereas the SeMet-balanced serum elevated the mRNA levels of only Caspase3 compared with the control serum. In the HTC116 cell line, both Se-Y and the MSA-balanced serum decreased (P < 0.05) the mRNA levels of the cell death genes (DP1, Cdc2, and Cyclin A). Meanwhile, only the Se-Y treatment increased (P < 0.05) mRNA levels of GADD153, p53, and p38. The selenite-balanced serum elevated (P < 0.05) mRNA levels of Caspase3 and FoxO1, and the SeMet-balanced serum increased (P < 0.05) mRNA levels of only FoxO1.
FIGURE 4.

Effects of Se-biofortified porcine serum (Se-Y) treatment on relative mRNA levels of cell cycle arrest and apoptosis-related signal proteins of human prostate cancer cells (DU145; A) and colon cancer cells (HTC116; B) compared with the control porcine serum or that serum was added with different Se compounds to match the Se concentration in Se-Y in Expt. 2. Data are means ± SEs, n = 4. Bars without a common letter differ, P < 0.05. MSA, methylseleninic acid; SeMet, seleno-dl-methionine; Se-Y, serum from pigs fed 1.0 mg Se/kg in Expt. 2.
Selenoprotein gene expression.
Among the 18 selenoprotein genes assayed, 8 genes in the DU145 cell line (Fig. 5A) and in the HCT116 cell line (Fig. 5B) each were affected (P < 0.05) by the high-Se serum treatments. In the DU145 cell line, the mRNA levels of 5 selenoprotein genes, including Gpx4, Selt, Gpx1, Sepp1, and Selm, were increased by both Se-Y and the MSA-balanced serum. Moreover, Se-Y increased (P < 0.05) mRNA levels of Sep15 and the MSA-balanced serum increased (P < 0.05) mRNA levels of Sels and Txnnd2. The SeMet-balanced sera increased (P < 0.05) mRNA levels of Sepp1 and SelS. In the HTC116 cell line, mRNA levels of Gpx4, Sels, and Sepn1 were increased by both Se-Y and the MSA-balanced serum. While Se-Y increased (P < 0.05) mRNA levels of Selh, Selk, and Txnrd2, the MSA-balanced serum elevated (P < 0.05) mRNA levels of Selt. Meanwhile, the mRNA levels of 4 selenoprotein genes, including Sepw1, Sels, Txnrd2 and Sepn1, were upregulated (P < 0.05) by both SeMet and selenite balanced sera. In addition, the SeMet-balanced serum also increased (P < 0.05) mRNA levels of Gpx4 and Selt.
FIGURE 5.

Effects of Se-biofortified porcine serum (Se-Y) treatment on relative mRNA levels of multiple selenoproteins of human prostate cancer cells (DU145; A) and colon cancer cells (HTC116; B) compared with the control porcine serum or that serum was added with different Se compounds to match the Se concentration in Se-Y in Expt. 2. Data are means ± SEs, n = 4. Bars without a common letter differ, P < 0.05. MSA, methylseleninic acid; SeMet, seleno-dl-methionine; Se-Y, serum from pigs fed 1.0 mg Se/kg in Expt. 2.
Western-blot analyses.
In the DU145 cells, both S-Y and the MSA-balanced serum resulted in greater (P < 0.05) protein levels of p53, c-Jun, Sepp1, and Sep15 than those of the control serum (Fig. 6A). The MSA-balanced serum decreased (P < 0.05) the proapoptotic protein Bcl-2 protein and the SeMet-balanced serum increased (P < 0.05) only the c-Jun protein. In the HCT116 cells, all 4 types of high-Se sera except for the SeMet-balanced serum induced (P < 0.05) p53 protein production (Fig. 6B). As in the case of DU145 cells, Bcl-2 protein was decreased (P < 0.05) by the MSA-balanced serum.
FIGURE 6.

Effects of Se-biofortified porcine serum (Se-Y) treatment on relative protein production of p53, Bcl-2, c-Jun, Sepp1, and Sep15 of human prostate cancer cells (DU145; A) and colon cancer cells (HTC116l; B) compared with the control porcine serum or that serum was added with different Se compounds to match the Se concentration in Se-Y in Expt. 2. The band was a representative image of 3–4 independent analyses. Values below the protein band were relative density and are expressed as means ± SEs, n = 3–4. Values without a common letter differ, P < 0.05. Bcl-2, B-cell lymphoma 2; c-Jun, cFos-binding protein; MSA, methylseleninic acid; p53, tumor suppressor protein 53; SeMet, seleno-dl-methionine; Sep15, 15 kDa selenoprotein; Sepp1, selenoprotein P; Se-Y, serum from pigs fed 1.0 mg Se/kg in Expt. 2.
Stepwise regression analysis of gene correlations.
Using the selenoprotein gene expression profiles as the independent variables and the pertaining mRNA levels of cell cycle arrest/apoptosis genes in the cells treated with the control, Se-Y, and MSA-balanced sera as the dependent variables, we conducted step-wise regression analysis for the responses of each cell cycle arrest/apoptosis gene (Supplemental Table 3). In the DU145 cells, high correlation with selenoprotein gene expression was found for Caspase3 (R2 = 0.96), GADD153 (R2 = 0.88), c-Jun (R2 = 0.78), Cdc25A (R2 = 0.77), and CyclinA (R2 = 0.73). Reciprocally, the expression of 7 selenoprotein genes, including Sep15, Selt, Gpx4, Gpx1, Sepp1, Selm and Txnrd2, were correlated with 7, 5, 3, 2, 2, 1, and 1 cell cycle arrest/apoptosis genes, respectively. In the HCT116 cells, CyclinA (R2 = 1.00), GADD153 (R2 = 1.00), FoxO1 (R2 = 1.00), and p38 (R2 = 0.93) had strong correlation (P < 0.02) with selenoprotein gene expression. Meanwhile, 6 selenoprotein genes, including Selh, Selk, Gpx4, Sels, Txnrd2, and Sepn1, were correlated with 3, 1, 1, 1, 1, and 1 cell cycle arrest/apoptosis genes, respectively.
Discussion
The two most novel findings from the present study were: 1) porcine serum could be biofortified with Se by feeding growing pigs with high dietary levels of sodium selenite or Se-enriched yeast; and 2) the Se-biofortified porcine sera effectively inhibited the growth and survival of 3 types of human cancer cells. Apparently, unique Se-related molecules or metabolites, rather than simply the Se content, in the Se-biofortified sera were responsible for this remarkable effectiveness. This is because elevating the Se concentration in the control sera with several selenocompounds other than MSA to the Se-Y level did not duplicate the antiproliferation action in any of the cell lines. The effective Se doses (<1 μmol/L) shown by the Se-biofortified serum were much lower than those of previously reported selenite (5–400 μmol/L) and SeMet or Se-enriched yeast (30–130 μmol/L) on various cancer cell lines (Supplemental Table 4) (10, 16, 30–35). Because these Se doses, in the form of sodium selenite or organic forms other than MSA, had no effect on the viability of epithelial DU145 and HCT116 cells or nonepithelial HepG2 cells compared with the respective culture media, the observed responses were not due to Se toxicity per se. In fact, previous research indicated that Se at these doses exerted no growth inhibition effect on primary or noncancerous cells (36, 37). Although Se-biofortified broccoli and milk were shown to protect rats against the induced formation of primary colon cancer (38, 39), our results reveal Se bio-fortified porcine serum as a novel form of Se in suppressing the growth of 3 types of human cancer cells at extremely low concentrations that are nutritionally or physiologically relevant. If the efficacy, safety, and utilities, including stability to resist heat inactivation during food-processing, are verified in future animal and human studies, the Se-enriched porcine serum may be used as a new treatment for cancer reoccurrence. The treatment will be available and convenient to the public, because porcine serum is a food ingredient in many parts of the world and the effective Se concentration in the porcine serum is extremely low.
The major potential functional form of Se in the Se-biofortified porcine serum shared similar characteristics with MSA in Expt. 2. Both Se-Y and the MSA-balanced serum resulted in similar growth inhibitions in the 2 tested cancer cell lines. In DU145 cells, the 2 sources of Se induced consistent decreases in expression of 4 cell cycle arrest genes (DP1, Cyclin A, Cdk2, and Cdk4) and increases of 3 apoptotic genes (p53, Caspase3, and c-Jun). Meanwhile, they exerted similar regulations on 6 of 8 selenoprotein genes altered by the treatments and 5 signal- and Se-dependent proteins assayed (p53, Bcl-2, c-Jun, Sepp1, and Sep15) in DU145 cells. Likewise, these 2 sources of Se shared similar effects on the expression of 3 cell cycle arrest genes (DP1, Cdc2, and Cyclin A) and 3 selenoprotein genes, along with p53 protein, in HTC116 cells. Seemingly, a large portion of the observed effect of the Se-biofortified porcine serum was mediated by MSA or MSA-like metabolites. As a stable monomethylated Se metabolite, MSA has shown excellent anticancer potency (10, 11). Because of the lack of methioninase or extensive metabolism of Se in the cultured cancer cells or media (41), the added selenite, selenate, SeCys, SeMet, or SeMC in the control porcine serum had no effect on the growth of either DU145 or HTC116 cells. However, Se-Y and the MSA-balanced serum did exhibit different impacts on a number of cell cycle arrest, apoptosis, and selenoprotein genes in both cell lines. One striking difference is that only Se-Y but not MSA upregulated the expression of apoptotic genes (GADD153, p53, and p38) in HTC116 cells. This implies the existence of other functional forms of Se in addition to MSA in Se-Y. In general, mammal tissue Se is categorized as nonprotein bound and protein bound (41). The former consists of intermediate metabolites of Se and constitutes 2–4% of total plasma Se (41), such as methylselenol, which is postulated to be an active anticancer selenocompound (19). The latter contains selenocysteine- and SeMet-containing proteins and Se-binding proteins (13, 19). These proteins constitute 96–98% of total plasma Se (41). Because GPX3 and Sepp1 are the 2 extracellular selenoproteins present in blood, the contributions of the 72% higher GPX3 activity in Se-Na and 56% higher Sepp1 activity in Se-Y than their respective control serum to their overall antiproliferation effect (42) should be assessed in future research. Similarly, systematic characterizations of nonprotein-bound and protein-bound Se metabolites in the Se-biofortified serum will help identify chemical forms of the most effective components of Se for its anticancer function.
A combined downregulation of cell cycle arrest genes and upregulation of apoptotic genes appeared to be the common signal mechanism for the Se-biofortified porcine serum in inhibiting proliferations of the 3 types of human cancer cells. The results of Expt. 1 demonstrated accelerated apoptosis and leaking of intracellular LDH in the NCI-H446 cancer cells treated with Se-Na. The enhanced programmed cell death was mediated by an upregulation of a tumor-suppressing protein p53 (43) and p38 (44), along with a downregulation of antiapoptotic inner mitochondrial membrane protein Bcl-2 (45). Involvements of p53 and p38 in the Se-mediated inhibition of prostate cancer cells were previously reported (39, 45). In Expt. 2, Se-Y downregulated the mRNA and/or protein levels of DP1, Cdc25A, Cdc2, Cyclin A, Cdk2, Cdk4, and Bcl-2 but upregulated those of p53, c-Jun, p38, Caspase3, FoxO1, and GADD15 in 1 or 2 cancer cell lines. Early studies have shown downregulation of DP1, Cdc25A, Cdk2, Cdc2, Cyclin A, and Cdk4 and/or upregulation of GADD153 along with cell cycle arrest in various lung, prostate, and breast cancer cells treated with MSA (14–16). Meanwhile, enhanced programmed cell death was mediated by upregulations of p53, p38, and c-Jun in prostate, cervical, or breast cancer cells treated with selenite, SeMet, or MSA (14, 40, 46) and downregulation of Bcl-2 in hepatoma cells treated with by selenium dioxide (47). Although regulated mRNA expression and/or protein production of all these genes by Se-Y were attributed to its cell proliferation inhibition in both DU145 and HTC116 cells, the actual contribution of signal molecules, including Caspase3 and FoxO1, was questionable, because SeMet and selenite affected their mRNA levels but showed no effect on cell viability.
Another new dimension of the present study was the mRNA and/or protein response profiles of 18 selenoprotein genes to the 4 different sources or forms of Se in the 2 human cancer cell lines. Those profiles enabled us to sort out and rank the most important selenoprotein gene expression in the Se-Y and MSA-mediated cell proliferation inhibition signaling via a systematic, stepwise regression analysis. While the control media (containing the control porcine serum) had an Se concentration of 0.42 μmol/L that was not Se deficient, adding Se-Y and MSA-balanced serum collectively upregulated the mRNA and/or protein levels of 8 and 7 selenoproteins in the DU145 and HTC116 cell lines, respectively. Compared with the remaining assayed selenoprotein genes, these differentially regulated genes responded to the Se supply higher than the adequate level and became the primary targets for the antiproliferation mediator of Se. Overall, selenoprotein genes that had a strong association with the cell cycle arrest/apoptosis signaling genes were ranked in the order (high to low) of Sep15, Selt, Gpx4, Gpx1, Sepp1, Selm, and Txnrd2 in DU145 cells and Selh, Gpx4, Selk, Sels, Txnrd2, and Sepn1 in HTC116 cells. Whereas Gpx1 (20, 48), Gpx4 (21), Sepp1 (49), Sels (50), Sep15 (51), and Txnrd2 (40) have been shown to possess anticancer functions (19, 42), the potential involvements of Selt, Selm, Selh, Selk, and Sepn1 in this regard are newly illustrated by us in the present study. Three of these proteins (Selm, Selk, and Sepn1) are residents of endoplasmic reticulum (19), and Selt plays a crucial role in cell growth (52). Although the exact functions of these novel selenoproteins remain to be determined, their strong associations with the Se-Y and MSA-mediated cancer cell death-related signaling offer a new target and strategy for cancer prevention and treatment.
Nevertheless, several seemingly conflicting or inconsistent scenarios were observed in the present study. Although all the selected 7 selenoprotein genes that had a strong correlation with the cell cycle arrest/apoptosis-related signal genes were indeed upregulated by only Se-Y and MSA-balanced serum in DU145 cells, only 2 of 6 such genes (Selh and Selk) in HTC116 cells fell into this category. The remaining 4 genes were also upregulated by the SeMet and selenite-balanced sera that did not produce significantly lower viability in these cancer cells.
These divergences suggest that regulations of selenoprotein genes as well as Se metabolism in the cancerous cells may not only be different from noncancerous cells (36, 37) but may also vary with cancer cell types. Comparatively, modulating functional gene expression of the target selenoproteins may contribute more to the antigrowth action by Se-Y and MSA in the human prostate cancer cells (DU145) than in the human colon cancer cells (HTC116). Meanwhile, another 7 selenoproteins, including Gpx2, Gpx3, Txnrd1, Selw1, Selo, Selv, and Dio1, were previously reported to be involved in the anticancer functions of Se (19, 42). However, none of them was correlated with the inhibition of cell proliferation in either DU145 or HTC116 cells. Intriguingly, we saw an upregulation of Sep15 protein by both Se-Y and the MSA-balanced serum and a strong correlation of Sep15 with 7 cell cycle arrest/apoptosis genes in DU145 cells. In contrast, an earlier study showed that knockdown of Sep15 inhibited colon and prostate cancer cell growth and decreased the likelihood of mice developing colon cancer by influencing the cell cycle (51). This discrepancy in the role of Sep15 may suggest that the anticancer function of individual selenoproteins is not simply unilateral or cumulative but rather conditional. Therefore, the particular matrix of coordinated expressions or responses of multiple selenoproteins, instead of any single selenoprotein status per se, dictates the anticancer effect of a given Se treatment. Overall, our findings are consistent with the view that outcomes of Se supplementation in cancer prevention or treatment depend on not only differential regulation of selenoprotein gene expression but also cell type, cancer type, and stage of carcinogenesis (5, 22, 53).
Supplementary Material
Acknowledgments
The authors thank Courtney Mills, Zhixiong Pan, Karl Roneker, Krystal Lum, Ji-Chang Zhou, Li-Wu Zhong, Jia-Yong Tang, Gang Jia, Li-Zhi Wang, Ling Huang, Xia Li, Yang Can, and Lian Tang for their technical assistance. X.G.L. and K.-N.W. designed the research; L.-H.S., J.-G.L., H.Z., J.S., J.Q.H. X.-J.X, and L.L. conducted the experiments and analyzed the data; L.-H.S., J.-G.L., and X.G.L. wrote the paper; and X.G.L. had primary responsibility for the final content. All authors read and approved the final manuscript.
Footnotes
Abbreviations used: AKT2, RAC-beta serine/threonine-protein kinase; Bcl-2, B-cell lymphoma 2; Cdc2, cyclin-dependent kinase 1; Cdc25A, cell division cycle 25 homolog A; Cdk2, cyclin-dependent kinase 2; Cdk4, cyclin-dependent kinase 4; c-Jun, cFos-binding protein; Dio1, deiodinase, iodothyronine, type I; DP1, transcription factor Dp-1; FoxO1, forkhead box O1; GADD153, growth arrest and DNA damage-inducible gene 153; Gpx1, cytosolic glutathione peroxidase; Gpx2, gastro-intestinal glutathione peroxidase; Gpx3, plasma glutathione peroxidase; Gpx4, phospholipidhydroperoxide glutathione peroxidase; HPRT, hypoxanthine-guanine phosphoribosyltransferase; LDH, lactate dehydrogenase; MSA, methylseleninic acid; p19, cyclin-dependent kinase inhibitor 2A; p21, cyclin-dependent kinase inhibitor 1; p38, mitogen-activated protein kinase p38; p53, tumor suppressor protein 53; SeCys, seleno-dl-cystine; Selh, selenoprotein H; Selk, selenoprotein K; Selm, selenoprotein M; Selo, selenoprotein O; Se-Na, serum from pigs fed 3.0 mg Se/kg in Expt. 1; Sels, selenoprotein S; Selt, selenoprotein T; Selv, selenoprotein V; SeMC, Se-methylseleno-l-cysteine; SeMet, seleno-dl-methionine; Sep15, 15 kDa selenoprotein; Sepn1, selenoprotein N; Sepp1, selenoprotein P; Sepw1, selenoprotein W; Se-Y, serum from pigs fed 1.0 mg Se/kg in Expt. 2; Txnrd1, thioredoxin reductase-1; Txnrd2, thioredoxin reductase-2.
Literature Cited
- 1.Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, Cooper D, Gansler T, Lerro C, Fedewa S, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62:220–41. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19:1893–907. [DOI] [PubMed] [Google Scholar]
- 3.Clark LC, Combs GF, Jr, Turnbull BW, Slate EH, Chalker DK, Chow J, Davis LS, Glover RA, Graham GF, Gross EG, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. JAMA. 1996;276:1957–63. [PubMed] [Google Scholar]
- 4.Duffield-Lillico AJ, Dalkin BL, Reid ME, Turnbull BW, Slate EH, Jacobs ET, Marshall JR, Clark LC. Selenium supplementation, baseline plasma selenium status and incidence of prostate cancer: an analysis of the complete treatment period of the Nutritional Prevention of Cancer Trial. BJU Int. 2003;91:608–12. [DOI] [PubMed] [Google Scholar]
- 5.Irons R, Carlson BA, Hatfield DL, Davis CD. Both selenoproteins and low molecular weight selenocompounds reduce colon cancer risk in mice with genetically impaired selenoprotein expression. J Nutr. 2006;136:1311–7. [DOI] [PubMed] [Google Scholar]
- 6.Stranges S, Marshall JR, Natarajan R, Donahue RP, Trevisan M, Combs GF, Cappuccio FP, Ceriello A, Reid ME. Effects of long-term selenium supplementation on the incidence of type 2 diabetes: a randomized trial. Ann Intern Med. 2007;147:217–23. [DOI] [PubMed] [Google Scholar]
- 7.Stranges S, Laclaustra M, Ji C, Cappuccio FP, Navas-Acien A, Ordovas JM, Rayman M, Guallar E. Higher selenium status is associated with adverse blood lipid profile in British adults. J Nutr. 2010;140:81–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schrauzer GN. Selenomethionine: a review of its nutritional significance, metabolism and toxicity. J Nutr. 2000;130:1653–6. [DOI] [PubMed] [Google Scholar]
- 9.Patterson JK, Lei XG, Miller DD. The pig as an experimental model for elucidating the mechanisms governing dietary influence on mineral absorption. Exp Biol Med (Maywood). 2008;233:651–64. [DOI] [PubMed] [Google Scholar]
- 10.Siwek B, Bahbouth E, Serra MA, Sabbioni E, de Pauw-Gillet MC, Bassleer R. Effect of selenium compounds on murine B16 melanoma cells and pigmented cloned pB16 cells. Arch Toxicol. 1994;68:246–54. [DOI] [PubMed] [Google Scholar]
- 11.Ip C, Thompson HJ, Zhu Z, Ganther HE. In vitro and in vivo studies of methylseleninic acid: evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000;60:2882–6. [PubMed] [Google Scholar]
- 12.Ip C, Dong Y, Ganther HE. New concepts in selenium chemoprevention. Cancer Metastasis Rev. 2002;21:281–9. [DOI] [PubMed] [Google Scholar]
- 13.Sinha R, El-Bayoumy K. Apoptosis is a critical cellular event in cancer chemoprevention and chemotherapy by selenium compounds. Curr Cancer Drug Targets. 2004;4:13–28. [DOI] [PubMed] [Google Scholar]
- 14.Dong Y, Ganther HE, Stewart C, Ip C. Identification of molecular targets associated with selenium-induced growth inhibition in human breast cells using cDNA microarrays. Cancer Res. 2002;62:708–14. [PubMed] [Google Scholar]
- 15.Swede H, Dong Y, Reid M, Marshall J, Ip C. Cell cycle arrest biomarkers in human lung cancer cells after treatment with selenium in culture. Cancer Epidemiol Biomarkers Prev. 2003;12:1248–52. [PubMed] [Google Scholar]
- 16.Dong Y, Zhang H, Hawthorn L, Ganther HE, Ip C. Delineation of the molecular basis for selenium-induced growth arrest in human prostate cancer cells by oligonucleotide array. Cancer Res. 2003;63:52–9. [PubMed] [Google Scholar]
- 17.Zhang H, Fang J, Yao D, Wu Y, Ip C, Dong Y. Activation of FOXO1 is critical for the anticancer effect of methylseleninic acid in prostate cancer cells. Prostate. 2010;70:1265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigó R, Gladyshev VN. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–43. [DOI] [PubMed] [Google Scholar]
- 19.Davis CD, Tsuji PA, Milner JA. Selenoproteins and cancer prevention. Annu Rev Nutr. 2012;32:73–95. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Hinkhouse MM, Sun W, Weydert CJ, Ritchie JM, Oberley LW, Cullen JJ. Redox regulation of pancreatic cancer cell growth: role of glutathione peroxidase in the suppression of the malignant phenotype. Hum Gene Ther. 2004;15:239–50. [DOI] [PubMed] [Google Scholar]
- 21.Heirman I, Ginneberge D, Brigelius-Flohé R, Hendrickx N, Agostinis P, Brouckaert P, Rottiers P, Grooten J. Blocking tumor cell eicosanoid synthesis by GP x 4 impedes tumor growth and malignancy. Free Radic Biol Med. 2006;40:285–94. [DOI] [PubMed] [Google Scholar]
- 22.Novoselov SV, Calvisi DF, Labunskyy VM, Factor VM, Carlson BA, Fomenko DE, Moustafa ME, Hatfield DL, Gladyshev VN. Selenoprotein deficiency and high levels of selenium compounds can effectively inhibit hepatocarcinogenesis in transgenic mice. Oncogene. 2005;24:8003–11. [DOI] [PubMed] [Google Scholar]
- 23.Zhou JC, Zhao H, Li JG, Xia XJ, Wang KN, Zhang YJ, Liu Y, Zhao Y, Lei XG. Selenoprotein gene expression in thyroid and pituitary of young pigs is not affected by dietary selenium deficiency or excess. J Nutr. 2009;139:1061–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Zhao H, Zhang Q, Tang J, Li K, Xia XJ, Wang KN, Li K, Lei XG. Prolonged dietary selenium deficiency or excess does not globally affect selenoprotein gene expression and/or protein production in various tissues of pigs. J Nutr. 2012;142:1410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeng MS, Li X, Liu Y, Zhao H, Zhou JC, Li K, Huang JQ, Sun LH, Tang JY, Xia XJ, et al. A high-selenium diet induces insulin resistance in gestating rats and their offspring. Free Radic Biol Med. 2012;52:1335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yasuda K, Dawson HD, Wasmuth EV, Roneker CA, Chen C, Urban JF, Welch RM, Miller DD, Lei XG. Supplemental dietary inulin influences expression of iron and inflammation related genes in young pigs. J Nutr. 2009;139:2018–23. [DOI] [PubMed] [Google Scholar]
- 27.Todor DR, Lewis I, Bruno G, Chyatte D. Identification of a serum gelatinase associated with the occurrence of cerebral aneurysms as pro-matrix metalloproteinase-2. Stroke. 1998;29:1580–3. [DOI] [PubMed] [Google Scholar]
- 28.Babson AL, Babson SR. Kinetic colorimetric measurement of serum lactate dehydrogenase activity. Clin Chem. 1973;19:766–9. [PubMed] [Google Scholar]
- 29.Beilstein MA, Whanger PD. Effects of vitamin B-6 deficiency on selenium metabolism in the rat. J Nutr. 1989;119:1962–72. [DOI] [PubMed] [Google Scholar]
- 30.Ganther HE. Selenium metabolism, selenoproteins and mechanisms of cancer prevention: complexities with thioredoxin reductase. Carcinogenesis. 1999;20:1657–66. [DOI] [PubMed] [Google Scholar]
- 31.Redman C, Scott JA, Baines AT, Basye JL, Clark LC, Calley C, Roe D, Payne CM, Nelson MA. Inhibitory effect of selenomethionine on the growth of three selected human tumor cell lines. Cancer Lett. 1998;125:103–10. [DOI] [PubMed] [Google Scholar]
- 32.Baines A, Taylor-Parker M, Goulet AC, Renaud C, Gerner EW, Nelson MA. Selenomethionine inhibits growth and suppresses cyclooxygenase-2 (COX-2) protein expression in human colon cancer cell lines. Cancer Biol Ther. 2002;1:370–4. [PubMed] [Google Scholar]
- 33.Zhao H, Whitfield ML, Xu T, Botstein D, Brooks JD. Diverse effects of methylseleninic acid on the transcriptional program of human prostate cancer cells. Mol Biol Cell. 2004;15:506–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang C, Wang Z, Ganther H, Lü J. Distinct effects of methylseleninic acid versus selenite on apoptosis, cell cycle, and protein kinase pathways in DU145 human prostate cancer cells. Mol Cancer Ther. 2002;1:1059–66. [PubMed] [Google Scholar]
- 35.Guan L, Huang F, Li Z, Han B, Jiang Q, Ren Y, Yang Y, Xu C. P53 transcription- independent activity mediates selenite-induced acute promyelocytic leukemia NB4 cell apoptosis. BMB Rep. 2008;41:745–50. [DOI] [PubMed] [Google Scholar]
- 36.Menter DG, Sabichi AL, Lippman SM. Selenium effects on prostate cell growth. Cancer Epidemiol Biomarkers Prev. 2000;9:1171–82. [PubMed] [Google Scholar]
- 37.Zeng H, Briske-Anderson M, Wu M, Moyer MP. Methylselenol, a selenium metabolite, plays common and different roles in cancerous colon HCT116 cell and noncancerous NCM460 colon cell proliferation. Nutr Cancer. 2012;64:128–35. [DOI] [PubMed] [Google Scholar]
- 38.Finley JW, Davis CD, Feng Y. Selenium from high selenium broccoli protects rats from colon cancer. J Nutr. 2000;130:2384–9. [DOI] [PubMed] [Google Scholar]
- 39.Hu Y, McIntosh GH, Le Leu RK, Woodman R, Young GP. Suppression of colorectal oncogenesis by selenium-enriched milk proteins: apoptosis and K-ras mutations. Cancer Res. 2008;68:4936–44. [DOI] [PubMed] [Google Scholar]
- 40.Zhao R, Domann FE, Zhong W. Apoptosis induced by selenomethionine and methioninase is superoxide mediated and p53 dependent in human prostate cancer cells. Mol Cancer Ther. 2006;5:3275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Combs GF, Jr, Clark LC, Turnbull BW. An analysis of cancer prevention by selenium. Biofactors. 2001;14:153–9. [DOI] [PubMed] [Google Scholar]
- 42.Zhuo P, Diamond AM. An analysis of molecular mechanisms by which selenoproteins affect cancer risk and progression. Cancer prevention by selenium. Biochim Biophys Acta. 2009;1790:1546–54. [DOI] [PMC free article] [PubMed]
- 43.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. [DOI] [PubMed] [Google Scholar]
- 44.Berra E, Diaz-Meco MT, Moscat J. The activation of p38 and apoptosis by the inhibition of Erk is antagonized by the phosphoinositide 3-kinase/Akt pathway. J Biol Chem. 1998;273:10792–7. [DOI] [PubMed] [Google Scholar]
- 45.Hockenbery D, Nuñez G, Milliman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–6. [DOI] [PubMed] [Google Scholar]
- 46.Rudolf E, Rudolf K, Cervinka M. Selenium activates p53 and p38 pathways and induces caspase-independent cell death in cervical cancer cells. Cell Biol Toxicol. 2008;24:123–41. [DOI] [PubMed] [Google Scholar]
- 47.Wei Y, Cao X, Ou Y, Lu J, Xing C, Zheng R. SeO(2) induces apoptosis with down-regulation of Bcl-2 and up-regulation of P53 expression in both immortal human hepatic cell line and hepatoma cell line. Mutat Res. 2001;490:113–21. [DOI] [PubMed] [Google Scholar]
- 48.Nasr MA, Fedele MJ, Esser K, Diamond AM. GPx-1 modulates Akt and P70S6K phosphorylation and Gadd45 levels in MCF-7 cells. Free Radic Biol Med. 2004;37:187–95. [DOI] [PubMed] [Google Scholar]
- 49.Gonzalez-Moreno O, Boque N, Redrado M, Milagro F, Campion J, Endermann T, Takahashi K, Saito Y, Catena R, Schomburg L, et al. Selenoprotein-P is down-regulated in prostate cancer, which results in lack of protection against oxidative damage. Prostate. 2011;71:824–34. [DOI] [PubMed] [Google Scholar]
- 50.Shibata T, Arisawa T, Tahara T, Ohkubo M, Yoshioka D, Maruyama N, Fujita H, Kamiya Y, Nakamura M, Nagasaka M, et al. SEPS1) gene -105G>A promoter polymorphism influences the susceptibility to gastric cancer in the Japanese population. BMC Gastroenterol. 2009;9:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsuji PA, Naranjo-Suarez S, Carlson BA, Tobe R, Yoo MH, Davis CD. Deficiency in the 15 kDa selenoprotein inhibits human colon cancer cell growth. Nutrients. 2011;3:805–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanguy Y, Falluel-Morel A, Arthaud S, Boukhzar L, Manecka DL, Chagraoui A, Prevost G, Elias S, Dorval-Coiffec I, Lesage J, et al. The PACAP-regulated gene selenoprotein T is highly induced in nervous, endocrine, and metabolic tissues during ontogenetic and regenerative processes. Endocrinology. 2011;152:4322–35. [DOI] [PubMed] [Google Scholar]
- 53.Hu Y, McIntosh GH, Le Leu RK, Young GP. Selenium-enriched milk proteins and selenium yeast affect selenoprotein activity and expression differently in mouse colon. Br J Nutr. 2010;104:17–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.